

Abstract
Given the rise in drug-resistant Streptococcus pneumoniae, there is an urgent need to discover new antimicrobials targeting this pathogen and an equally urgent need to characterize new drug targets. A promising antibiotic target is dihydrodipicolinate synthase (DHDPS), which catalyzes the rate-limiting step in lysine biosynthesis. In this study, we firstly show by gene knock out studies that S. pneumoniae (sp) lacking the DHDPS gene is unable to grow unless supplemented with lysine-rich media. We subsequently set out to characterize the structure, function and stability of the enzyme drug target. Our studies show that sp-DHDPS is folded and active with a k cat = 22 s-1, K M PYR = 2.55 ± 0.05 mM and K M ASA = 0.044 ± 0.003 mM. Thermal denaturation experiments demonstrate sp-DHDPS exhibits an apparent melting temperature (T M app) of 72 °C, which is significantly greater than Escherichia coli DHDPS (Ec-DHDPS) (T M app = 59 °C). Sedimentation studies show that sp-DHDPS exists in a dimer-tetramer equilibrium with a K D 4→2 = 1.7 nM, which is considerably tighter than its E. coli ortholog (K D 4→2 = 76 nM). To further characterize the structure of the enzyme and probe its enhanced stability, we solved the high resolution (1.9 Å) crystal structure of sp-DHDPS (PDB ID 3VFL). The enzyme is tetrameric in the crystal state, consistent with biophysical measurements in solution. Although the sp-DHDPS and Ec-DHDPS active sites are almost identical, the tetramerization interface of the s. pneumoniae enzyme is significantly different in composition and has greater buried surface area (800 Å2) compared to its E. coli counterpart (500 Å2). This larger interface area is consistent with our solution studies demonstrating that sp-DHDPS is considerably more thermally and thermodynamically stable than Ec-DHDPS. Our study describe for the first time the knock-out phenotype, solution properties, stability and crystal structure of DHDPS from S. pneumoniae, a promising antimicrobial target.
Introduction
Streptococcus pneumoniae is a Gram-positive bacterium and human commensal inhabiting the upper respiratory tract [1]. The organism often causes pneumonia in children, the elderly and immunocompromized, and if left untreated can result in death [2]. Pneumonia accounts for the death of approximately 1 million children per annum under the age of 5, making this disease the leading cause of childhood mortality worldwide [2,3]. In recent years S. pneumoniae has received considerable attention due to the emergence of multi-drug resistant strains, commonly referred to as drug-resistant Streptococcus pneumoniae (DRSP) [3,4]. Standard antimicrobial treatment options, such as the β-lactam and macrolide antibiotics, are becoming less effective due to the rise in DRSP [4-7]. This is in part due to the promiscuous nature of S. pneumoniae in acquiring genetic resistance elements from other bacteria, accompanied by selective pressure as a result of high antibiotic usage [8,9]. Alongside the use of antibiotic agents to combat infection, vaccination is available as a preventative measure [10]; however, current pneumococcal vaccines do not offer protection against all infectious strains. Thus there is an urgent need to discover new therapeutics targeting appropriate biomolecules from S. pneumoniae.
A promising antimicrobial target is the enzyme dihydrodipicolinate synthase (DHDPS), which catalyzes the first committed step in the lysine biosynthetic pathway of bacteria, namely the condensation of pyruvate and (S)-aspartate semialdehyde [(S)-ASA] to form the product, hydroxytetrahydrodipicolinic acid (HTPA) (Figure 1A) [11-14]. Humans do not synthesize lysine de novo and thus acquire this essential amino acid from dietary sources; whereas bacteria, such as S. pneumoniae, synthesize lysine de novo for both protein and cell-wall synthesis [11-14]. The absence of a lysine biosynthetic pathway in humans and the fact that lysine is a fundamental building block of proteins and peptidoglycan in bacteria, highlights the potential for targeting the enzymatic machinery involved in this pathway for novel antibiotic discovery [11-15].
Figure 1. Enzymatic reaction and multiple sequence alignment of DHDPS.
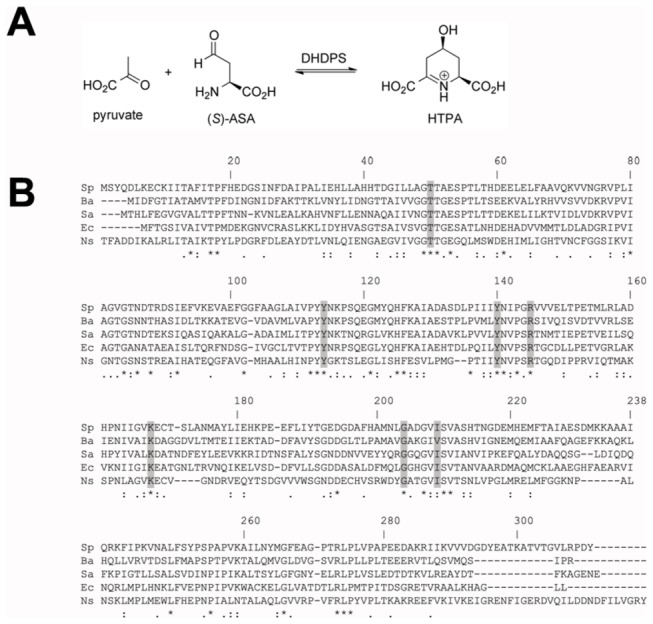
(A) Condensation reaction catalyzed by DHDPS. (B) Multiple sequence alignment of DHDPS sequences from bacteria, namely S. pneumoniae (Sp), B. anthracis (Ba), S. aureus (Sa), and E. coli (Ec), and also the plant species N. sylvestris (Ns). Conserved active-site residues are shaded grey.
To date, almost all characterized DHDPS enzymes, excluding notable exceptions from Staphylococcus aureus [16,17] and Pseudomonas aeruginosa [18,19], adopt a homotetrameric structure [20-36]. Each monomeric unit folds to form a TIM-barrel, or (β/α)8 topology, which subsequently self-associates to form a tetramer or dimer of ‘tight’ dimers [20-36]. Tetramerization of DHDPS is shown to be important for stabilizing conformational dynamics of the ‘tight’ dimer interface where the key active-site residues are located [26,27,36]. These include K161 (E. coli numbering), which forms a Schiff base with the first substrate to bind the enzyme (i.e. pyruvate), and a catalytic triad comprised of Y107, T44 and Y133, which are strongly conserved in all DHDPS enzymes characterized to date [25,31] including S. pneumoniae (Figure 1B).
Given the clinical importance of S. pneumoniae and the rise in multi-drug resistance in this Gram-positive pathogen, the aims of this study were to (i) determine the phenotype of a DHDPS gene knock mutant of S., pneumoniae; (ii) characterize the solution properties, stability and catalytic activity of Sp-DHDPS; and (iii) determine the high-resolution crystal structure of the enzyme to afford structure-based drug design strategies in future studies.
Materials and Methods
Bacterial strains, media, and chemicals
Escherichia coli K-12 TOP10 cells (Invitrogen, Carlsbad, CA), grown in Luria-Bertani (LB) medium, were used for preparation of plasmid DNA. Escherichia coli BL21(DE3) strain grown in LB medium was used for recombinant protein expression. S. pneumoniae 774A, isolated from CSF of a child with meningitis [37] was grown routinely in Brain Heart Infusion (BHI) broth or on Horse Blood Agar (HBA) plates, at 37°C in an atmosphere of 5% CO2. The chemically defined medium with (CDM+), or without (CDM-), (S)-lysine (200 µg ml-1) was that described by van de Rijn and Kessler [38], supplemented with choline chloride (5 µg ml-1), asparagine (50 µg ml-1), and sodium pyruvate 250 µg ml-1) as described by Moscoso et al. [1]. Ampicillin was used at a final concentration of 50 µg ml-1, chloramphenicol at 10 µg ml-1, erythromycin at 0.1 µg ml-1, and 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-Gal) at 25 µg ml-1. For growth studies, cells from freshly grown single colonies on HBA plates were inoculated into 3 ml of CDM+ and 3 ml CDM-. Cultures were incubated statically for 16 hours in an atmosphere of 5% CO2 at 37°C. Growth was assessed by monitoring the optical density (Absorbance) of duplicate cultures on an hourly basis using a Klett-Summerson photoelectric colorimeter (filter no. 54, spectral range 520-580 nm).
Recombinant DNA techniques
Routine DNA manipulations were performed using standard techniques [39]. Plasmid DNA was purified using Wizard plus SV DNA purification system (Promega, Madison, WI). PCR amplifications were performed with Phusion High-Fidelity DNA Polymerase (Finnzymes Oy, Finland) or high-fidelity Platinum Taq DNA polymerase (Invitrogen, Carlsbad, CA). DNA derived from PCR reactions was purified using the UltraClean PCR Clean-up Kit (Mo Bio Laboratories, Inc.). Oligonucleotides were purchased from GeneWorks Pty. Ltd. (Hindmarsh, South Australia, Australia).
Construction of the S. pneumoniae 447A ΔdapA mutant strain
Overlapping extension PCR [40] was used to generate a DNA fragment carrying the erythromycin resistance (EmR) gene flanked by regions up and downstream of S. pneumoniae 447A dapA. Briefly, primer pairs dap.F/dapERM.R and dapERM.F/dap.R (Table 1) were used to amplify DNA flanking the region to be deleted from the chromosome of S. pneumoniae 447A, and primers pVA838.F/pVA838.R (Table 1) were used to amplify the EmR gene from plasmid pVA838 [41]. The products of these three PCR reactions (100 ng each) served as template in overlapping extension PCR using primers dap.F/dap.R (Table 1) to generate a linear construct, which was cloned into pGEM-T Easy (Promega, Madison,WI), introduced into E. coli K-12 TOP10 cells and confirmed by sequencing. The pGEM-T Easy construct was used as a template in a PCR with primers dap.F/dap.R, to amplify the linear allelic replacement DNA fragment, which was introduced into S. pneumoniae 447A by transformation. The ΔdapA mutation was confirmed by PCR using primer pairs in which one primer flanked the targeted region and the other primed within the EmR gene (OCD52/dapERM.R and OCD53/dapERM.F). The PCR products were sequenced using primers OCD52 and OCD53 (Table 1).
Table 1. Sequences of primers employed in the S. pneumoniae dapA knock out experiments.
| Primer | Sequence |
|---|---|
| pVA838.F | CACAAGTGATTTGTGATTGTTG |
| pVA838.R | GCGCTTAGTGGGAATTTGTAC |
| dapERM.F | GTACAAATTCCCACTAAGCGCGTCGTAGATGGCGACTACGAAGC |
| dapERM.R | CAACAATCACAAATCACTTGTGCAATCAAGGCTGGAATAGCATC |
| dap.F | CGAAGAGATGAAGATGACCAAGG |
| dap.R | GAATCAACAACCTCTTCTTTGAAAATGC |
| OCD52 | CACGTGATTTGCATGCGGAA |
| OCD53 | ATCGGTGTTGAGCGTTCGAA |
Transformation of S. pneumoniae 447A
Bacteria were grown in c-CAT medium (1% w/v Casamino acids, 0.5% w/v Tryptone, 0.5% w/v NaCl, 1% w/v Yeast Extract, 16 mM K2HPO4, 0.2 % w/v glucose, 15 μg ml-1 glutamine) at 37°C to OD600 of 0.25-0.30. Cells were diluted 1/10 in 10 ml CTM medium (c-CAT containing 0.2% BSA and 1 mM CaCl2), grown at 37°C to OD600 of 0.10, collected by centrifugation and resuspended in 1 ml of 15% v/v glycerol prepared in CTM adjusted to pH 7.8. 100 μl aliquots of cell suspension were stored at -80°C until required. For transformation, 100 µl of cells were thawed on ice, 1 ml of CTM-pH 7.8 and 100 ng of synthetic competence-stimulating peptide 1 (CSP-1) [42] were added and cells incubated at 37°C for 13 min. DNA was added and cells were incubated at 32°C for 35 min then incubation continued for 3 hours at 37°C. Transformation mixture was plated out on HBA containing 0.1 µg ml-1 erythromycin and incubated overnight at 37°C in an atmosphere of 5% CO2.
Expression and purification of Sp-DHDPS
Recombinant Sp-DHDPS was expressed, purified and assessed by SDS-PAGE and mass spectrometry to be >95% homogeneous as described previously [43].
Coupled kinetics assay
Kinetic studies were conducted using the coupled-assay method as previously described [16,36,44,45]. Data were collected on a Cary UV-Vis spectrophotometer (Varian) connected to a Peltier cell to maintain a constant temperature of 30°C. Assays were performed in 1.5 ml semi-micro acrylic cuvettes with a path length of 10 mm. (S)-ASA was synthesized according to the methods of Roberts et al. [46]. A standard assay contained 20 nM of DHDPS, 250 mM HEPES pH 8.0, 0.2 mM NADPH, varied concentrations of pyruvate and (S)-ASA, 75 µg ml-1 of DHDPR (purified from E. coli) in a final volume of 0.8 ml. Cuvettes were pre-incubated at 30°C for 8 min before the reaction was initiated via the addition of (S)-ASA. Rates were determined from the initial linear portion of the data collected. The background degradation of NADPH was factored into rate calculations and each data point was measured in triplicate within a <10% error margin.
Circular dichroism (CD) spectroscopy
CD spectroscopy experiments were performed on an Aviv Model 420SF CD spectrometer using a 1.0 nm bandwidth. CD spectra of Sp-DHDPS were recorded at a protein concentration of 4.5 µM solubilized in 20 mM Tris-HCl pH 8.0 and 150 mM NaCl. Wavelength scans spanning 195-240 nm were measured using a step size of 0.5 nm with a 2 sec averaging time in a 1 mm stoppered quartz cuvette as reported previously [16,36,47]. The resulting spectra were analyzed using the CONTINLL algorithm and SP43 database employing the CDPRO software package [48,49]. Thermal denaturation experiments were monitored at 222 nm over a temperature range of 4–90°C collecting data at 1°C intervals with a 5 s averaging time. Given that DHDPS requires chaperones to fold [50], the denaturation of DHDPS enzymes is irreversible in vitro, and therefore the apparent melting temperature (T M app), or midpoint of the transition between folded to unfolded state, was determined empirically from the ordinate maximum of the first derivative of the thermal denaturation profile [36].
Analytical ultracentrifugation
Analytical ultracentrifugation (AUC) studies were conducted in a XL-I analytical ultracentrifuge (Beckman Coulter) using 12 mm double sector cells with quartz windows loaded into either an An-60 Ti 4-hole rotor or An-50 Ti 8-hole rotor at a temperature of 20°C. Sp-DHDPS samples for all centrifugation runs were solubilized in 20 mM Tris-HCl pH 8.0 and 150 mM NaCl. For sedimentation velocity experiments, 380 µL of sample and 400 µL of reference (20 mM Tris-HCl pH 8.0 and 150 mM NaCl) were employed and absorbance versus radial profiles were generated at 40,000 rpm in continuous mode using a step size of 0.003 cm and a radial range of 5.8-7.3 cm without averaging. Data were collected at wavelengths of 210 nm (296-740 nM Sp-DHDPS) or 230 nm (4.5 µM Sp-DHDPS). The absorbance versus radii profiles at different time points were fitted to single discrete species or a continuous size-distribution model using the program SEDFIT [51,52] (available from www.analyticalultracentrifugation.com). The program SEDNTRP [53,54] was used to determine the partial specific volume of Sp-DHDPS (0.7475 ml g-1), buffer density (1.005 g ml-1) and buffer viscosity (1.021 cp) at 20°C. For sedimentation equilibrium experiments, 120 µL of reference solution and 100 µL of sample at three initial protein concentrations (i.e. 296 nM, 355 nM and 740 nM) were centrifuged at rotor speeds of 10,000 rpm and 18,000 rpm. Initial absorbance versus radial profiles were measured at 210 nm between 6.8 and 7.2 cm in step mode using a 0.001 cm step size and 3 averages until sedimentation equilibrium was attained (t ~ 24 hours). At sedimentation equilibrium, detailed absorbance versus radius scans were taken using a step size of 0.001 cm over a radial range of 6.8 to 7.2 cm with 15 replicates. The resulting absorbance versus radial position profiles at multiple sample concentrations and rotor speeds were globally fitted to a single discrete species model or various self-associating models (including monomer-dimer, monomer-trimer, dimer-tetramer, trimer-hexamer and monomer-dimer-tetramer models) using the program SEDPHAT [55] (available from www.analyticalultracentrifugation.com).
Dynamic light scattering
Dynamic light scattering measurements were made on an ALV 5022F DLS (ALV, Germany). Protein samples were analyzed at a final concentration of 59 μM. Measurements were taken over a 30 s time period with 10 replicates at 20°C. The samples were illuminated with a HeNe laser (633 nm) and experiments were conducted at a scattering angle of 90 degrees. The in-built ALV analysis software was used to determine average radii.
Crystallization
Crystallization of Sp-DHDPS was performed as previously described [40]. Briefly, crystals were obtained by employing the hanging-drop vapor-diffusion method. 1 µl protein solution (10 mg ml-1) and 1 µl precipitant solution were equilibrated against 1 ml reservoir solution [0.2 M ammonium chloride, 20% (w/v) PEG 6000, 0.1 M MES pH 6.0] in 24-well Linbro plates at 20°C. Crystals were soaked briefly in cryoprotectant composed of 0.2 M ammonium chloride, 20% (w/v) PEG 6000, 0.1 M MES pH 6.0 and 20%(w/v) glycerol and were then flash-frozen using liquid nitrogen.
Structure determination
X-ray diffraction experiments were carried out at the Australian Synchrotron, Victoria, Australia on the MX1 beamline using Blu-Ice [56]. A 1.9 Å resolution data set was integrated and merged with HKL2000 [57] and scaled with SCALA [58]. The crystals were initially assigned to the tetragonal crystal system, space group P42212 with unit cell dimensions a = b = 105.5 Å and c = 62.4 Å, and an overall R merge of 6%. Molecular replacement was carried out using the program PHASER [59] with the structure of dihydrodipicolinate synthase from B. anthracis (PDB ID: 1XL9) [22] as the search model. One molecule was located in the asymmetric unit. The structure was refined using the program PHENIX [60] with rigid body refinement, simulated annealing and atomic displacement parameters (ADP) refinement. Iterative model building was carried out using the program COOT [61] followed by ADP refinement. Even though the 2F o -F c electron density map was clear, there were spurious streaks in the F o -F c difference maps and refinement stalled with a R work of 24 % and a R free of 32 %. Examination of the cumulative intensity plot from SCALA suggested that the data were affected by crystal twinning. The data were inspected further by running the program phenix.xtriage. Since there are no twin laws possible in the above crystal symmetry, over-merging of pseudo-symmetric or twinned data may have been present. The diffraction data were re-integrated in space group P1 using XDS [62] and further analyzed with phenix.xtriage. The presence of seven pseudo-merohedral twin operators were found, two 4-fold and five 2-fold. The twin law giving the lowest R obs was -h,l,k superimposing on h,k,l. The data were scaled in space group P2 (cell dimensions a = 64.4 Å, b = 105.3, Å, c = 105.5 Å, β = 90.0° degrees) and refined as above with the inclusion of the above two-fold twin law and four crystallographically-independent subunits (residues 2-299 and 2-297) in the asymmetric unit leading to values for R work of 19.7 % and R free of 22.9 % (this model has been deposited with PDB ID: 3H5D). However, inspection of the diffraction data and the structural model provided clear evidence for 21 screw axes perpendicular to the monoclinic unique b axis. The twinning axis is parallel to the crystallographic two-fold axis (which is a pseudo-42 or 21 screw axis with significant intensity violations), leading to reflections −k,h,l being overlapped with h,k,l and to the observed pseudo-tetragonal 4/mmm Laue symmetry. The final model, now in space group P22121, was solved by molecular replacement using MOLREP [63] with the model PDB ID: 3HIJ [36] together with amplitude-based twin refinement with NCS restraints using REFMAC5 [64]. The final model (R work = 16.5% and R free = 21.2%) comprises of two monomers (residues A2 – A299 and B2 – B297, and A302 – A311 and B302 – B311), 235 water molecules, 4 potassium ions, 4 glycerol molecules and 2 MES ions. The pseudo-tetragonal symmetry leads to two possible choices of asymmetric unit, one comprising a pair of subunits forming the ‘weak’ dimer interface, the other comprising a pair of subunits forming the ‘tight’ interface – the usual asymmetric unit seen in other tetrameric DHDPS structures. Both were tested, the latter being confirmed as correct. The crystallographic two-fold axis generates the tetramer. Residues 300-301 of chain A and residues 298-301 of chain B were not observed in electron density maps, and as a result of crystal packing, it is not unequivocally clear whether the C-terminal residues 302-311, which were very clearly defined in electron density maps once the twinning and space group problems were solved, indulge in domain swapping to a neighboring tetramer or remain with their own tetramer, However, electrospray ionization mass spectrometry analysis of dissolved crystals of recombinant Sp-DHDPS indicates that the entire protein (residues 1-311) is intact. A summary of the crystallographic data collection and refinement statistics is provided in Table 2.
Table 2. Data collection and refinement statistics for the X-ray structure of Sp-DHDPS (PDB ID: 3VFL).
| Temperature (K) |
100 | ||
|---|---|---|---|
| Space group | P22121 | ||
| Cell dimensions Å (a, b, c) | 62.4, 105.3, 105.5 | ||
| Resolution (Å) | 74.6 -1.9 | ||
| No. of observations | 757223 | ||
| No. of unique observations | 51243 | ||
| Completeness (%) | 99.6 (97.7)1 | ||
| I/σI | 35.7 (4.7) | ||
| R sym (%) 2 | 6.3 (56.2) | ||
| Twin operator | -h, l, k | ||
| Twin fraction | 0.46 | ||
| Wilson B factor Å2 | 22.6 | ||
| REFINEMENT STATISTICS | |||
| Non-hydrogen atoms | |||
| Protein | 4725 | ||
| Water | 235 | ||
| Ligands | 52 | ||
| R work (%) 3 | 16.5 | ||
| R free (%) 4 | 21.2 | ||
| RMSD values from ideal value | |||
| Bond lengths (Å) | 0.02 | ||
| Bond angles (°) | 2.4 | ||
| Ramachandran plot | |||
| Most favored and allowed region (%) | 99.3 | ||
| B factors (Å2) | |||
| Average main chain | 27.8 | ||
| Average side chain | 29.6 | ||
| Average water molecule | 29.9 | ||
1 Values in parentheses represent the highest resolution shell (2.1–1.9).
2 R sym = Σ|I - (I)|/ΣI.
3 R work = Σ||F o| - |F c||/Σ|F o|.
4 R free is based on 5% of the total reflections excluded from refinement.
Coordinates
Coordinates and structure factors for the final model are accessible via the Protein Data Bank (PDB ID: 3VFL).
Results
S. pneumoniae 447A ΔdapA mutant requires (S)-lysine for growth
To validate DHDPS as a promising drug target in Streptococcus pneumoniae, the gene encoding this enzyme (i.e. dapA) was deleted from strain 447A. The ΔdapA mutant was generated by homologous recombination as described in the Materials and Methods. Cultivation of the ΔdapA and wild-type strains on nutrient-rich media, such as HBA, showed that their size and morphology were indistinguishable (data not shown). To determine whether the ΔdapA mutant required (S)-lysine for growth, wild-type and mutant strains were grown in Chemically Defined Medium CDM+ and CDM- (where + and - indicates the presence and absence of 200 µg ml-1 (S)-lysine, respectively) at 37°C in an atmosphere of 5% CO2. Analysis of growth rates of wild-type and ΔdapA mutant strains showed that the rate of growth of the mutant in CDM+ was not significantly different to that of the wild-type strain (Figure 2). However, the ΔdapA mutant was unable to grow in CDM- whereas the wild-type strain reached comparable cell density in CDM+ and CDM- (Figure 2). This demonstrates that the dapA gene, encoding DHDPS, is essential for the growth of S. pneumoniae in the absence of lysine.
Figure 2. Growth phenotype of the dapA knockout strain of S. pneumoniae.
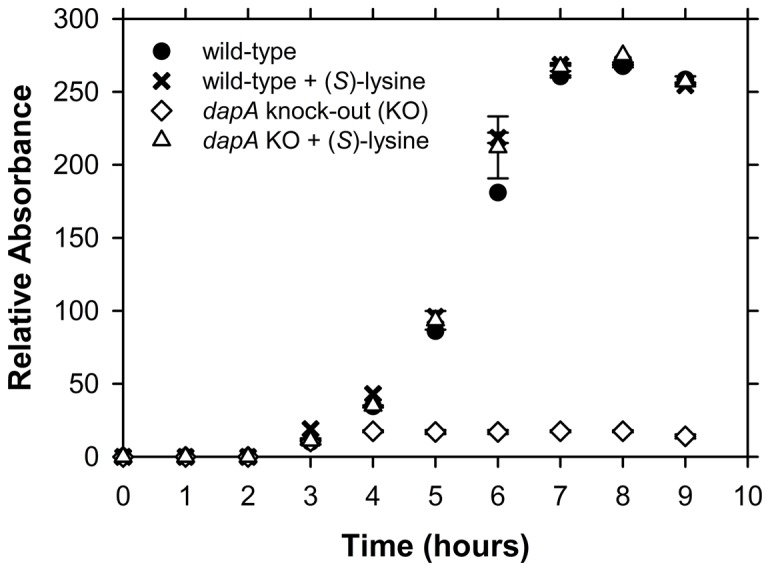
Growth of wt S. pneumoniae 447A (control) and the ΔdapA mutant strain in the presence and absence of 20 mM (S)-lysine. Growth was assessed by monitoring the optical density (Relative Absorbance) of duplicate cultures on an hourly basis as described in the Materials and Methods.
Secondary structure and stability of Sp-DHDPS
Sp-DHDPS was expressed and purified to >95% homogeneity as described previously [43]. The enzyme was subjected to circular dichroism (CD) spectroscopy in aqueous solution to assess the secondary structure of the recombinant product. The CD spectrum (Figure 3A) shows a broad minimum spanning 208 nm to 222 nm, suggesting Sp-DHDPS adopts a mixed α/β secondary structure in solution [16,36,47]. This assertion was confirmed by fitting the CD spectrum of Sp-DHDPS to the CONTINLL algorithm and SP43 database using the CDPRO software suite [48,49]. The nonlinear best-fit demonstrates a significant proportion of α-helix and β-strand (Table 3). The calculated secondary structure composition of Sp-DHDPS is comparable to that of DHDPS enzymes from other bacteria, including E. coli (Table 3). Next we assessed the stability of recombinant Sp-DHDPS in solution by conducting thermal denaturation experiments monitored by CD at 222 nm (Figure 3B). The resulting thermal denaturation profile reveals that the enzyme unfolds via a single transition with an apparent melting temperature (T M app) of 72°C (Figure 3B and Table 4). Recombinant DHDPS from E. coli (Ec-DHDPS) also unfolds via a single transition, but with a significantly lower T M app of 59°C (Figure 3B and Table 4). These data therefore indicate that Sp-DHDPS is markedly more stable in solution than Ec-DHDPS. We were thus interested in unraveling the molecular mechanism for this enhanced thermostability by assessing the quaternary structure of Sp-DHDPS in solution.
Figure 3. Circular dichroism spectroscopy of Sp-DHDPS.
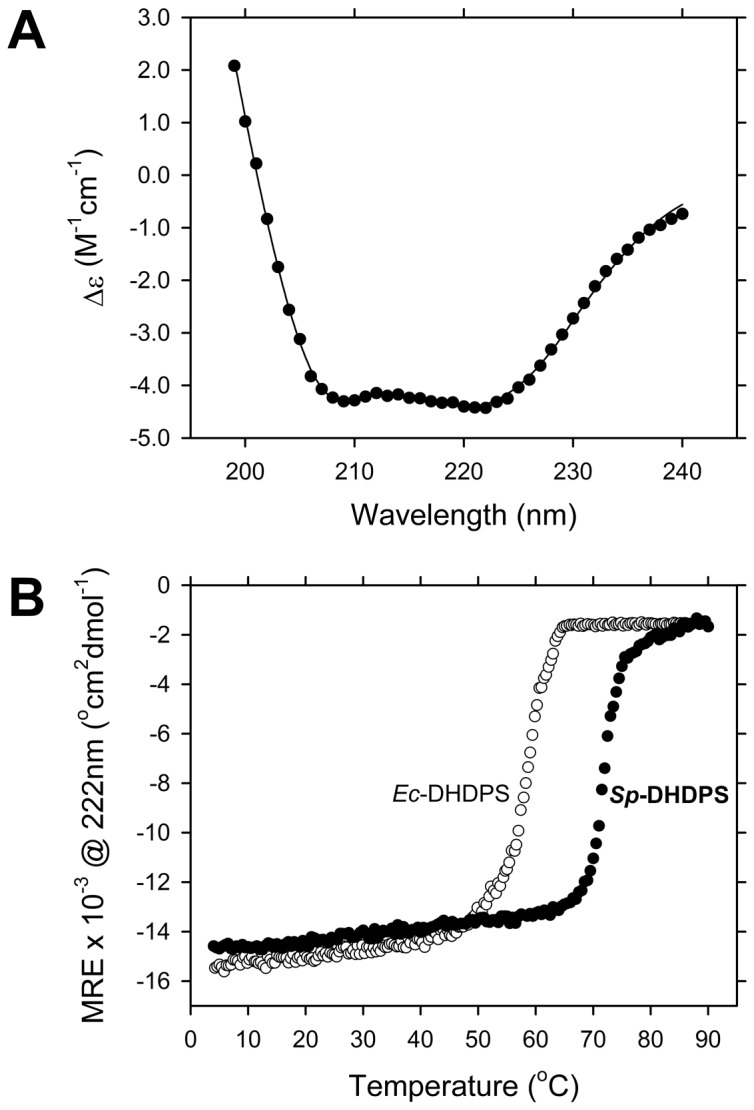
(A) CD spectrum of Sp-DHDPS () plotted as the molar circular dichroism (Δε) as a function of wavelength. The solid line represents the nonlinear least squares fit using the CONTINLL algorithm and SP43 database within the CDPRO software suite [48.49]. The best-fit resulted in the secondary structure composition reported in Table 3. (B) Thermal denaturation profiles of Sp-DHDPS () and Ec-DHDPS () plotted as mean residue ellipticity (MRE) versus temperature. The apparent melting temperature (T M app), or midpoint of the transition between folded and unfolded states, was determined from the ordinate maximum of a plot of the first derivative of the MRE as a function of temperature to yield 59°C for Ec-DHDPS and 72°C for Sp-DHDPS.
Table 3. Secondary structure composition of Sp-DHDPS and Ec-DHDPS determined by CD spectroscopy 1 .
| Enzyme | α-helix | β-strand | turn | unordered | RMSD |
|---|---|---|---|---|---|
| Sp-DHDPS | 36 | 15 | 20 | 29 | 0.05 |
| Ec-DHDPS | 34 | 17 | 20 | 29 | 0.04 |
Table 4. Comparison of the thermostability and hydrodynamic properties of Sp-DHDPS and Ec-DHDPS derived from circular dichroism spectroscopy and analytical ultracentrifugation studies.
| Enzyme | TMapp1 °C | s20,w (S) | Mr4 (kDa) | M (kDa) | f/f0 6 | a/b7 | KD42 (nM) |
|---|---|---|---|---|---|---|---|
| Sp-DHDPS | 72 | 7.22 | 135 | 1335 | 1.256 | 2.2 | 1.78 |
| Ec-DHDPS | 59 | 6.93 | 125 | 1283 | 1.263 | 2.6 | 763 |
1 The apparent melting temperature (T M app), or midpoint of the transition between folded and unfolded states, was determined from the ordinate maximum of a plot of the first derivative of the MRE as a function of temperature.
2 Value determined experimentally from the ordinate maximum of the c(s) distribution best-fit shown in Figure 4A.
3 Hydrodynamic properties reported in [34].
4 Molecular mass calculated from the amino acid sequence.
5 Value calculated experimentally from the apparent molecular mass taken from the ordinate maximum of the c(M) distribution best-fit shown in Figure 4B.
6 Frictional ratio calculated using the partial specific volume () method employing SEDNTERP software [53,54].
7 Axial-ratio as calculated from the program SEDNTERP using the method assuming a prolate ellipsoid.
8 The tetramer-dimer dissociation constant calculated from the global nonlinear least squares best-fit described in Figure 5
Quaternary structure of Sp-DHDPS in aqueous solution
To gain further insights into the solution stability of Sp-DHDPS, sedimentation velocity studies were conducted in the analytical ultracentrifuge. Absorbance versus radial data profiles at different time points were fitted to a continuous size-distribution, c(s), model [51.52,65,66]. The resulting c(s) distribution for Sp-DHDPS at an initial concentration of 4.5 µM is shown in Figure 4A. The c(s) distribution reveals a single peak with a s 20,w value of 7.2 S that is consistent with a tetrameric species (Table 4) [34,36]. The corresponding c(M) distribution (Figure 4B) confirmed this assertion and shows a single peak with a molar mass of 133 kDa, which closely matches the theoretical mass of the Sp-DHDPS tetramer (135 kDa). Sedimentation studies at a 6-fold lower protein concentration (i.e. 740 nM) were conducted and the resulting c(s) nonlinear least-squares fit also reveals the presence of a single peak (Figure 4B) with a s 20,w value of 7.0 S, consistent with the enzyme existing primarily as a tetramer [26,27,34,36]. These sedimentation velocity analyses suggest that Sp-DHDPS exists as a very stable tetramer in solution. Sedimentation equilibrium experiments were subsequently performed in the analytical ultracentrifuge to examine the strength of subunit interactions and the resulting data at multiple protein concentrations fitted to various equilibrium schemes. Not surprisingly, the optimal global nonlinear least-squares fit was obtained for a dimer-tetramer equilibrium model (Figure 5, solid lines) with a tetramer-dimer dissociation constant (K D 42) of 1.7 nM. Interestingly, the calculated K D 42 for Sp-DHDPS is considerably tighter than that obtained for the previously characterized Ec-DHDPS tetramer (Table 4) [34]. We next set out to determine the average hydrodynamic radius of the Sp-DHDPS tetramer in solution using dynamic light scattering.
Figure 4. Sedimentation velocity and dynamic light scattering analyses of Sp-DHDPS.
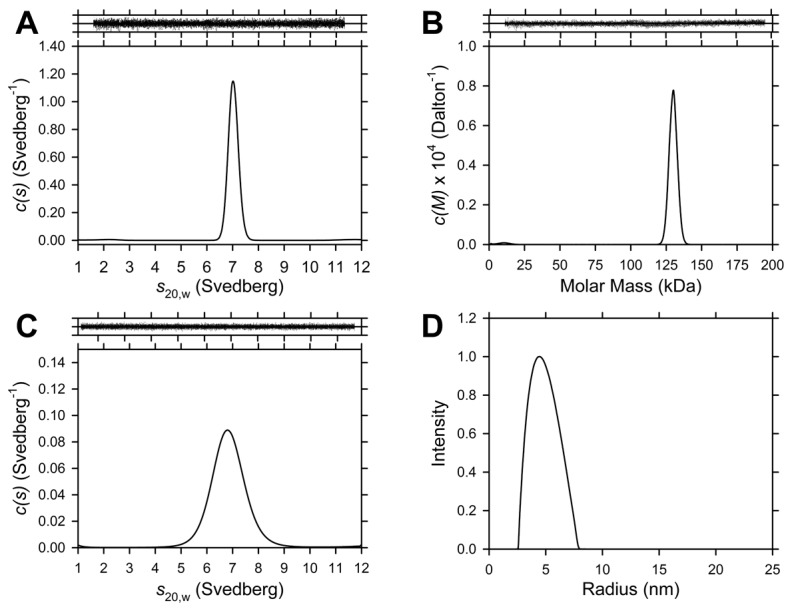
(A) Continuous sedimentation coefficient, c(s), distribution of Sp-DHDPS at 4.5 µM plotted as a function of standardized sedimentation coefficient (s 20,w) with RMSD and runs test Z values for the best-fit of 0.008 and 6.6, respectively. (B) Continuous mass, c(M), distribution of Sp-DHDPS at a concentration of 4.5 µM plotted as a function of molar mass with RMSD and runs test Z values of 0.008 and 6.6 respectively. (C) c(s) distribution of Sp-DHDPS at 740 nM, with RMSD and runs test Z values of 0.02 and 1.64, respectively. Data were analyzed employing the program SEDFIT [51,52,65,66] using a resolution (N) of 200 and a sedimentation coefficient range of 0.1-12 S or molar mass range 1.0-250 kDa with a P-value of 0.95. Above Panels A-C: Residuals plotted as a function of radial position resulting from continuous size-distribution best-fits shown in panels A-C. (D) A distribution plot of the average unweighted hydrodynamic radii of Sp-DHDPS at a concentration of 59 µM determined using dynamic light scattering (DLS). The results of the distribution plot reveal an average hydrodynamic radius of 4.5 ± 0.2 nm.
Figure 5. Sedimentation equilibrium analyses of Sp-DHDPS.
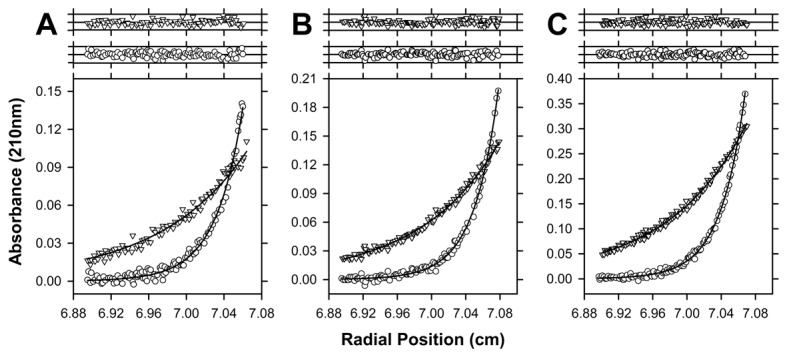
Absorbance at sedimentation equilibrium is plotted as a function of radial position for Sp-DHDPS at an initial concentration of (A) 296 nM; (B) 355 nM and (C) 740 nM. Data was measured at 210 nm and rotor speeds of 10,000 rpm () and 18,000 rpm (). The resulting global nonlinear least squares fit to a dimer-tetramer equilibrium model is shown as solid lines and yielded a KD 4à2 of 1.7 nM with a global reduced χ2 of 0.5. Above Panels A-C: Residuals plotted as a function of radial position resulting from the global nonlinear best-fit analysis to a dimer-tetramer model for data at 10,000 rpm () and 18,000 rpm ().
The hydrodynamic radius of the Sp-DHDPS tetramer
The quaternary structure and hydrodynamic radius of Sp-DHDPS were studied by dynamic light scattering (DLS) experiments at an enzyme concentration of 59 μM [3.5 × 104-fold above the K D 4→2 of Sp-DHDPS (Table 4)]. The resulting data were analyzed by the method of cumulants (second-order) and were found to be reproducible between runs and independent of the choice of fit range. Figure 4D shows a regularized distribution fit employing the in-built software (ALV). Analysis of data resulted in a range of hydrodynamic radii that centered on an average value of 4.5 ± 0.2 nm. The average hydrodynamic radius was consistent between runs and was not affected by the choice of fit parameters (correlation function limits or radius limits). By comparison, the Stokes radius of the tetramer calculated from sedimentation velocity studies is 4.3 nm, which is in excellent agreement with the DLS experiment.
Enzyme kinetic properties of Sp-DHDPS
To characterize the catalytic properties of the Sp-DHDPS tetramer, we employed the DHDPS-DHDPR coupled assay [45] at an enzyme concentration of 20 nM, which is ~12-fold greater than the K D 4→2 (Table 4). Initial rates were measured with fixed pyruvate concentrations of 0.5 mM, 1.0 mM, 2.0 mM, 4.0 mM, 8.0 mM and 16.0 mM and varying ASA concentrations (0-0.48 mM). The resulting data were expressed initially as Lineweaver-Burk plots (Figure 6A), which displayed a characteristic series of parallel lines indicating that Sp-DHDPS follows a Ping-Pong kinetic mechanism [67]. This mechanism has been demonstrated for DHDPS orthologs from other bacterial species, including Ec-DHDPS [45,68,69]. The data were subsequently plotted to produce Michaelis-Menten profiles (Figure 6B) and fitted to bi-substrate kinetic models (with and without substrate inhibition), namely the ternary complex and Ping-Pong mechanism employing ENZFITTER software (Biosoft). The global nonlinear regression analysis yielded a best-fit to a Ping-Pong model with no substrate inhibition (Figure 6B, solid lines) that resulted in a R 2 = 0.98 and the kinetic parameters reported in Table 5. The K M of Sp-DHDPS for (S)-ASA is similar that for the E. coli enzyme (Table 5). However, the K M for pyruvate is 10-fold higher than that for Ec-DHDPS, and the catalytic turnover (k cat) is 6-fold lower for Sp-DHDPS compared to the E. coli ortholog (Table 5).
Figure 6. Enzyme kinetic profiles of recombinant Sp-DHDPS.

Initial velocity was measured as a function of (S)-ASA concentration. Experiments were conducted at fixed pyruvate concentrations of () 0. 5 mM, () 1.0 mM, () 2.0 mM, () 4.0 mM, () 8.0 mM and () 16.0 mM. (A) Lineweaver-Burk plots showing multiple parallel lines; diagnostic of a ping pong kinetic mechanism [25,67,69]. (B) Michaelis-Menten plots of data shown in A, where solid lines represent the global nonlinear best-fit to a Ping-Pong mechanism (without substrate inhibition) using ENZFITTER software, resulting in a R 2 = 0.98 and the enzyme kinetic parameters summarized in Table 5.
Table 5. Summary of the enzyme kinetic parameters of Sp-DHDPS compared to Ec-DHDPS.
| Enzyme | kcat (sec-1) | KMASA (mM) | KMPYR (mM) |
|---|---|---|---|
| Sp-DHDPS | 22 | 0.044 ± 0.003 | 2.55 ± 0.05 |
| Ec-DHDPS 1 | 124 | 0.11 ± 0.01 | 0.26 ± 0.03 |
1 Kinetic parameters reported in [25].
Crystal structure of Sp-DHDPS
In order to gain further insight into the stability and kinetic behavior at the atomic level, we next sought to determine the crystal structure of Sp-DHDPS. The collection of high-resolution synchrotron X-ray data has been reported recently [43]. Crystals were pseudo-merohedrally twinned to give the illusion of tetragonal symmetry, with a twin fraction of 0.46 (Table 2). Moreover, the apparent 42 screw axis was characterized by significant systematic absence violations, which led after abortive attempts in P42212 to solution and refinement in the monoclinic space group P21 where there is ambiguity with respect to the twin law. However, with two well-defined 21 screw axes perpendicular to the pseudo-tetragonal axis, final refinements proceeded successfully in the orthorhombic space group P22121 with the twinning axis being parallel to the pseudo-tetrad. Only in this final assignment of crystallographic symmetry did the C-terminal residues 302-311 (see below) become well defined. The structure has been refined at 1.9 Å resolution (Table 2) to an R work of 16.5 % (R free of 21.2 %). Two residues, V147 and Y114 in both chains A and B lie in disallowed Ramachandran conformers, but are clearly defined in the electron density. Both residues are located at the ‘tight’ dimer interface (Figure 7) and are held in strained conformations by hydrogen bonding and hydrophobic packing at this interface. The strained conformation of the highly conserved Y114 (Figure 1B) has been observed in all DHDPS structures determined to date, including those from plants [20-36]. However, the strained conformation of V147, which is poorly conserved (Figure 1B), appears to be unique to Sp-DHDPS.
Figure 7. Crystal structure of Sp-DHDPS determined at a resolution of 1.9 Å.

The enzyme crystallizes as a tetramer comprised of four identical subunits labeled A, B, C & D with subunits AB or CD connected via the ‘tight’ dimer interface, and subunits AC or BD connected via the ‘weak’ dimer interface.
Consistent with the solution studies, Sp-DHDPS forms a homotetrameric structure (PDB ID: 3VFL) that is depicted in Figure 7. Each monomer unit is folded to form an N-terminal (β/α)8-barrel (residues 2-229) with a C-terminal extension comprised of three helices (residues 232-292). The C-terminal residues 302-311 lie in a groove formed by the ‘tight’ dimer interface (i.e. the interface between chains A & B or C & D in Figure 7). As with other DHDPS structures, including E. coli DHDPS [25,31], the active site of Sp-DHDPS is located within the β-barrel of each subunit. Many of the residues adopt a similar spatial arrangement to that of the Ec-DHDPS structure [PDB ID: 1YXC] [25] (Figure 8A). The key lysine residue, K168, which forms a Schiff base with the pyruvate substrate, is present alongside residues Y140, R145, and G192, which play a pivotal role in the cyclization after reaction with the second substrate (S)-ASA [25,70] (Figure 8A). Two of the residues forming the catalytic triad, which serves to shuttle protons to and from the active site (i.e. Y140 and T50), are also present in a similar orientation to the equivalent residues from the E. coli ortholog (Figure 8A). However, the third residue of the catalytic triad, Y114, undergoes a ~70° rotation in the active site of Sp-DHDPS relative to the equivalent residue of the E. coli enzyme (i.e. Y107), although the functionally important –OH group is similarly positioned. This residue interdigitates across the ‘tight’ dimer interface forming a hydrophobic stack with Y113 from the adjacent monomer (Figure 8B). This 70° rotation serves to change the interaction to a π stacking between the two tyrosine residues.
Figure 8. Comparison of the active sites of Sp-DHDPS (yellow) and Ec-DHDPS (pink).
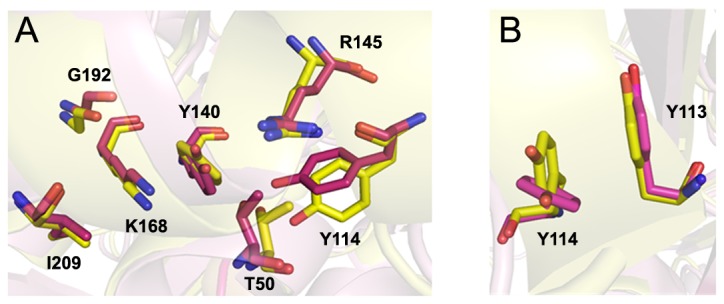
(A) An overlay of the active site with residues labeled according to Sp-numbering. (B) Image demonstrating the approximate 70° rotation of Y114 in the active site of Sp-DHDPS compared to the equivalent residue in Ec-DHDPS (Y107).
Relative to the Ec-DHDPS structure (PDB ID: 1YXC) [26], insight into the enhanced stability observed for Sp-DHDPS in solution is revealed by close inspection of the interactions stabilizing the ‘tight’ dimer interface, and in particular, the ‘weak’ dimer interface (Figure 7, interface AC or BD). Analysis of the solvent inaccessible surface areas (SISA), calculated using the Protein Interfaces, Surfaces and Assemblies (PISA) program [71], reveals that the ‘tight’ dimer interface of Sp-DHDPS (Figure 7, interface AB or CD) buries 1350 Å2 (Table 6, Figure 9A), slightly more than that calculated (1290 Å2) for the equivalent interface of the Ec-DHDPS structure (Table 6, Figure 9C). Although both enzymes contain the same number of residues (i.e. 38 in total) at this interface, Sp-DHDPS contains a larger proportion of residues forming hydrogen bonds. In contrast, the ‘weak’ dimer interface has a total SISA of 800 Å2 (Table 6, Figure 9B), which is significantly greater than the equivalent interface of the E. coli structure (500 Å2) (Table 6 & Figure 9D). The substantially larger buried surface area in Sp-DHDPS is due to a greater number of residues forming contacts at this interface. In addition, it is interesting to note that this enzyme also contains three residues that form salt bridge interactions, a feature that is absent in Ec-DHDPS (Table 6 & Figure 9D).
Table 6. Comparison of the ‘tight’ dimer and ‘weak’ dimer interfaces of Sp-DHDPS and Ec-DHDPS.
| Enzyme | Tight Dimer SISA 1 (Å2) | Residues involved in Hydrophobic contacts | Residues involved in Hydrogen bonding | Residues involved in Ion-Ion interactions | Weak Dimer SISA 1 (Å2) | Residues involved in Hydrophobic contacts | Residues involved in Hydrogen bonding | Residues involved in Ion-Ion interactions |
|---|---|---|---|---|---|---|---|---|
| Sp-DHDPS | 1350 | 27 | 11 | 0 | 800 | 20 | 4 | 3 |
| Ec-DHDPS | 1290 | 31 | 7 | 0 | 500 | 11 | 6 | 0 |
1 SISA = solvent inaccessible surface area. Calculated employing PISA [71] analysis (http://www.ebi.ac.uk/msd-srv/prot_int/pistart) and PDB coordinates for Sp-DHDPS (PDB ID: 3VFL) and Ec-DHDPS (PDB ID: 1YXC).
Figure 9. Comparison of the oligomeric interfaces of DHDPS from S. pneumoniae (panels A & B) and E. coli (panels C & D).
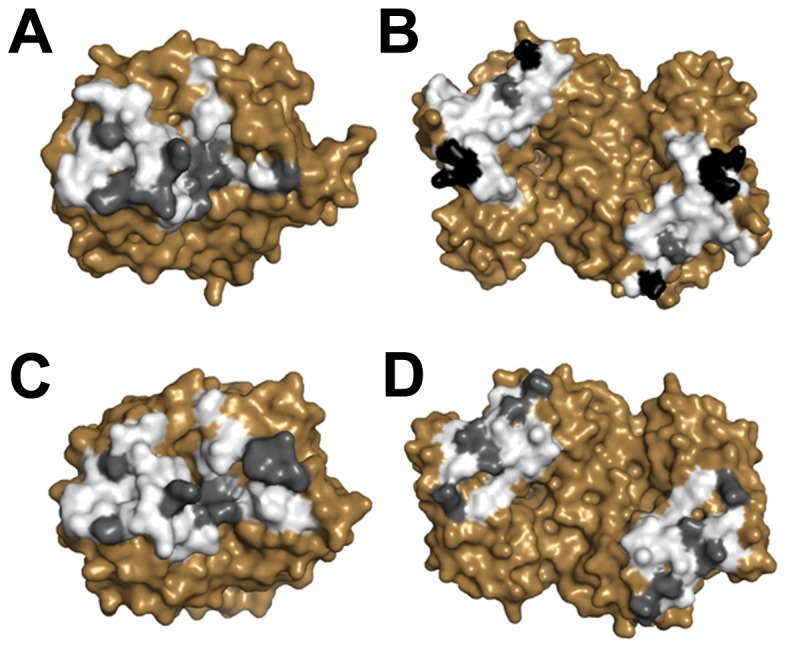
Noncovalent interactions and solvent-inaccessible surface area (SISA) were calculated using the program PISA [71]. The ‘tight’ dimer interface of Sp-DHDPS (panel A) buries 1350 Å2 of SISA compared to Ec-DHDPS (panel C), which buries 1290 Å2 of SISA. The ‘weak’ dimer interface of Sp-DHDPS (panel B) buries 800 Å2 of SISA compared to 500 Å2 for Ec-DHDPS (panel D). White, grey and black shaded regions correspond to interfacing, hydrogen bonding and ion bonding residues, respectively.
Discussion
Streptococcus pneumoniae is one of the leading causes of disease in the world. In the United States, for example, the organism is responsible for 500,000 cases of pneumonia, 50,000 cases of bacteremia, and 3,000 cases of meningitis per annum [3,72]. Current treatment relies primarily upon the use of penicillin-based antibiotics. However, this approach has had significant limitations given the emergence of drug-resistant S. pneumoniae (DRSP). The appearance of multiple drug resistant (MDR) strains in recent times and the occurrence of community acquired infection are also of particular concern [72,73]. Accordingly, there is an urgent need to develop novel antimicrobials and an equally urgent need to discover new antibiotic targets. A promising pneumococcal drug target is DHDPS, given the enzymes function in producing lysine required for protein and cell wall synthesis. However, our knock-out studies show that the ΔdapA strain of S. pneumoniae is a lysine auxotroph (Figure 2), which suggests DHDPS attenuated strains may be able to survive in vivo by scavenging exogenous lysine from the host. Further studies are required to ascertain the viability of the ΔdapA strain in vivo. Nevertheless, having established the ΔdapA strain was essential to S. pneumoniae in minimal media, we set out to characterize the structure, function and stability of the enzyme to afford insight into rational drug design strategies to afford the discovery of Sp-DHDPS inhibitors in future work.
Our studies show that Sp-DHDPS is similar in some respects to the well characterized ortholog from E. coli, which shares 36 % sequence identity. For instance, CD spectroscopy and AUC studies reveals that Sp-DHDPS, like Ec-DHDPS, has high α/β secondary structure (Table 3, Figure 3A) and resides in a dimer-tetramer equilibrium (Table 4, Figures 4 and 5). Likewise, enzyme kinetic studies demonstrate both Sp-DHDPS and Ec-DHDPS operate via a Ping-Pong catalytic mechanism. However, the enzyme kinetic parameters calculated for Sp-DHDPS are significantly different to the E. coli enzyme (Table 5). The greater K M PYR for Sp-DHDPS suggests a lower thermodynamic affinity for this substrate (assuming the coupling of pyruvate with (S)-ASA is rate-determining), consistent with the very strongly associated, and presumably more rigid tetramer, that hinders access of pyruvate to the binding site and relaxation of the protein to accommodate pyruvate. This is consistent with our prior conclusions that protein dynamics control enzyme kinetics [26,27,36,74].
It is worthwhile noting that pyruvate is a central metabolite, serving as a substrate for several other enzymes. Pyruvate-utilizing enzymes, such as pyruvate dehydrogenase and lactate dehydrogenase from Gram-positive bacterial species, including Bacillus subtilis, Corynebacterium glutamicum and Lactococcus lactis, display K M PYR ranging 1 - 15 mM [75,76]. These values are considerably higher than orthologs from Gram-negative species, such as E. coli and Thermotoga maritima, where K M PYR values range from 0.018 - 0.43 mM [77-79]. These differences, found also in comparison of DHDPS from Gram-positive and Gram-negative species, may indicate that pyruvate is present at a higher intracellular steady-state concentration in S. pneumoniae (and other Gram-positive bacteria) than in E. coli (and other Gram-negative bacteria). The origins of these differences in K M PYR are not apparent from the structures of these DHDPS enzymes.
Solution studies also showed Sp-DHDPS possesses significantly greater thermostability compared to Ec-DHDPS (Figure 3B). We subsequently demonstrated using analytical ultracentrifugation that the enhanced thermostability of Sp-DHDPS is contributed by a 45-fold tighter tetramer-dimer dissociation constant (Table 4) [34]. We therefore determined the three-dimensional structure of Sp-DHDPS using X-ray crystallography (Figure 7) to provide insight into the enhanced thermal and thermodynamic stability. The origin of the enhanced thermal stability is clearly revealed in the 1.9 Å resolution crystal structure of Sp-DHDPS (Figure 7) that shows a tetrameric structure of the enzyme, consistent with our solution studies (Table 4, Figures 4 and 5). The tertiary and quaternary structure architecture of Sp-DHDPS is very similar to Ec-DHDPS [25,31,34] with an overall RMSD for superposition of the tetramers of 1.1 Å (alpha carbon atoms), as well as to other structurally characterized bacterial DHDPS enzymes [16-19,22,24-27,29-36];. However, significant structural differences are observed between Sp-DHDPS and Ec-DHDPS at the subunit interfaces (Figure 9). We show that there is an increase of 60 Å2 and 300 Å2 in solvent-inaccessible surface area (SISA) at the ‘tight’ dimer and ‘weak’ dimer interfaces of Sp-DHDPS, respectively (Table 6, Figure 9). This significant increase in SISA, in combination with the greater proportion of hydrogen bonding residues at the ‘tight’ dimer interface, as well as the presence of residues participating in salt bridge interactions at the ‘weak’ dimer interface (also referred to as the tetramerization interface [36]), is consistent with the 45-fold lower tetramer-dimer dissociation constant for Sp-DHDPS (Table 4, Figure 5), and to its considerably higher thermal stability (Figure 3B). Consistent with previous studies of other DHDPS enzymes, the residues that form interactions at the ‘weak’ dimer interface are poorly conserved in Sp-DHDPS, whereas strong conservation is observed at the ‘tight’ dimer interface where the active sites are located [16-36]. Given that recent studies show dimeric mutants of DHDPS have significantly attenuated catalytic function compared to the wild-type tetramers [26,27,36], the poor conservation at the ‘weak’ dimer interface offers potential for the design of pathogen-specific antimicrobial agents [34], particularly given that protein-protein interfaces represent highly specific drug targets [80,81]. Indeed, with the increase in drug-resistant bacteria linked to the overuse and misuse of broad spectrum antibiotics [8,9], exploiting the ‘weak’ dimer interface of Sp-DHDPS may provide a means to negate the incidence of broad spectrum drug resistance.
In conclusion, through gene knock-out studies, circular dichroism spectroscopy, analytical ultracentrifugation, dynamic light scattering, enzyme kinetics and X-ray crystallography studies, we demonstrate that Sp-DHDPS is an essential, active and thermostable tetramer. Our work offers insight into rational drug design strategies targeting multiple sites of the enzyme to afford the discovery of novel antibiotic agents with potential to negate drug resistance.
Acknowledgments
We acknowledge Geoff Hogg (Microbiological Diagnostic Unit, University of Melbourne) for providing genomic DNA, the friendly staff of the Bio21-C3, the MX beamline scientists at the Australian Synchrotron, Suresh. K. Bhargava (School of Applied Sciences, RMIT University) for access to dynamic light scattering equipment and Craig A. Hutton (School of Chemistry, University of Melbourne) and Renwick C.J. Dobson (School of Biological Sciences, University of Canterbury) for useful discussion during preparation of this manuscript.
Funding Statement
Funding provided by (1) Australian Research Council Future Fellowship FT0991245, (2) National Health & Medical Research Council (NHMRC) Project Grant 628681l (3) NHMRC Fellowship APP1021645. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Moscoso M, García E, López R (2006) Biofilm formation by Streptococcus pneumoniae: role of choline, extracellular DNA, and capsular polysaccharide in microbial accretion. J Bacteriol 188: 7785-7795. doi: 10.1128/JB.00673-06. PubMed: 16936041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Janssens JP, Krause KH (2004) Pneumonia in the very old. Lancet Infect Dis 4: 112-124. doi: 10.1016/S1473-3099(04)00931-4. PubMed: 14871636. [DOI] [PubMed] [Google Scholar]
- 3. Cartwright K (2002) Pneumococcal disease in western Europe: burden of disease, antibiotic resistance and management. Eur J Pediatr 161: 188-195. doi: 10.1007/s00431-001-0907-3. PubMed: 12014384. [DOI] [PubMed] [Google Scholar]
- 4. Whitney CG, Farley MM, Hadler J, Harrison LH, Bennett NM et al. (2000) Increasing prevalence of multidrug-resistant Streptococcus pneumoniae in the United States. N Engl J Med 343: 1917-1924. doi: 10.1056/NEJM200012283432603. PubMed: 11136262. [DOI] [PubMed] [Google Scholar]
- 5. Nunes S, Sá-Leão R, Carriço J, Alves CR, Mato R et al. (2005) Trends in drug resistance, serotypes, and molecular types of Streptococcus pneumoniae colonizing preschool-age children attending day care centers in Lisbon, Portugal: a summary of 4 years of annual surveillance. J Clin Microbiol 43: 1285-1293. doi: 10.1128/JCM.43.3.1285-1293.2005. PubMed: 15750097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schrag SJ, McGee L, Whitney CG, Beall B, Craig AS et al. (2004) Emergence of Streptococcus pneumoniae with very-high-level resistance to penicillin. Antimicrob Agents Chemother 48: 3016-3023. doi: 10.1128/AAC.48.8.3016-3023.2004. PubMed: 15273115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stephens DS, Zughaier SM, Whitney CG, Baughman WS, Barker L et al. (2005) Incidence of macrolide resistance in Streptococcus pneumoniae after introduction of the pneumococcal conjugate vaccine: population-based assessment. Lancet 365: 855-863. doi: 10.1016/S0140-6736(05)71043-6. PubMed: 15752529. [DOI] [PubMed] [Google Scholar]
- 8. Sørum H, L'Abée-Lund TM (2002) Antibiotic resistance in food-related bacteria-a result of interfering with the global web of bacterial genetics. Int J Food Microbiol 78: 43-56. doi: 10.1016/S0168-1605(02)00241-6. PubMed: 12222637. [DOI] [PubMed] [Google Scholar]
- 9. Wegener HC (2003) Antibiotics in animal feed and their role in resistance development. Curr Opin Microbiol 6: 439-445. doi: 10.1016/j.mib.2003.09.009. PubMed: 14572534. [DOI] [PubMed] [Google Scholar]
- 10. Kyaw MH, Lynfield R, Schaffner W, Craig AS, Hadler J et al. (2006) Effect of introduction of the pneumococcal conjugate vaccine on drug-resistant Streptococcus pneumoniae . N Engl J Med 354: 1455-1463. doi: 10.1056/NEJMoa051642. PubMed: 16598044. [DOI] [PubMed] [Google Scholar]
- 11. Dogovski C, Atkinson SC, Dommaraju SR, Hor L, Dobson RCJ, et al. (2009) Lysine biosynthesis in bacteria – an unchartered pathway for novel antibiotic design. In: Doelle H. Encyclopedia of life support systems, Volume 11 (Biotechnology Part I). Oxford: EOLSS Publishers; . pp 116–136 [Google Scholar]
- 12. Dogovski C, Atkinson SC, Dommaraju SR, Downton M, Hor L et al. (2012) Enzymology of bacterial lysine biosynthesis. In: Ekinci D. Biochemistry: InTech Open Access Publisher. pp 225-262. [Google Scholar]
- 13. Hutton CA, Perugini MA, Gerrard JA (2007) Inhibition of lysine biosynthesis: an emerging antibiotic strategy. Mol Biosyst 3: 458-465. doi: 10.1039/b705624a. PubMed: 17579770. [DOI] [PubMed] [Google Scholar]
- 14. Yugari Y, Gilvarg C (1965) The condensation step in diaminopimelate synthesis. J Biol Chem 240: 4710-4716. PubMed: 5321309. [PubMed] [Google Scholar]
- 15. Hutton CA, Southwood TJ, Turner JJ (2003) Inhibitors of lysine biosynthesis as antibacterial agents. Mini Rev Med Chem 3: 115-127. doi: 10.2174/1389557033405359. PubMed: 12570844. [DOI] [PubMed] [Google Scholar]
- 16. Burgess BR, Dobson RCJ, Bailey MF, Atkinson SC, Griffin MDW et al. (2008) Structure and evolution of a novel dimeric enzyme from a clinically-important bacterial pathogen. J Biol Chem 283: 27598-27603. doi: 10.1074/jbc.M804231200. PubMed: 18684709. [DOI] [PubMed] [Google Scholar]
- 17. Girish TS, Sharma E, Gopal B (2008) Structural and functional characterization of Staphylococcus aureus dihydrodipicolinate synthase. FEBS Lett 582: 2923-2930. doi: 10.1016/j.febslet.2008.07.035. PubMed: 18671976. [DOI] [PubMed] [Google Scholar]
- 18. Kaur N, Gautam A, Kumar S, Singh A, Singh N et al. (2011) Biochemical studies and crystal structure determination of dihydrodipicolinate synthase from Pseudomonas aeruginosa . Int J Biol Macromol 48: 779-787. doi: 10.1016/j.ijbiomac.2011.03.002. PubMed: 21396954. [DOI] [PubMed] [Google Scholar]
- 19. Schnell R, Oehlmann W, Sandalova T, Braun Y, Huck C et al. (2012) Tetrahydrodipicolinate N-succinyltransferase and dihydrodipicolinate synthase from Pseudomonas aeruginosa: structure analysis and gene deletion. PLOS ONE 7: e31133. doi: 10.1371/journal.pone.0031133. PubMed: 22359568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Atkinson SC, Dogovski C, Downton MT, Pearce FG, Reboul CF et al. (2012) Crystal, solution and in silico structural studies of dihydrodipicolinate synthase from the common grapevine. PLOS ONE 7: e38318. doi: 10.1371/journal.pone.0038318. PubMed: 22761676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Atkinson SC, Dogovski C, Downton MT, Czabotar PE, Dobson RCJ et al. (2013) Structural, kinetic and computational investigation of Vitis vinifera DHDPS reveals new insight into the mechanism of lysine-mediated allosteric inhibition. Plant Mol Biol 81: 431-446. doi: 10.1007/s11103-013-0014-7. PubMed: 23354837. [DOI] [PubMed] [Google Scholar]
- 22. Blagova E, Levdikov V, Milioti N, Fogg MJ, Kalliomaa AK et al. (2006) Crystal structure of dihydrodipicolinate synthase (BA3935) from Bacillus anthracis at 1.94 Å resolution. Proteins 62: 297-301. PubMed: 16287120. [DOI] [PubMed] [Google Scholar]
- 23. Blickling S, Beisel HG, Bozic D, Knäblein J, Laber B et al. (1997) Structure of dihydrodipicolinate synthase of Nicotiana sylvestris reveals novel quaternary structure. J Mol Biol 274: 608-621. doi: 10.1006/jmbi.1997.1393. PubMed: 9417939. [DOI] [PubMed] [Google Scholar]
- 24. Devenish SRA, Huisman FHA, Parker EJ, Hadfield AT, Gerrard JA (2009) Cloning and characterisation of dihydrodipicolinate synthase from the pathogen Neisseria meningitides . Biochim Biophys Acta 1794: 1168-1174. doi: 10.1016/j.bbapap.2009.02.003. PubMed: 19236959. [DOI] [PubMed] [Google Scholar]
- 25. Dobson RC, Griffin MD, Jameson GB, Gerrard JA (2005) The crystal structures of native and (S)-lysine-bound dihydrodipicolinate synthase from Escherichia coli with improved resolution show new features of biological significance. Acta Crystallogr D Biol Crystallogr 61: 1116-1124. doi: 10.1107/S0907444905016318. PubMed: 16041077. [DOI] [PubMed] [Google Scholar]
- 26. Griffin MDW, Dobson RCJ, Pearce FG, Antonio L, Whitten AE et al. (2008) Evolution of quaternary structure in a homotetrameric protein. J Mol Biol 380: 691-703. doi: 10.1016/j.jmb.2008.05.038. PubMed: 18556019. [DOI] [PubMed] [Google Scholar]
- 27. Griffin MDW, Dobson RCJ, Gerrard JA, Perugini MA (2010) Exploring the dimer-dimer interface of the dihydrodipicolinate synthase tetramer: how resilient is the interface? Arch Biochem Biophys 494: 58-63. doi: 10.1016/j.abb.2009.11.014. PubMed: 19919824. [DOI] [PubMed] [Google Scholar]
- 28. Griffin MDW, Billakanti JM, Wason A, Keller S, Mertens HDT et al. (2012) Characterisation of lysine biosynthetic enzymes in Arabidopsis thaliana . PLOS ONE 7: e40318. doi: 10.1371/journal.pone.0040318. PubMed: 22792278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kang BS, Kim YG, Ahn JW, Kim KJ (2010) Crystal structure of dihydrodipicolinate synthase from Hahella chejuensis at 1.5 Å resolution. Int J Biol Macromol 46: 512-516. doi: 10.1016/j.ijbiomac.2010.03.005. PubMed: 20227435. [DOI] [PubMed] [Google Scholar]
- 30. Kefala G, Evans GL, Griffin MD, Devenish SR, Pearce FG et al. (2008) Crystal structure and kinetic study of dihydrodipicolinate synthase from Mycobacterium tuberculosis . Biochem J 411: 351-360. doi: 10.1042/BJ20071360. PubMed: 18062777. [DOI] [PubMed] [Google Scholar]
- 31. Mirwaldt C, Korndörfer I, Huber R (1995) The crystal structure of dihydrodipicolinate synthase from Escherichia coli at 2.5 Å resolution. J Mol Biol 246: 227-239. doi: 10.1006/jmbi.1994.0078. PubMed: 7853400. [DOI] [PubMed] [Google Scholar]
- 32. Padmanabhan B, Strange RW, Antonyuk SV, Ellis MJ, Hasnain SS et al. (2009) Structure of dihydrodipicolinate synthase from Methanocaldococcus jannaschii . Acta Crystallogr Sect F Struct Biol Cryst Commun 65: 1222-1226. doi: 10.1107/S0907444909037421. PubMed: 20054116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pearce FG, Perugini MA, McKerchar HJ, Gerrard JA (2006) Dihydrodipicolinate synthase from Thermotoga maritima . Biochem J 400: 359-366. doi: 10.1042/BJ20060771. PubMed: 16872276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Perugini MA, Griffin MDW, Smith BJ, Webb LE, Davis AJ et al. (2005) Insight into the self-association of key enzymes from pathogenic species. Eur Biophys J 34: 469-476. doi: 10.1007/s00249-005-0491-y. PubMed: 15981001. [DOI] [PubMed] [Google Scholar]
- 35. Rice EA, Bannon GA, Glenn KC, Jeong SS, Sturman EJ et al. (2008) Characterization and crystal structure of lysine insensitive Corynebacterium glutamicum dihydrodipicolinate synthase (cDHDPS) protein. Arch Biochem Biophys 480: 111-121. doi: 10.1016/j.abb.2008.09.018. PubMed: 18930704. [DOI] [PubMed] [Google Scholar]
- 36. Voss JE, Scally SW, Taylor NL, Atkinson SC, Griffin MDW et al. (2010) Substrate-mediated stabilization of a tetrameric drug target reveals achilles heel in anthrax. J Biol Chem 285: 5188-5195. doi: 10.1074/jbc.M109.038166. PubMed: 19948665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wei BP, Shepherd RK, Robins-Browne RM, Clark GM, O'Leary SJ (2006) Pneumococcal meningitis: development of a new animal model. Otol Neurotol 27: 844-854. doi: 10.1097/01.mao.0000231603.25961.f1. PubMed: 16936571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Van de Rijn I, Kessler RE (1980) Growth characteristics of group A streptococci in a new chemically defined medium. Infect Immun 27: 444-448. PubMed: 6991416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: A Laboratory Manual. Cold Spring Harbor: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 40. Chalker AF, Minehart HW, Hughes NJ, Koretke KK, Lonetto MA et al. (2001) Systematic identification of selective essential genes in Helicobacter pylori by genome prioritization and allelic replacement mutagenesis. J Bacteriol 183: 1259-1268. doi: 10.1128/JB.183.4.1259-1268.2001. PubMed: 11157938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Macrina FL, Tobian JA, Jones KR, Evans RP, Clewell DB (1982) A cloning vector able to replicate in Escherichia coli and Streptococcus sanguis . Gene 19: 345-353. doi: 10.1016/0378-1119(82)90025-7. PubMed: 6295886. [DOI] [PubMed] [Google Scholar]
- 42. Pozzi G, Masala L, Iannelli F, Manganelli R, Håy Varstein LS et al. (1996) Competence for genetic transformation in encapsulated strains of Streptococcus pneumoniae: two allelic variants of the peptide pheromone. J Bacteriol 178: 6087-6090. PubMed: 8830714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sibarani NE, Gorman MA, Dogovski C, Parker MW, Perugini MA (2010) Crystallization of dihydrodipicolinate synthase from a clinical isolate of Streptococcus pneumoniae . Acta Crystallogr Sect F Struct Biol Cryst Commun 66: 32-36. doi: 10.1107/S0108767309041749. PubMed: 20057065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Domigan LJ, Scally SW, Fogg MJ, Hutton CA, Perugini MA et al. (2009) Characterisation of dihydrodipicolinate synthase from Bacillus anthracis . Biochim Biophys Acta 1794: 1510-1516. doi: 10.1016/j.bbapap.2009.06.020. PubMed: 19595801. [DOI] [PubMed] [Google Scholar]
- 45. Karsten WE (1997) Dihydrodipicolinate synthase from Escherichia coli: pH dependent changes in the kinetic mechanism and kinetic mechanism of allosteric inhibition by L-lysine. Biochemistry 36: 1730-1739. doi: 10.1021/bi962264x. PubMed: 9048556. [DOI] [PubMed] [Google Scholar]
- 46. Roberts SJ, Morris JC, Dobson RCJ, Gerrard JA (2003) The preparation of (S)-aspartate semi-aldehyde appropriate for use in biochemical studies. Bioorg Med Chem Lett 13: 265-267. PubMed: 12482436. [DOI] [PubMed] [Google Scholar]
- 47. Davis AJ, Perugini MA, Smith BJ, Stewart JD, Ilg T et al. (2004) Properties of GDP-mannose pyrophosphorylase, a critical enzyme and drug target in Leishmania mexicana . J Biol Chem 279: 12462-12468. PubMed: 14718535. [DOI] [PubMed] [Google Scholar]
- 48. Sreerama N, Venyaminov SY, Woody RW (2000) Estimation of protein secondary structure from circular dichroism spectra: inclusion of denatured proteins with native proteins in the analysis. Anal Biochem 287: 243-251. doi: 10.1006/abio.2000.4879. PubMed: 11112270. [DOI] [PubMed] [Google Scholar]
- 49. Sreerama N, Woody RW (2000) Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal Biochem 287: 252-256. doi: 10.1006/abio.2000.4880. PubMed: 11112271. [DOI] [PubMed] [Google Scholar]
- 50. McLennan N, Masters M (1998) GroE is vital for cell-wall synthesis. Nature 392: 139. doi: 10.1038/32317. PubMed: 9515958. [DOI] [PubMed] [Google Scholar]
- 51. Schuck P (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys J 78: 1606-1619. doi: 10.1016/S0006-3495(00)76713-0. PubMed: 10692345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schuck P, Perugini MA, Gonzales NR, Howlett GJ, Schubert D (2002) Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys J 82: 1096-1111. doi: 10.1016/S0006-3495(02)75469-6. PubMed: 11806949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hayes DB, Laue T, Philo J (2003) Sedimentation Interpretation Program, Version 1.08. Durham, New Hampshire: University of New Hampshire. [Google Scholar]
- 54. Laue TM, Shah BD, Ridgeway TM, Pelletier SL (1992) Computer-aided interpretation of analytical sedimentation data for proteins. In Harding SE, Rowe AJ, Horton JC, Analytical ultracentrifugation in biochemistry and polymer science. Royal Society of Chemistry, Cambridge, UK: pp 90–125. [Google Scholar]
- 55. Vistica J, Dam J, Balbo A, Yikilmaz E, Mariuzza RA et al. (2004) Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal Biochem 326: 234-256. doi: 10.1016/j.ab.2003.12.014. PubMed: 15003564. [DOI] [PubMed] [Google Scholar]
- 56. McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM et al. (2002) Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. J Synchrotron Radiat 9: 401-406. doi: 10.1107/S0909049502015170. PubMed: 12409628. [DOI] [PubMed] [Google Scholar]
- 57. Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307-326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 58. Evans P (2006) Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 62: 72-82. doi: 10.1107/S0907444905036693. PubMed: 16369096. [DOI] [PubMed] [Google Scholar]
- 59. McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ (2005) Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr 61: 458-464. doi: 10.1107/S0907444905001617. PubMed: 15805601. [DOI] [PubMed] [Google Scholar]
- 60. Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ et al. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58: 194-1954. PubMed: 12393927. [DOI] [PubMed] [Google Scholar]
- 61. Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126-2132. doi: 10.1107/S0907444904019158. PubMed: 15572765. [DOI] [PubMed] [Google Scholar]
- 62. Kabsch W (2010) XDS. Acta Crystallogr D Biol Crystallogr 66: 125-132. doi: 10.1107/S0907444909047337. PubMed: 20124692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Vagin A (1997) MOLREP: an automated program for molecular replacement. J Appl Crystallography 30: 1022-1025. doi: 10.1107/S0021889897006766. [DOI] [Google Scholar]
- 64. Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53: 240-255. doi: 10.1107/S0907444996012255. PubMed: 15299926. [DOI] [PubMed] [Google Scholar]
- 65. Perugini MA, Schuck P, Howlett GJ (2000) Self-association of human apolipoprotein E3 and E4 in the presence and absence of phospholipid. J Biol Chem 275: 36758-36765. doi: 10.1074/jbc.M005565200. PubMed: 10970893. [DOI] [PubMed] [Google Scholar]
- 66. Perugini MA, Schuck P, Howlett GJ (2002) Differences in the binding capacity of human apolipoprotein E3 and E4 to size-fractionated lipid emulsions. Eur J Biochem 269: 5939-5949. doi: 10.1046/j.1432-1033.2002.03319.x. PubMed: 12444983. [DOI] [PubMed] [Google Scholar]
- 67. Cornish-Bowden A (2004) Fundamental enzyme kinetics. 3rd edition. Portland Press Ltd, London. [Google Scholar]
- 68. Dobson RCJ, Valegård K, Gerrard JA (2004) The crystal structure of three site-directed mutants of Escherichia coli dihydrodipicolinate synthase: further evidence for a catalytic triad. J Mol Biol 338: 329-339. doi: 10.1016/j.jmb.2004.02.060. PubMed: 15066435. [DOI] [PubMed] [Google Scholar]
- 69. Laber B, Gomis-Rüth FX, Romão MJ, Huber R (1992) Escherichia coli dihydrodipicolinate synthase. Identification of the active site and crystallization. Biochem J 288: 691-695. PubMed: 1463470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Blickling S, Renner C, Laber B, Pohlenz H-D, Holak TA et al. (1997) Reaction mechanism of Escherichia coli dihydrodipicolinate synthase investigated by X-ray crystallography and NMR spectroscopy. Biochemistry 36: 24–33. doi: 10.1021/bi962272d. PubMed: 8993314. [DOI] [PubMed] [Google Scholar]
- 71. Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372: 774-797. doi: 10.1016/j.jmb.2007.05.022. PubMed: 17681537. [DOI] [PubMed] [Google Scholar]
- 72. Nordberg P, Monnet DL, Cars O (2005) Antibacterial resistance. Background document for the WHO project: priority medicines for Europe and the World. a public health approach to innovation. Geneva: WHO. [Google Scholar]
- 73. Novak R, Henriques B, Charpentier E, Normark S, Tuomanen E (1999) Emergence of vancomycin tolerance in Streptococcus pneumoniae . Nature 399: 590-593. doi: 10.1038/21202. PubMed: 10376600. [DOI] [PubMed] [Google Scholar]
- 74. Reboul CF, Porebski BT, Griffin MDW, Dobson RCJ, Perugini MA et al. (2012) Structural and dynamic requirements for optimal activity of the essential bacterial enzyme dihydrodipicolinate synthase. PLoS Comput Biol 8: e1002537 PubMed: 22685390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dietrich C, Nato A, Bost B, Le Maréchal P, Guyonvarch A (2009) Regulation of ldh expression during biotin-limited growth of Corynebacterium glutamicum . Microbiology 155: 1360-1375. doi: 10.1099/mic.0.022004-0. PubMed: 19332837. [DOI] [PubMed] [Google Scholar]
- 76. Garvie EI (1980) Bacterial lactate dehydrogenases. Microbiol Rev 44: 106-139. PubMed: 6997721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dowd SR, Pratt EA, Sun Z-Y, Ho C (1995) Nature and environment of the sulfhydryls of membrane-associated D-lactate dehydrogenase of Escherichia coli . Biochim Biophys Acta 1252: 278-283. doi: 10.1016/0167-4838(95)00121-A. PubMed: 7578234. [DOI] [PubMed] [Google Scholar]
- 78. Ostendorp R, Auerbach G, Jaenicke R (1996) Extremely thermostable L (+)-lactate dehydrogenase from Thermotogamaritima: cloning, characterization, and crystallization of the recombinant enzyme in its tetrameric and octameric state. Protein Sci 5: 862-873 [DOI] [PMC free article] [PubMed]
- 79. Sun Z, Do PM, Rhee MS, Govindasamy L, Wang Q et al. (2012) Amino acid substitutions at glutamate-354 in dihydrolipoamide dehydrogenase of Escherichia coli lower the sensitivity of pyruvate dehydrogenase to NADH. Microbiology 158: 1350-1358. doi: 10.1099/mic.0.055590-0. PubMed: 22343352. [DOI] [PubMed] [Google Scholar]
- 80. Gerrard JA, Hutton CA, Perugini MA (2007) Inhibiting protein-protein interactions as an emerging paradigm for drug discovery. Mini Rev Med Chem 7: 151-157. doi: 10.2174/138955707779802561. PubMed: 17305589. [DOI] [PubMed] [Google Scholar]
- 81. Wells JA, McClendon CL (2007) Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 450: 1001-1009. doi: 10.1038/nature06526. PubMed: 18075579. [DOI] [PubMed] [Google Scholar]