

Abstract
Bisphenol AF (BPAF), an endocrine disrupting chemical, can induce estrogenic activity through binding to estrogen receptor (ER). However, the metabolism of BPAF in vivo and the estrogenic activity of its metabolites remain unknown. In the present study, we identified four metabolites including BPAF diglucuronide, BPAF glucuronide (BPAF-G), BPAF glucuronide dehydrated and BPAF sulfate in the urine of Sprague-Dawley (SD) rats. BPAF-G was further characterized by nuclear magnetic resonance (NMR). After treatment with a single dose of BPAF, BPAF was metabolized rapidly to BPAF-G, as detected in the plasma of SD rats. Biotransformation of BPAF to BPAF-G was confirmed with human liver microsomes (HLM), and Vmax of glucuronidation for HLM was 11.6 nmol/min/mg. We also found that BPAF glucuronidation could be mediated through several human recombinant UDP-glucuronosyltransferases (UGTs) including UGT1A1, UGT1A3, UGT1A8, UGT1A9, UGT2B4, UGT2B7, UGT2B15 and UGT2B17, among which UGT2B7 showed the highest efficiency of glucuronidation. To explain the biological function of BPAF biotransformation, the estrogenic activities of BPAF and BPAF-G were evaluated in ER-positive breast cancer T47D and MCF7 cells. BPAF significantly stimulates ER-regulated gene expression and cell proliferation at the dose of 100 nM and 1 μM in breast cancer cells. However, BPAF-G did not show any induction of estrogenic activity at the same dosages, implying that formation of BPAF-G is a potential host defense mechanism against BPAF. Based on our study, biotransformation of BPAF to BPAF-G can eliminate BPAF-induced estrogenic activity, which is therefore considered as reducing the potential threat to human beings.
Introduction
With a similar structure to the synthetic estrogen bisphenol A (BPA), bisphenol AF (4,4´-hexafluoroisopropylidene-2-diphenol, BPAF) is used primarily as a monomer for polyimides, polyamides, polyesters and other specialty polymers and as a cross linker for certain fluoroelastomers [1,2]. In 2008, BPAF was nominated by the National Institute of Environmental Health Sciences (NIEHS) for comprehensive toxicological characterization based on its moderate production [1]. The presence of BPAF was reported in the environmental samples collected around a manufacturing plant which is one of the largest BPAF manufacturers in China [3]. It has been well-documented that BPAF could bind strongly to estrogen receptor (ER) in vitro, and function as an endocrine disrupting chemical (EDC) by acting as an agonist for ERα and antagonist of ERβ to mediate physiological processes [4-6]. BPAF also could activate the human pregnane X receptor, a nuclear receptor that functions as a regulator of xenobiotic metabolism [7]. Our recent study demonstrated that BPAF can cause reduction of testosterone by directly affecting testis function in adult male rats [8].
UDP-glucuronosyltransferases (UGTs) that were constitutively expressed in a tissue-specific manner play a key role in the glucuronide biotransformation of xenobiotics and endogenous substances [9-11]. It has been reported that UGTs were closely associated with the metabolism and toxicity of BPA in mammals [12,13]. The major metabolite of BPA is the monoglucuronide conjugate in the urine and plasma of rats [13]. In the in vitro metabolism studies, BPA could be metabolized to BPA glucuronide by UGT2B1 in rat liver microsomes [14,15] and by human recombinant UGT isoforms [11]. Moreover, BPA also could be metabolized to 3-hydroxy BPA and BPA o-quinone by cytochrome P450s in vitro [16,17]. Recently, Schmidt et al reported that P450 could mediate biotransformation of BPAF to hydroxylated BPAF, followed by the central carbon bridge degradation which product 4-hexafluorohydroxyisopropylidene-phenol as the main metabolite in the presence of human liver microsomes (HLM) with NADPH and GSH in vitro [18]. However, the biotransformation of BPAF in vivo and the estrogenic effect of its metabolites remain unknown.
The information on potential toxicities, metabolism, environmental presence and environmental fate of BPAF is limited. It is important to understand BPAF’s biotransformation to better estimate the potential threat to human beings. Therefore, our aim is to identify and characterize the metabolites of BPAF both in vivo and in vitro. Moreover, we evaluate the estrogenic activities of BPAF and its metabolites to explain the biological function of BPAF biotransformation in vivo.
Materials and Methods
Chemicals and reagents
BPAF (98% purity) was purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan); HPLC grade acetonitrile and methanol (MeOH) were purchased from Fisher Scientific (Fair Lawn, NJ, USA); and corn oil and DMSO-d6 was obtained from Sigma Aldrich (St. Louis, MO, USA). UGT reaction mix solution A, UGT reaction mix solution B, NADPH reaction buffer, pooled HLM and human recombinant UGTs (UGT1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 1A10, 2B4, 2B7, 2B15 and 2B17) were obtained from BD Biosciences (San Jose, CA, USA).
Animal studies
Male Sprague-Dawley (SD) rats 8 weeks of age (250-300 g) were purchased from the experimental animal center of the Academy of Military Medical Sciences, China. Animals were housed in steel metabolism cages for acclimation 3 days before the experiment. All rats were allowed free access to standard rat chow and water. Due to the limited solubility in saline solution, BPAF was dissolved in corn oil at the concentration of 100 mg/mL for animal experiments. A dose of 200 mg/kg BPAF was administered daily by oral gavage for 2 consecutive weeks. Total urine samples were collected and pooled everyday, and samples were chilled on ice.
For evaluating the metabolites of BPAF in plasma, 6 rats randomly divided into two dose groups were orally administrated by a single dose of 20 mg/kg or 100 mg/kg BPAF. 100 µL blood samples were collected at 0.5, 1, 2, 3, 6, 12, 24 and 48 h in heparinized tubes by direct puncture of the tail vein, chilled immediately on ice and centrifuged for 10 min at 3,000 g at 4 °C. The resulting plasma was stored at -20 °C until analysis. All of the experiments procedures using animals were approved by Animal Care and Use Committee of Beijing Center for Disease Control and Prevention.
Isolation and Purification of BPAF metabolites
Because of the lack of commercial standards for BPAF metabolites, four metabolites were isolated and purified from the urine of SD rats by semi-preparative HPLC as described in detail in Method S1 in File S1.
UPLC/ESI-QTOF-MS analysis
Metabolites of BPAF were identified by ultra-performance liquid chromatography/electrospray ionization quadrupole time-of-flight mass spectrometry (UPLC/ESI-QTOF-MS). Frozen urine samples were thawed, vortex-mixed, then centrifuged at 7,000 g for 10 min. 5 μL raw urine was directly injected into the UPLC. Liquid chromatographic separations were conducted using a Waters Acquity UPLCTM system (Milford, MA, USA) with a BEH C18 column (2.1 mm ×100 mm; particle size, 1.7 µm) from Waters (Milford, MA, USA). The UPLC conditions for separating metabolites are described in Method S2 in File S1.
The MSE analysis, which provided information on the intact and fragment ions of products and metabolites in a single sample injection, was performed on a Waters SynaptTM Q-TOF mass spectrometer (Milford, MA, USA) under negative electrospray ionization condition in V-mode. The capillary, sampling cone and extraction cone voltages were set to 3.1 kV, 35 V and 4 V, respectively. Source and desolvation temperatures were held at 110 °C and 400 °C, respectively. The cone gas and desolvation gas used was nitrogen, and they were set at 50 L/h and 800 L/h, respectively. Their functions were run for data acquisition in centroid mode with m/z 50-1,000. For MS scan, trap collision energy was set to 6.0 eV, 20 eV, and 30 eV. An external reference solution containing 1 mg/L of leucine enkephalin (m/z 554.2615) was used for mass lock.
UPLC/ESI-MS/MS analysis
The quantification of BPAF and BPAF-G was conducted by ultra-high-pressure liquid chromatography/electrospray ionization tandem mass spectrometry (UPLC/ESI-MS/MS) in negative ionization mode. 400 μL acetonitrile was added to 100 μL plasma sample. The mixture was sonicated at room temperature for 15 min, then centrifuged at 7,000 g for 10 min to precipitate proteins. The supernatant was dried under a gentle stream of nitrogen, and the residual was reconstituted with 500 μL MeOH/H2O (50/50, v/v) for UPLC/ESI-MS/MS analysis. Liquid chromatographic separations were performed using a Waters Acquity UPLCTM system (Milford, MA, USA) with a BEH C18 column (2.1 mm ×50 mm; particle size, 1.7 µm) from Waters (Milford, MA, USA). The mobile phase was solvents A (methanol) and B (water). With a flow rate of 0.4 mL/min, gradient elution was operated with 20% A, followed by a 4 min linear gradient to 100% A and kept for 2 min. The system was re-equilibrated for 3 min between runs. The MS used was a Xevo triple quadrupole mass spectrometer (Milford, MA, USA). The capillary voltage and source temperature were set at 2.7 kV and 150 °C, respectively. The desolvation temperature and nitrogen flow rate were set at 400 °C and 1,000 L/h, respectively. Argon was used as the collision gas at a flow rate of 0.16 mL/min. The MS/MS acquisition parameters were optimized in ESI negative mode for maximum sensitivity. The quantification of BPAF and BPAF-G was performed by Multiple Reaction Monitoring (MRM) mode, MRM transitions and collision energies (Ecoll) for quantification were 335.2 > 265.0 Ecoll = 25 eV for BPAF, 510.8 > 112.9 Ecoll = 20 eV for BPAF-G; MRM transition and Ecoll for conformation were 335.2 >197.0 Ecoll = 35 eV for BPAF, 510.8 > 265.0 Ecoll = 40 eV for BPAF-G. Quantification of BPAF and BPAF-G was based on six-point matrix-fortified curves obtained after addition of known amounts of BPAF and BPAF-G to blank plasma samples from untreated control rats. Calibration curves were linear from 1 µg/L to 250 µg/L for BPAF and from 10 µg/L to 2500 µg/L for BPAF-G. The correlation coefficients (r 2) of the matrix-fortified calibration curves were all greater than 0.99. Method accuracy evaluated by spiked recovery at three levels (n = 5) were ranged from 82.8% to 106.7%, with precision (RSD %) no more than 10.9%. The method limits of quantization (MLOQ) of BPAF and BPAF-G in the plasma were 1 µg/L and 10 µg/L, respectively. More details were described and summarized in Method S3 and Table S1 in File S1.
In vitro metabolism
UGT activity assay followed the general procedure in the data sheets provided by BD Biosciences with minor modifications. Briefly, 10 µL pooled HLM (5 mg/mL) or individual human recombinant UGT (5 mg/mL) was pre-incubated with 100 µL UGT reaction mix solution B and 340 µL purified water at 37°C for 5 min in a dry bath incubator. Reactions were initiated by the addition of substrates containing 40 µL UGT reaction mix solution A and 10 µL BPAF (500 µM). Vmax represents the maximum velocity achieved by UGT reaction system at the saturating concentrations of all substrates. For hydrolysis assay, 2 µL BPAF (5 mM) was incubating with 15 µL HLM (5 mg/mL) in the presence of NADPH systems, including 713 µL purified water, 200 µL potassium phosphate (0.5 M and PH 7.4), 50 µL NADPH regenerating solution A and 20 µL NADPH regenerating solution B. Reactions were stopped by adding equal volume of acetonitrile to the reaction mixture. After centrifugation at 10,000 g for 3 min, supernatant samples after 1,000-fold dilution by MeOH/H2O (50:50, v/v) were analyzed as described above.
Cell culture
T47D and MCF7 cells were purchased from Cell Bank (Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences) and cultured in RPMI-1640 medium supplemented with 10% (v/v) fetal bovine serum (FBS) (Gibco) at 37°C and 5% CO2/95% air (v/v). At 70% confluence, the medium was changed to RPMI-1640 medium supplemented with 10% charcoal-stripped fetal bovine serum (SERANA, Australia) for 3 days before treatment in order to minimize the estrogen activity from FBS. BPAF was dissolved in DMSO at a concentration of 1 M as a stock solution, and working solutions were prepared freshly by diluting the stock in cell culture medium.
RNA isolation and real-time PCR analysis
Total RNA from breast cancer cells was extracted using the RNeasy Mini Kit in accordance with the manufacturer’s protocol (QIAGEN). 1 µg total RNA was reverse-transcribed using using Superscript reverse transcriptase (Promega). Primers for Cathepsin D (CTSD) were 5’-CAGCCAGGCATCACCTTCAT-3’ and 5’-CAGGTAGAAGGAGAAGATGT-3’, primers for growth regulation by estrogen in breast cancer 1 (GREB1) were 5’-ATTGGTGGACCGATTGCTCA-3’ and 5’-GCTGATGAGGGTGTGCTGTGT-3’, primers for trefoil factor 1 (TFF1) were 5’-GTGCAAATAAGGGCTGCTGTT-3’ and 5’-CACACTCCTCTTCTGGAGGGA-3’ and primers for GAPDH were 5’-CACCCACTCCTCCACCTTTGA-3’ and 5’-ACCACCCTGTTGCTGTAGCCA-3’. The reactions were performed using Power SYBR Green PCR Master Mix (Applied Biosystems) in an ABI Prism 7300 Sequence Detection system for 40 cycles (95 °C 15 sec, 60 °C for 60 sec) after an initial 10 min incubation at 95 °C. The fold change in the expression of each gene was calculated using 2-ΔΔCt method.
Cell proliferation assay
Cell proliferation was determined by the WST-1 dye (#C0038 Beyotime, China). Briefly, T47D and MCF7 cells were seeded in 96-well plate at the concentration of 3,000 cells per well. After treatment with BPAF or BPAF-G for 7 days, cell proliferation was determined using WST-1 according to the manufacturer’s instruction.
Statistical analysis
Statistical analysis was performed using one-way ANOVA followed by a Tukey-Kramer multiple comparison test. A p value < 0.05 was considered statistically significant.
Results
Identification and characterization of the metabolites of BPAF in SD rats
To identify BPAF metabolites, we performed full-scan UPLC/ESI-QTOF-MS to analyze the possible metabolites by comparing the chromatogram and mass spectrum data of BPAF-treated urine samples (Figure 1A) with those of untreated control samples (Figure 1B). Four metabolites with the retention times of 2.89 min, 7.68 min, 8.44 min and 9.37 min, marked as M1, M2, M3 and M4, were identified in the urine of BPAF-treated SD rats. The electrospray spectra of the four metabolites were analyzed by the MSE mode of UPLC/ESI-QTOF-MS (Figure 1C). The most polar metabolite, M1, yielded a m/z 686.9947, corresponding to the diglucuronide of BPAF ([M−H] −). The fragmentation of this ion led to the formation of ions at m/z 510.9669 and m/z 334.9472, resulting from the cleavage of either one or two glucuronic acid moiety from the precursor ion. Thus, this metabolite was identified as BPAF diglucuronide. The metabolite M2 gave an [M−H] − ion at m/z 510.9673. The fragmentation of this ion resulted in the loss of the glucuronic acid moiety and led to the formation of the m/z 334.9468. The fragment m/z 264.9511 corresponded to the loss of CF3 from the BPAF moiety. Fragments m/z 174.9404 and m/z 112.9536 corresponded to the glucuronic acid and the loss of CO2 and H2O from glucuronic acid moiety. Thus, the metabolite characterized as a glucuronide conjugation of BPAF was considered as BPAF glucuronide (BPAF-G), which produced the strongest responses among the four metabolites in the UPLC/ESI-QTOF-MS. The metabolite M3 had an [M−H] − ion at m/z 492.9583 with a retention time of 8.44 min and generated similar fragments to M2. The only different ion was found at m/z 156.9341, corresponding to the loss of H2O from the glucuronic acid moiety. M3 was characterized as BPAF glucuronide dehydrated, but its structure could not be determined because of the uncertainty of the dehydration position on the glucuronic acid ring. The precursor ion of M4 was [M−H] − at m/z 414.8993. The product ions m/z 334.9478 and 79.8173 corresponded to the BPAF moiety and sulfuric acid, enabling the precursor ion to be identified as the sulfate conjugation of BPAF. The data and structure of four metabolites and BPAF analyzed by mass spectrometry are summarized in Figure 1D.
Figure 1. Identification of the metabolites of BPAF in vivo.

SD rats were orally administrated with 200 mg/kg/day of BPAF for 2 weeks. We analyzed the metabolites of BPAF in the urine of BPAF-treated SD rats (A) and control rats (B) by UPLC/ESI-QTOF-MS. TIC, total ion chromatogram. (C) UPLC/ESI-QTOF-MSE electrospray spectra of BPAF diglucuronide conjugation (M1), BPAF glucuronide (M2), BPAF glucuronide dehydrated (M3) and BPAF sulfate (M4) in the urine of BPAF-treated SD rats. (D) Mass spectrometric data and structure of BPAF metabolites and BPAF.
Because standards of BPAF metabolites were not commercially available, we isolated and purified the metabolites from the urine of SD rats to further confirm the structural characterization by nuclear magnetic resonance (NMR). Due to the limited amount of M1 in the urine, amounts of M2, M3 and M4 components were sufficient for NMR analysis. However, M3 and M4 components collected with poor purity could not be applied for NMR. Only the M2 component was further characterized by 1H and 13C NMR (Figure S1 in File S1). In the 1H and 13C NMR spectra, the resonances for carbons and protons of the aglycone moiety had a close resemblance to those of BPAF [19,20]. In addition, the characteristic 1H and 13C NMR data of glucuronic acid with signals for an anomeric proton (δ5.06, 1H, d) and a carboxylic group at C-6″ at δ171.0, were present. The full assignments of carbon and proton signals are summarized in Table S2 in File S1. Our data finally verified that M2 component was BPAF-4-O-glucuronide.
To further evaluate the biotransformation of BPAF in vivo, we also detected the concentrations of BPAF and BPAF-G in the plasma of SD rats using UPLC/ESI-MS/MS. After treatment with a single dose of 20 mg/kg or 100 mg/kg BPAF, the peak of BPAF-G in plasma was observed at 30 min followed by rapid decline in the next three hours, representing quick clearance of BPAF in the rats (Figure 2A). However, the peak of BPAF was achieved at 1 h and eliminated completely at 48 h (Figure 2B). We also found a non-significant decline of BPAF-G concentration from the 6 h to 24 h and the second peak of BPAF at 12 h, which may be as a result of enterohepatic circulation. Moreover, we observed that the maximum concentration of BPAF-G is about 30-40-fold more than that of BPAF. Our data indicated that biotransformation of BPAF to BPAF-G occurred rapidly in vivo. However, M1, M3 and M4 were not observed in the plasma of rats exposed with 100 mg/kg BPAF. Moreover, after isolation and purification of the four metabolites from the urine, M2 was the only metabolite available for NMR analysis indicating its high abundance in the urine. Taken together, BPAF-G is therefore considered as the major metabolite in vivo.
Figure 2. Biotransformation of BPAF in the plasma of SD rats.
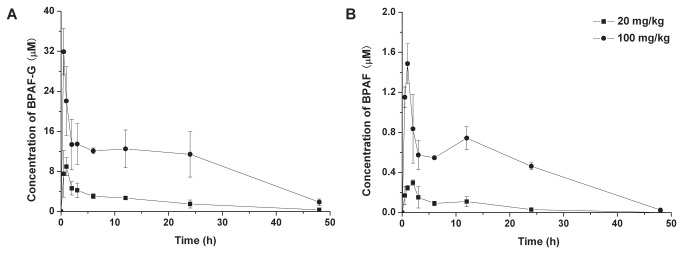
(A) The concentrations of BPAF-G in the plasma. (B) The concentrations of BPAF in the plasma. A single dose of 20 mg/kg or 100 mg/kg of BPAF was administered orally to SD rats, the concentrations of BPAF and BPAF-G in the plasma were detected by UPLC/ESI-MS/MS at the indicated time point. Data represent mean ± SD, n = 3.
Biotransformation of BPAF in vitro
Full-scan UPLC/ESI-QTOF-MS was performed to check the possible metabolites of BPAF in the incubation with HLM. BPAF-G was the only metabolite identified after incubation for 90 min (Figure S2 in File S1). Then, we quantified the concentrations of BPAF and BPAF-G in the glucuronidation reaction system using UPLC/ESI-MS/MS. We observed that the amount of BPAF decrease is equal to that of BPAF-G increase for the indicated time (Table 1), suggesting that BPAF is transformed completely to BPAF-G by HLM in the presence of UGT reaction buffer. The Vmax of BPAF glucuronidation for HLM was 11.6 nmol/min/mg of microsomal protein.
Table 1. The concentrations of BPAF and BPAF-G in the incubation with HLM.
| Time (min) | BPAF (µM) | BPAF-G (µM) |
|---|---|---|
| 0 | 9.9 ± 0.26 | 0 |
| 1 | 8.80 ± 0.32 | 1.22 ± 0.08 |
| 2 | 7.79 ± 0.25 | 2.34 ± 0.15 |
| 5 | 4.21 ± 0.34 | 5.66 ± 0.40 |
| 10 | 1.21 ± 0.10 | 8.72 ± 0.69 |
| 15 | 0.91 ± 0.03 | 9.37 ± 0.61 |
| 20 | 0.31 ± 0.01 | 9.52 ± 0.84 |
| 30 | 0.11 ± 0.01 | 9.89 ± 0.84 |
Data represent mean ± SD, n = 3
In addition, we observed that HLM could catalyze the formation of hydroxylated BPAF in the presence of NADPH (Figure 3A), which is consistent with Schmidt et al’s report [18]. However, the rate of hydroxylation is obviously slower than that of glucuronidation, because the response value of BPAF did not change substantially from 0 to 90 min (Figure 3B). These results suggest that glucuronidation was more efficient than hydroxylation for BPAF biotransformation.
Figure 3. Biotransformation BPAF to hydroxylated BPAF by HLM.
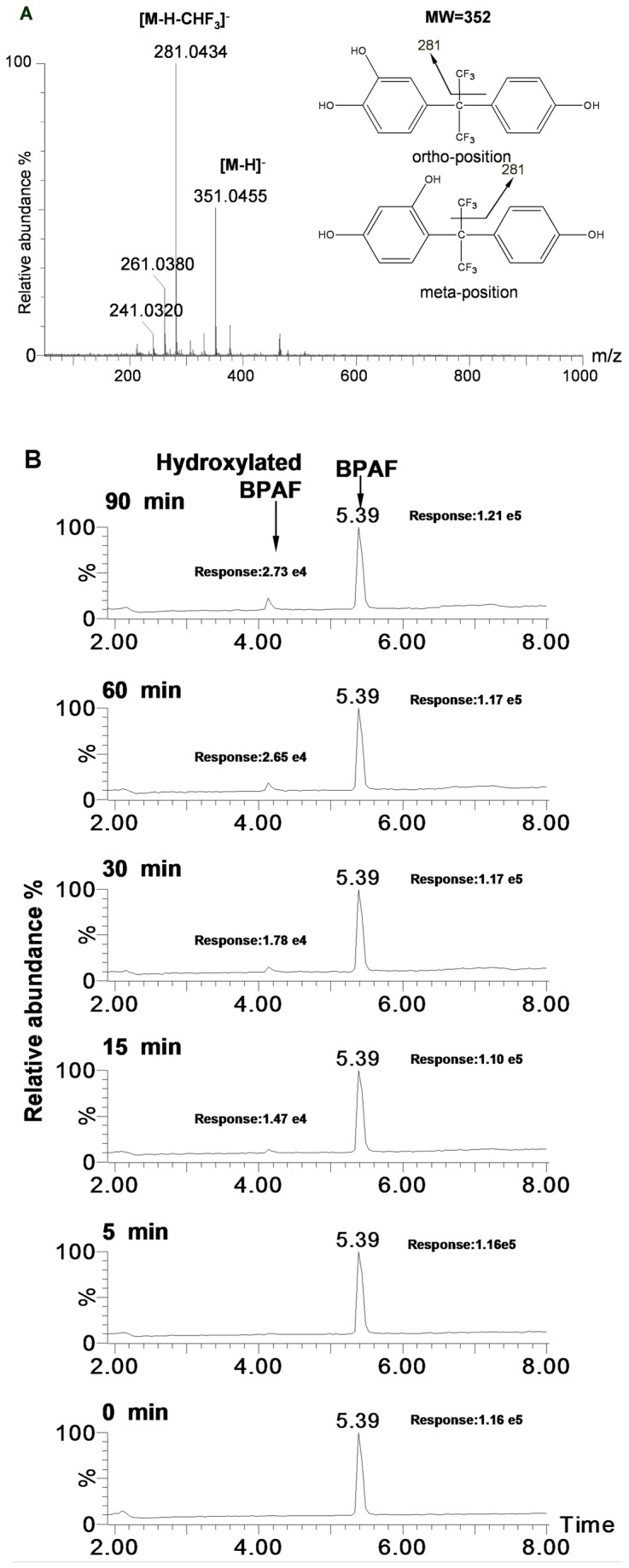
(A) Identification of hydroxylated BPAF. Typical mass spectrum of BPAF metabolite that yielded in the presence of NADPH was identified as hydroxylated BPAF. (B) Chromatograms of BPAF and hydroxylated BPAF at indicated time. BPAF (10 µM) was incubated with HLM (100 µg/mL) in the presence of NADPH reaction buffer, the samples were collected at indicated time points from the reaction mixture, both BPAF and hydroxylated BPAF were detected by UPLC/ESI-QTOF-MS.
Next, we also investigated whether glucuronidation of BPAF is regulated by UGT isoforms, which is the specific enzyme family responsible for the process of glucuronidation [21]. As shown in Figure 4, several UGTs were capable of mediating BPAF biotransformation, and the glucuronidation efficiency of UGTs for BPAF is UGT2B7 > UGT1A3 > UGT2B15 and UGT1A9 > UGT2B17 and UGT1A1 > UGT1A8 and UGT2B4. However, we did not detect any generation of BPAF-G in the reaction mix with the other UGT isoforms including UGT1A4, UGT1A6, UGT1A7 and UGT1A10. The capability of UGTs examined to catalyze BPAF glucuronidation (Vmax of UGT2B7 is 1.25 nmol/min/mg) is over 9 times lower than that of HLM, suggesting that other enzymes existed in HLM could facilitate BPAF glucuronidation.
Figure 4. Biotransformation of BPAF to BPAF-G by human recombinant UGTs.

BPAF (10 µM) was incubated with 12 individual human recombinant UGT and UGT reaction buffer, the samples were collected at 20 min and detected by UPLC/ESI-MS/MS. Data present mean ± SD of three independent experiments. ND = not detected.
Estrogenic activities of BPAF and BPAF-G
To determine whether biotransformation could modulate estrogenic activity of BPAF, we evaluated the estrogenic activities of BPAF and its major metabolite BPAF-G via gene expression (Figure 5) and cell proliferation (Figure 6) in ER-positive breast cancer T47D and MCF7 cells. Regulation of estrogenic activities is a multifactorial and complex process, involving both genomic and nongenomic actions. In the classical genomic action, ERα-regulated transcription occurs in a ligand-dependent manner that ERα binds to the estrogen responsive elements (EREs) within the promoter proximal regions of known estrogen-responsive genes, such as CTSD, GREB1 and TFF1 [22]. Expression of the lysosomal protease CTSD is induced by ERα via proximal promoter region and distal ERE binding sites located at 9 and 33 kb upstream of the transcription start site [22]. Regulation of GREB1 expression, which plays a role in estrogen-induced proliferation of breast cancer cells, is mediated by binding of ERα to three consensus EREs spread over approximately 20 kb of upstream flanking sequences of GREB1 [23]. Transcription of TFF1 is commonly used to evaluate the estrogenic activity in human breast cancers [5]. In the present study, expression of ER-regulated genes was measured by real-time PCR. BPAF significantly induced expression of CTSD, GREB1 and TFF1 genes in a dose-dependent manner at concentrations of 100 nM to 1 μM in both T47D (Figure 5A-C) and MCF7 (Figure 5D-F) cells. In contrast, BPAF-G did not stimulate ER-regulated gene expression compared to BPAF. In consistence with gene expression, proliferations of T47D (Figure 6A) and MCF7 (Figure 6B) cells were stimulated by BPAF at the dose of 100 nM and 1 μM, but not by BPAF-G at the same doses. It is believed that BPAF-G does not show any estrogenic activity in ER-positive breast cancer cells. Therefore, biotransformation of BPAF to BPAF-G completely eliminated BPAF-induced estrogenic activities, which implies that formation of BPAF-G is a potential defense mechanism against BPAF toxicity in vivo.
Figure 5. Effects of BPAF and BPAF-G on gene expression in T47D and MCF7 cells.

mRNA levels of CTSD (A, D), GREB1 (B, E) and TFF1 (C, F) were measured by real-time PCR in T47D (A, B, C) and MCF7 (D, E, F) cells after treatment with different concentrations of BPAF and BPAF-G for 24 h. Data present mean ± SD of three independent experiments. ** p < 0.01 compared with control.
Figure 6. Effects of BPAF and BPAF-G on the proliferation in T47D and MCF7 cells.

Proliferation of T47D (A) and MCF7 (B) cells was measured at day 7 after treatment with different concentrations of BPAF and BPAF-G. Data present mean ± SD of three independent experiments. ** p < 0.01 compared with control.
Discussion
Increasing evidence shows that exposure to BPAF can cause adverse effects both in animal models and in vitro [6,8,24]. In an effort to better understand the health effects of BPAF to humans, this study was undertaken to clarify the biotransformation of BPAF under physiological conditions. Four metabolites, including diglucuronide conjugated (M1), glucuronide conjugated (M2), glucuronide dehydrated (M3) and sulfate conjugated (M4), were identified in the urine of SD rats. M1, M2 and M3 were related to glucuronidation indicating that glucuronidation is an important reaction for BPAF metabolism. Among the four metabolites, BPAF-G was the only metabolite detected in the plasma of SD rats administrated with a single dose of BPAF, implying that M1 and M3 may be the byproducts or reactive intermediates produced during BPAF glucuronidation. Sulfate conjugation (M4) is also detected, suggesting that there are other metabolism pathways and different enzymes responsible for BPAF biotransformation in vivo. However, the value of mass spectrometry response for M4 was substantially lower than that for M2, which suggests that sulfate conjugation is not likely the major pathway for BPAF biotransformation in vivo.
Chemicals conjugated with glucuronic acid or sulfonate, which are known as Phase II reactions, tend to be less active than their parent chemicals. Biotransformation of BPAF to glucuronide and sulfate conjugation indicated that metabolic Phase II conjugation of BPAF does occur in vivo. Several studies on the biotransformation of BPA have been carried out both in vivo and in vitro [15,17,25-27]. BPA glucuronide was characterized as the major metabolite of BPA, while other metabolites such as BPA diglucuronide, BPA sulfate conjugate, and 5-hydroxybisphenol A were identified as well [28]. Based on previous studies and our results, both BPA and BPAF have the metabolites with glucuronide, diglucuronide and sulfate conjugation, which suggests that BPAF has similar metabolic pathway to BPA in vivo.
EDCs have been extensively studied for their potential adverse health effects, including infertility, birth defects, development and progression of breast cancer and islet disruption [29-33]. The rate of BPAF glucuronidation has implications for predicting the potential threat of BPAF in humans. According to our study, biotransformation of BPAF occurred quickly in vivo, which resulted in the lower level of BPAF compared to BPAF-G in plasma. Therefore, additional experiments are necessary to explore the toxicology of BPAF-G, which may reflect a more authentic burden on BPAF exposure.
UGT family plays an important role in the chemical reaction of glucuronidation [21]. It has been reported that UGT2B1 is mainly responsible for glucuronidation of BPA in rat liver microsomes [14]. Human UGT2B7 shares similar sequence homology to rat UGT2B1. Therefore, we speculated that BPAF could be metabolized to BPAF-G by human UGT2B7. Our results showed that human UGT2B7 has the highest efficiency for BPAF glucuronidation among the 12 human UGTs tested. This result suggests that the effects of BPAF in human beings may depend on the presence or absence of the UGT isoforms corresponding to UGT2B7 expressed in the liver, kidney and small intestine [34], and that overexpression of UGT2B7 could decrease the BPAF burden in humans. Expression of UGT2B7 in fetal liver is much lower than that in the adults [35], which suggests that the threat of BPAF to fetus can be more severe than that to adults, because BPAF is unable to be metabolized in the fetus following the maternal exposure. On the other hand, the glucuronidation rate of UGT2B7 is markedly lower than that of HLM, suggesting other UGT isoforms with higher efficiency of BPAF glucuronidation than UGT2B7 could exist in HLM.
Metabolism could play an essential role in modulating the estrogenic activities of EDCs [17,36]. It has been reported that metabolism of BPA to BPA glucuronide will decrease BPA-induced estrogenic potency [28]. According to our results, BPAF-G did not show any estrogenic activation compared to BPAF, indicating that BPAF could be metabolized to the inactive form, which is similar to BPA. These results suggest that glucuronidation by UGT family in vivo may serve as a general defensive mechanism against BPAF and other EDCs.
Chronic exposure to multiple EDCs is considered as a cause of increasing the risk of developing breast cancer [37-39]. Experimental studies showed that BPA could stimulate carcinogenesis [39,40] and promote the proliferation of cancer cells by activating the ER [41,42]. BPA can also induce tumor aggressiveness in clinical tissue of breast cancer patients [43]. Since BPAF exhibits more estrogenic potency than BPA [4,44], BPAF should pose more severe threats to human beings, especially to hormone-sensitive tumor patients. According to our study, biotransformation of BPAF to BPAF-G could be a general mechanism to decrease BPAF-induced threat to humans.
In the present study, we identified four metabolites of BPAF including BPAF diglucuronide, BPAF glucuronide (BPAF-G), BPAF glucuronide dehydrated and BPAF sulfate in the urine of SD rats. After determination of BPAF metabolites in the plasma of SD rats, we found that BPAF glucuronidation is a rapid and efficient pathway for the biotransformation of BPAF, and BPAF-G was considered as a major metabolite in vivo. In addition, biotransformation of BPAF to BPAF-G was also confirmed in the incubation with HLM as well as UGTs, including UGT1A1, UGT1A3, UGT1A8, UGT1A9, UGT2B4, UGT2B7, UGT2B15 and UGT2B17 in vitro. In ER-positive breast cancer T47D and MCF7 cells, ER-regulated gene expression and cell proliferation were stimulated by BPAF, not by its metabolite BPAF-G. These results clearly show that biotransformation of BPAF to BPAF-G could eliminate BPAF-induced estrogenic activity, implying that biotransformation of BPAF has a key biological significance to decrease the potential risk to humans, especially to estrogen-sensitive cancer patients. Our findings thus provide a possible defensive mechanism against BPAF in vivo.
Supporting Information
Figure S1, NMR spectrum of BPAF-G. Figure S2, Identification of BPAF metabolites in the incubation with HLM and UGT reaction mixture. Table S1, Method validation for BPAF and BPAF-G. Table S2, NMR data of BPAF-G (DMSO-d6). Method S1, Isolation and purification of BPAF metabolites. Method S2, UPLC conditions for UPLC/ESI-QTOF-MS analysis. Method S3, Method validation for BPAF and BPAF-G.
(DOC)
Acknowledgments
The authors thank Dr. Bin Zhao (Research Center for Eco-environmental Sciences, Chinese Academy of Sciences) for critical reading of the manuscript.
Funding Statement
This work was supported by National Natural Science Foundation of China (grant number 41273132 and 21207008, http://www.nsfc.gov.cn/Portal0/default166.htm) and the Beijing Municipal Senior Technical Training Plan for the Health System. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. National Toxicology Program (NTP) (2008). Available: http://ntp.niehs.nih.gov/ntp/htdocs/Chem_Background/ExSumPdf/BisphenolAF_093008_508.pdf#search=BisphenolAF. Accessed 10 July 2013
- 2. LaFleur AD, Schug KA (2011) A review of separation methods for the determination of estrogens and plastics-derived estrogen mimics from aqueous systems. Anal Chim Acta 696 (1): 6-26. PubMed: 21621029. [DOI] [PubMed] [Google Scholar]
- 3. Song S, Ruan T, Wang T, Liu R, Jiang G (2012) Distribution and preliminary exposure assessment of Bisphenol AF (BPAF) in various environmental matrices around a manufacturing plant in China. Environ Sci Technol 46: 13136-13143. doi: 10.1021/es303960k. PubMed: 23151244. [DOI] [PubMed] [Google Scholar]
- 4. Matsushima A, Liu X, Okada H, Shimohigashi M, Shimohigashi Y (2010) Bisphenol AF is a full agonist for the estrogen receptor ERalpha but a highly specific antagonist for ERbeta. Environ Health Perspect 118 (9): 1267-1272. doi: 10.1289/ehp.0901819. PubMed: 20427257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li Y, Luh CJ, Burns KA, Arao Y, Jiang Z et al. (2013) Endocrine-Disrupting Chemicals (EDCs): In Vitro Mechanism of Estrogenic Activation and Differential Effects on ER Target Genes. Environ Health Perspect 121 (4): 459-466. PubMed: 23384675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li Y, Burns KA, Arao Y, Luh CJ, Korach KS (2012) Differential estrogenic actions of endocrine-disrupting chemicals bisphenol A, bisphenol AF, and zearalenone through estrogen receptor alpha and beta in vitro. Environ Health Perspect 120 (7): 1029-1035. doi: 10.1289/ehp.1104689. PubMed: 22494775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sui Y, Ai N, Park SH, Rios-Pilier J, Perkins JT et al. (2012) Bisphenol A and its analogues activate human pregnane X receptor. Environ Health Perspect 120 (3): 399-405. doi: 10.1289/ehp.1104426. PubMed: 22214767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feng Y, Yin J, Jiao Z, Shi J, Li M et al. (2012) Bisphenol AF may cause testosterone reduction by directly affecting testis function in adult male rats. Toxicol Lett 211 (2): 201-209. doi: 10.1016/j.toxlet.2012.03.802. PubMed: 22504055. [DOI] [PubMed] [Google Scholar]
- 9. Ritter JK (2000) Roles of glucuronidation and UDP-glucuronosyltransferases in xenobiotic bioactivation reactions. Chem Biol Interact 129 (1-2): 171-193. doi: 10.1016/S0009-2797(00)00198-8. PubMed: 11154740. [DOI] [PubMed] [Google Scholar]
- 10. Sneitz N, Krishnan K, Covey DF, Finel M (2011) Glucuronidation of the steroid enantiomers ent-17beta-estradiol, ent-androsterone and ent-etiocholanolone by the human UDP-glucuronosyltransferases. J Steroid Biochem Mol Biology 127 (3-5): 282-288. doi: 10.1016/j.jsbmb.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Doerge DR, Twaddle NC, Woodling KA, Fisher JW (2010) Pharmacokinetics of bisphenol A in neonatal and adult rhesus monkeys. Toxicol Appl Pharmacol 248 (1): 1-11. doi: 10.1016/j.taap.2010.07.009. PubMed: 20655935. [DOI] [PubMed] [Google Scholar]
- 12. Kang JH, Katayama Y, Kondo F (2006) Biodegradation or metabolism of bisphenol A: from microorganisms to mammals. Toxicology 217 (2): 81-90. [DOI] [PubMed] [Google Scholar]
- 13. Pottenger LH, Domoradzki JY, Markham DA, Hansen SC, Cagen SZ et al. (2000) The relative bioavailability and metabolism of bisphenol A in rats is dependent upon the route of administration. Toxicol Sci 54 (1): 3-18. doi: 10.1093/toxsci/54.1.3. PubMed: 10746927. [DOI] [PubMed] [Google Scholar]
- 14. Yokota H, Iwano H, Endo M, Kobayashi T, Inoue H et al. (1999) Glucuronidation of the environmental oestrogen bisphenol A by an isoform of UDP-glucuronosyltransferase, UGT2B1, in the rat liver. Biochem J 340 (2): 405-409. doi: 10.1042/0264-6021:3400405. PubMed: 10333482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yoshihara S, Makishima M, Suzuki N, Ohta S (2001) Metabolic activation of bisphenol A by rat liver S9 fraction. Toxicol Sci 62 (2): 221-227. doi: 10.1093/toxsci/62.2.221. PubMed: 11452134. [DOI] [PubMed] [Google Scholar]
- 16. Niwa T, Fujimoto M, Kishimoto K, Yabusaki Y, Ishibashi F et al. (2001) Metabolism and interaction of bisphenol A in human hepatic cytochrome P450 and steroidogenic CYP17. Biol Pharm Bull 24 (9): 1064-1067. doi: 10.1248/bpb.24.1064. PubMed: 11558570. [DOI] [PubMed] [Google Scholar]
- 17. Yoshihara Si Mizutare T, Makishima M, Suzuki N, Fujimoto N et al. (2004) Potent estrogenic metabolites of bisphenol A and bisphenol B formed by rat liver S9 fraction: their structures and estrogenic potency. Toxicol Sci 78 (1): 50-59. doi: 10.1093/toxsci/kfh047. PubMed: 14691209. [DOI] [PubMed] [Google Scholar]
- 18. Schmidt J, Kotnik P, Trontelj J, Knez Z, Mašič LP (2013) Bioactivation of bisphenol A and its analogs (BPF, BPAF, BPZ and DMBPA) in human liver microsomes. Toxicology in Vitro 27 (4): 1267-1276. doi: 10.1016/j.tiv.2013.02.016. PubMed: 23470418. [DOI] [PubMed] [Google Scholar]
- 19. Arewång CJ, Lahmann M, Oscarson S, Tidén AK (2007) Synthesis of urine drug metabolites: glucuronic acid glycosides of phenol intermediates. Carbohydr Res 342 (7): 970-974. doi: 10.1016/j.carres.2007.01.014. PubMed: 17324385. [DOI] [PubMed] [Google Scholar]
- 20. Hayasaka T, Katsuhara Y, Kume T, Yamazaki T (2011) HF-mediated equilibrium between fluorinated ketones and the corresponding α-fluoroalcohols. Tetrahedron 67 (12): 2215-2219. doi: 10.1016/j.tet.2011.01.087. [DOI] [Google Scholar]
- 21. Tang L, Ye L, Singh R, Wu B, Lv C et al. (2010) Use of glucuronidation fingerprinting to describe and predict mono-and dihydroxyflavone metabolism by recombinant UGT isoforms and human intestinal and liver microsomes. Mol Pharmaceutics 7 (3): 664-679. doi: 10.1021/mp900223c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR et al. (2006) Genome-wide analysis of estrogen receptor binding sites. Nat Genet 38 (11): 1289-1297. doi: 10.1038/ng1901. PubMed: 17013392. [DOI] [PubMed] [Google Scholar]
- 23. Deschênes J, Bourdeau V, White JH, Mader S (2007) Regulation of GREB1 transcription by estrogen receptor α through a multipartite enhancer spread over 20 kb of upstream flanking sequences. J Biol Chem 282 (24): 17335-17339. doi: 10.1074/jbc.C700030200. PubMed: 17463000. [DOI] [PubMed] [Google Scholar]
- 24. Lee S, Kim YK, Shin T-Y, Kim S-H (2013) Neurotoxic Effects of Bisphenol AF on Calcium-Induced ROS and MAPKs. Neurotox Res 23 (3): 249-259. PubMed: 22996013. [DOI] [PubMed] [Google Scholar]
- 25. Doerge DR, Twaddle NC, Vanlandingham M, Brown RP, Fisher JW (2011) Distribution of bisphenol A into tissues of adult, neonatal, and fetal Sprague-Dawley rats. Toxicol Appl Pharmacol 255 (3): 261-270. doi: 10.1016/j.taap.2011.07.009. PubMed: 21820460. [DOI] [PubMed] [Google Scholar]
- 26. Fisher JW, Twaddle NC, Vanlandingham M, Doerge DR (2011) Pharmacokinetic modeling: prediction and evaluation of route dependent dosimetry of bisphenol A in monkeys with extrapolation to humans. Toxicol Appl Pharmacol 257 (1): 122-136. doi: 10.1016/j.taap.2011.08.026. PubMed: 21920375. [DOI] [PubMed] [Google Scholar]
- 27. Upmeier A, Degen GH, Diel P, Michna H, Bolt HM (2000) Toxicokinetics of bisphenol A in female DA/Han rats after a single i.v. and oral administration. Arch Toxicol 74 (8): 431-436. doi: 10.1007/s002040000144. PubMed: 11097379. [DOI] [PubMed] [Google Scholar]
- 28. Elsby R, Maggs JL, Ashby J, Park BK (2001) Comparison of the modulatory effects of human and rat liver microsomal metabolism on the estrogenicity of bisphenol A: implications for extrapolation to humans. J Pharmacol Exp Ther 297 (1): 103-113. PubMed: 11259533. [PubMed] [Google Scholar]
- 29. Vandenberg LN, Maffini MV, Sonnenschein C, Rubin BS, Soto AM (2009) Bisphenol-A and the great divide: a review of controversies in the field of endocrine disruption. Endocr Rev 30 (1): 75-95. PubMed: 19074586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alonso-Magdalena P, Morimoto S, Ripoll C, Fuentes E, Nadal A (2006) The estrogenic effect of bisphenol A disrupts pancreatic β-cell function in vivo and induces insulin resistance. Environ Health Perspect 114 (1): 106-112. PubMed: 16393666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ben-Jonathan N, Steinmetz R (1998) Xenoestrogens: the emerging story of bisphenol A. Trends Endocrinol Metab 9 (3): 124-128. doi: 10.1016/S1043-2760(98)00029-0. PubMed: 18406253. [DOI] [PubMed] [Google Scholar]
- 32. Davis DL, Webster P, Stainthorpe H, Chilton J, Jones L et al. (2007) Declines in sex ratio at birth and fetal deaths in Japan, and in US whites but not African Americans. Environ Health Perspect 115 (6): 941-946. doi: 10.1289/ehp.9540. PubMed: 17589604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nadal A, Alonso-Magdalena P, Soriano S, Quesada I, Ropero AB (2009) The pancreatic β-cell as a target of estrogens and xenoestrogens: Implications for blood glucose homeostasis and diabetes. Mol Cell Endocrinol 304 (1): 63-68. [DOI] [PubMed] [Google Scholar]
- 34. Ohno S, Nakajin S (2009) Determination of mRNA expression of human UDP-glucuronosyltransferases and application for localization in various human tissues by real-time reverse transcriptase-polymerase chain reaction. Drug Metab Dispos 37 (1): 32-40. doi: 10.1124/dmd.108.023598. PubMed: 18838504. [DOI] [PubMed] [Google Scholar]
- 35. King CD, Rios GR, Assouline JA, Tephly TR (1999) Expression of UDP-glucuronosyltransferases (UGTs) 2B7 and 1A6 in the human brain and identification of 5-hydroxytryptamine as a substrate. Arch Biochem Biophys 365 (1): 156-162. doi: 10.1006/abbi.1999.1155. PubMed: 10222050. [DOI] [PubMed] [Google Scholar]
- 36. Mercado-Feliciano M, Bigsby RM (2008) Hydroxylated metabolites of the polybrominated diphenyl ether mixture DE-71 are weak estrogen receptor-α ligands. Environ Health Perspect 116 (10): 1315-1321. doi: 10.1289/ehp.11343. PubMed: 18941571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Safe SH (1997) Xenoestrogens and breast cancer. N Engl J Med 337 (18): 1303-1304. doi: 10.1056/NEJM199710303371809. PubMed: 9345081. [DOI] [PubMed] [Google Scholar]
- 38. Soto AM, Vandenberg LN, Maffini MV, Sonnenschein C (2008) Does breast cancer start in the womb? Basic Clin Pharmacol Toxicol 102 (2): 125-133. doi: 10.1111/j.1742-7843.2007.00165.x. PubMed: 18226065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Weber Lozada K, Keri RA (2011) Bisphenol A increases mammary cancer risk in two distinct mouse models of breast cancer. Biol Reprod 85 (3): 490-497. doi: 10.1095/biolreprod.110.090431. PubMed: 21636739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Keri RA, Ho S-M, Hunt PA, Knudsen KE, Soto AM et al. (2007) An evaluation of evidence for the carcinogenic activity of bisphenol A. Reprod Toxicol 24 (2): 240-252. doi: 10.1016/j.reprotox.2007.06.008. PubMed: 17706921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Diel P, Olff S, Schmidt S, Michna H (2002) Effects of the environmental estrogens bisphenol A, o,p'-DDT, p-tert-octylphenol and coumestrol on apoptosis induction, cell proliferation and the expression of estrogen sensitive molecular parameters in the human breast cancer cell line MCF-7. J Steroid Biochem Mol Biology 80 (1): 61-70. doi: 10.1016/S0960-0760(01)00173-X. [DOI] [PubMed] [Google Scholar]
- 42. Olsen CM, Meussen-Elholm ET, Samuelsen M, Holme JA, Hongslo JK (2003) Effects of the environmental oestrogens bisphenol A, tetrachlorobisphenol A, tetrabromobisphenol A, 4-hydroxybiphenyl and 4, 4'-dihydroxybiphenyl on oestrogen receptor binding, cell proliferation and regulation of oestrogen sensitive proteins in the human breast cancer cell line MCF7. Pharmacol Toxicol 92 (4): 180-188. doi: 10.1034/j.1600-0773.2003.920408.x. PubMed: 12753421. [DOI] [PubMed] [Google Scholar]
- 43. Dairkee SH, Seok J, Champion S, Sayeed A, Mindrinos M et al. (2008) Bisphenol A induces a profile of tumor aggressiveness in high-risk cells from breast cancer patients. Cancer Res 68 (7): 2076-2080. doi: 10.1158/0008-5472.CAN-07-6526. PubMed: 18381411. [DOI] [PubMed] [Google Scholar]
- 44. Kitamura S, Suzuki T, Sanoh S, Kohta R, Jinno N et al. (2005) Comparative study of the endocrine-disrupting activity of bisphenol A and 19 related compounds. Toxicol Sci 84 (2): 249-259. doi: 10.1093/toxsci/kfi074. PubMed: 15635150. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1, NMR spectrum of BPAF-G. Figure S2, Identification of BPAF metabolites in the incubation with HLM and UGT reaction mixture. Table S1, Method validation for BPAF and BPAF-G. Table S2, NMR data of BPAF-G (DMSO-d6). Method S1, Isolation and purification of BPAF metabolites. Method S2, UPLC conditions for UPLC/ESI-QTOF-MS analysis. Method S3, Method validation for BPAF and BPAF-G.
(DOC)