

Abstract
The authors describe a 16-year-old boy, previously healthy, who was admitted to our hospital for left-sided spontaneous pneumothorax. On physical examination he presented with marfanoid habitus. Pneumothorax was managed conservatively with resolution. Four months later he had a recurrence of left-sided pneumothorax and 1 week after that he presented with contralateral pneumothorax. He underwent video-assisted thoracoscopic surgery twice for bullectomy and pleurodesis. No further recurrence was stated. Additional investigation showed a prolapsed cardiac mitral valve and Marfan syndrome was confirmed genetically.
Background
Spontaneous pneumothorax (SP) is a relatively rare condition in the paediatric population with a bimodal peak age of occurrence, with most cases occurring either in the neonatal period or in late adolescence. In children, SP is often caused by a tear in the visceral pleura due to the rupture of a subpleural bulla, and often affects tall individuals with an asthenic habitus.1
Marfan syndrome is a common inherited connective tissue disorder with typical skeletal, ocular and cardiovascular manifestations. Pulmonary involvement occurs less frequently, with SP being the most frequently reported (4–11%).2–4
Marfan syndrome should be suspected in patients with SP and marfanoid habitus. The authors highlight the importance of an early diagnosis of this condition and a multidisciplinary approach to these patients.
Case presentation
We report a 16-year-old male, previously healthy, who was admitted to our hospital with left thoracic pain over the last 2 weeks. There was no history of previous trauma, smoking or illicit drug use, fever, cough or shortness of breath; however, he had myopia. The family history was negative for lung disorders.
On physical examination there were no signs of respiratory distress, but he presented decreased breath sounds on the left hemithorax apex. The patient was tall and thin, with long tapered extremities. He presented arachnodactyly, joint hypermobility with positive wrist (Walker) and thumb (Steinberg) signs (figures 1 and 2). Pneumothorax of the left side was confirmed by a chest radiograph. He was admitted for investigation and treatment (figure 3).
Figure 1.
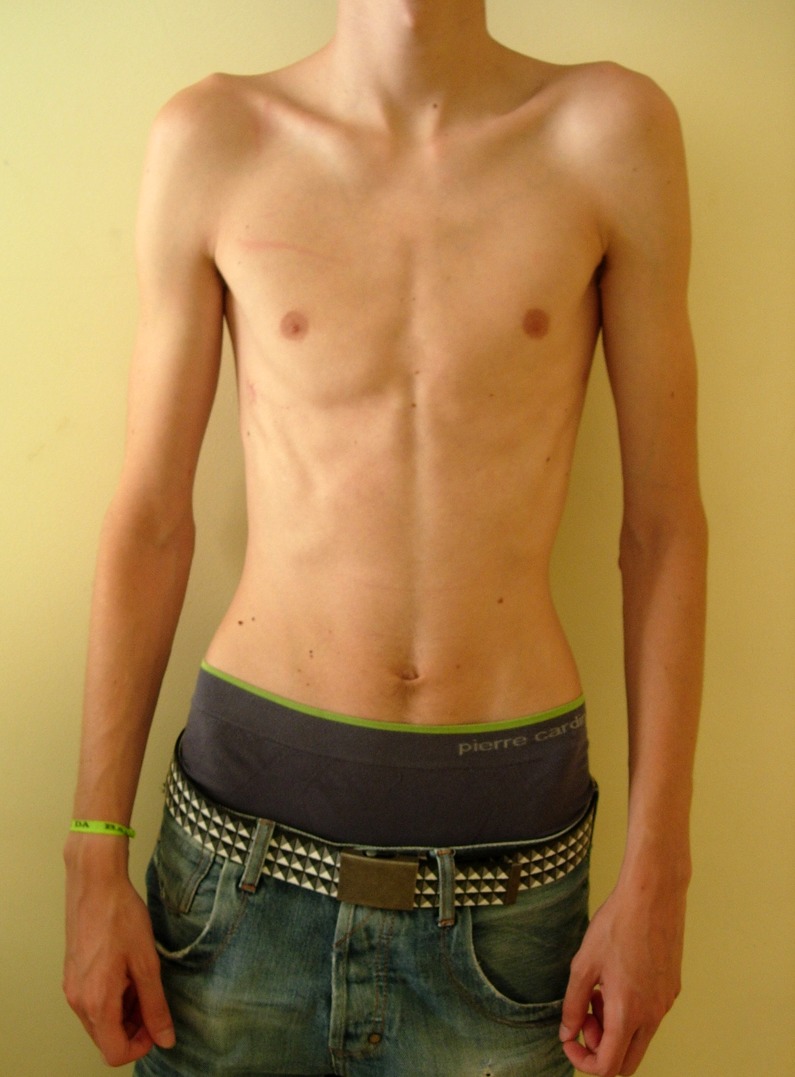
Marphanoid habitus: the patient was tall and thin, with long tapered extremities.
Figure 2.
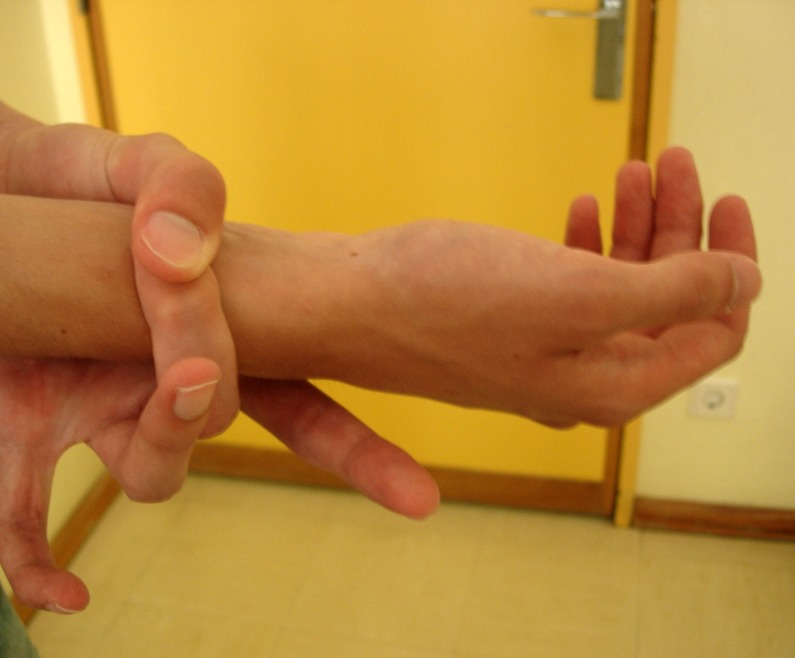
Walker sign.
Figure 3.
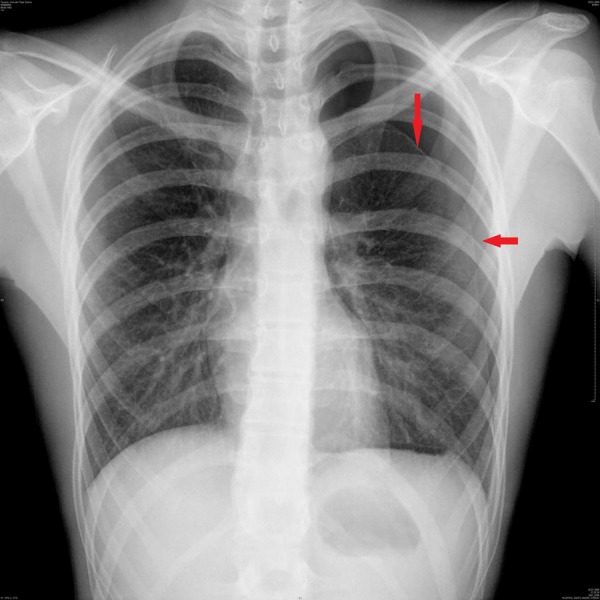
Lung X-ray. The red arrow indicates the pneumothorax.
The pneumothorax was managed with conservative measures (oxygen by facemask and rest), and the patient was discharged clinically well.
Four months later he was admitted to the emergency ward with chest pain, and a recurrence of his left-sided pneumothorax with an identical localisation was stated. He underwent video-assisted thoracoscopic surgery (VATS), with resection of newly identified enfisematous blebs and pleurodesis.
One week after surgery he presented with right-sided pneumothorax. A contralateral VATS was performed for bullectomy and pleurodesis and no further recurrence was stated.
Investigations
During the investigation of the first episode, thoracic CT scan showed a residual left-sided pneumothorax and a small-dimension pleural effusion, no mention to subpleural blebs. Sequential chest X-rays after discharge, confirmed pneumothorax resolution.
Mantoux tuberculin skin test was negative. He presented normal blood levels of α-1 antitrypsin.
Echocardiogram revealed mitral prolapse without regurgitation.
Abdominal ultrasonography presented no alterations.
Marfan’s syndrome was suspected on the basis of characteristic skeletal and cardiovascular findings. The diagnosis was confirmed by genetic testing for FBN1 (Mut. c.1027G>A (p.Gly343Arg) in heterozygoty at exon 9).
Differential diagnosis
At presentation, other lung diseases such as pulmonary tuberculosis, α-1 antitrypsin deficit and chronic lung diseases were ruled out.
Treatment
At the first episode, pneumothorax was managed conservatively. The patient was monitored constantly and several chest X-rays were taken. He was discharged clinically well.
Four months later he had two recurrences. The treatment of choice after recurrence was a surgical approach with VATS for bullectomy and pleurodesis without perioperative complications.
Outcome and follow-up
After the second surgery no further recurrences were stated.
After a 2-year follow-up he is asymptomatic and presents normal respiratory function tests. He is currently on β-blockers (bisoprolol) and maintains follow-up in cardiology with no deterioration of his heart condition.
Discussion
Marfan syndrome is a variable, autosomal dominant connective tissue disorder whose cardinal features affect the cardiovascular system, the eyes and the skeleton.5 Additionally, other systems can be affected (lungs, skin and dura). Pulmonary manifestations include honeycombing, congenital bronchial malformations, bronchiectatic changes in the lower lobes, bullous emphysema and SP.
Two studies published in the 1980s and the Karpman et al2 study in 2011 found a prevalence of pneumothorax in patients with Marfan syndromebetween 4% and 11%, being the most frequently described respiratory feature in those patients.
The increased risk of pneumothorax has been attributed to the presence of apical blebs, bullae, abnormal connective tissue constituents in the lung parenchyma or increased mechanical stresses in the lung apices due to the tall body habitus.2 The causal gene for Marfan’s syndrome, FBN1, encodes the extracellular matrix glycoprotein fibrillin-1, which can be found in the lung as a component of elastic fibres, it has been proposed that abnormalities of fibrillin result in connective tissue friability and laxity. These features can result in chest wall deformity or, within the lung, flaccidity of small airways and terminal bronchioles, predisposing to premature airway closure, obstruction and air trapping leading to degenerative changes and emphysema, which may be the main mechanism for pneumothorax in these individuals.2
Karpman et al2 study showed, however, that not all individuals with Marfan syndrome may have the same increased risk of SP and the use of available imaging studies such as CT scanning, may help physicians to stratify patients into higher and lower risk groups.
There is a lack of consensus between different international guidelines regarding many aspects of pneumothorax management in paediatric age. In patients with Marfan syndrome, those who have an increased risk of recurrence, treatment should be more aggressive, unless there is a very rapid and uncomplicated resolution of SP on medical management.6 According to British Thoracic Society (BTS) guidelines surgical treatment should be considered early in the first episode, within 48 h, in cases of persistent air leak or failure of the lung to re-expand.7
After the initial diagnosis of Marfan syndrome, management by a multidisciplinary team is recommended. Patients should be referred to an ophthalmologist to exclude lens subluxation, refraction and visual correction, to an orthopaedist, for evaluation of skeletal manifestations that may require attention (eg, severe scoliosis, severe pectus excavatum).
The existence of cardiovascular disease, namely aortic root dilation and valvular disease should be evaluated by echocardiography, and a referral to a cardiologist is mandatory even in the absence of severe disease, as medications that reduce hemodynamic stress on the aortic wall, such as β-blockers, are routinely prescribed.
A medical genetics consultation should be provided for genetic counselling and psychological support may be required in order to help patients cope with the fact that they have a chronic disease. Other consultations like cardiothoracic surgery may be required as manifestations of disease appear.8
Clinical evolution of patients with Marfan syndrome is highly variable. Prognosis is highly related to cardiovascular complications, particularly aortic root dilation, which is the main cause of death.9 Over the past 30 years, there has been a steady improvement in life expectancy of patients with Marfan syndrome—a consequence of the advances in treatments and, in particular, early detection of aortic dilation and prophylactic aortic root surgery.9 Hence the importance of monitoring these patients, especially the aortic disease progression.
Learning points.
Secondary spontaneous pneumothorax (SP) is rare in childhood.
Paediatricians should suspect Marfan syndrome when facing a patient with SP and marfanoid habitus.
Treatment of secondary SP is more aggressive, as to prevent recurrence.
A multidisciplinary approach is fundamental in these patients.
Monitoring illness progression, particularly aortic disease and prevention of complications are extremely important in these patients.
Footnotes
Contributors: CV and PR were involved in conception, design and the drafting of the article. CC and MMZ were involved in revising the article critically for intellectual content. MMZ was involved in final approval of the version to be published.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.O'Lone E, Elphick HE, Robinson PJ. Spontaneous pneumothorax in children: when is invasive treatment indicated? Pediatr Pulmonol 2008;43:41–6 [DOI] [PubMed] [Google Scholar]
- 2.Karpman C, Aughenbaugh GL, Ryu JH. Pneumothorax and bullae in Marfan syndrome. Respiration 2011;82:219–24 [DOI] [PubMed] [Google Scholar]
- 3.Hall JR, Pyeritz RE, Dudgeon DL, et al. Pneumothorax in the Marfan syndrome: prevalence and therapy. Ann Thorac Surg 1984;37:500–4 [DOI] [PubMed] [Google Scholar]
- 4.Wood JR, Bellamy D, Child AH, et al. Pulmonary disease in patients with Marfan syndrome. Thorax 1984;39:780–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dean JCS, Davies SJ, Loeys B. Marfan syndrome and related disorders. In: Kumar D, Elliott P. eds. Principles and practice of clinical cardiovascular genetics. chap 10 Oxford University Press, 2010: 157–70 [Google Scholar]
- 6.Rigante D, Segni S, Bush A. Persistent spontaneous pneumothorax in an adolescent with Marfan's syndrome and pulmonary bullous dysplasia. Respiration 2001;68:621–4 [DOI] [PubMed] [Google Scholar]
- 7.MacDuff A, Arnold A, Harvey J, BTS Pleural Disease Guideline Group Management of spontaneous pneumothorax: British Thoracic Society Pleural Disease Guideline 2010. Thorax 2010;65(Suppl 2):ii18–31 [DOI] [PubMed] [Google Scholar]
- 8.Dietz HC. Marfan syndrome. GeneReviews™ [Internet]. Last update: 1 Dec 2011. http://www.ncbi.nlm.nih.gov/books/NBK1335/ [Google Scholar]
- 9.Cañadas V, Vilacosta I, Bruna I, et al. Marfan syndrome. Part 2: treatment and management of patients. Nat Rev Cardiol 2010;7:266–76 [DOI] [PubMed] [Google Scholar]