

Abstract
Angiotensin II (AngII) is the main effector peptide of the renin–angiotensin system (RAS), and contributes to the pathogenesis of cardiovascular disease by exerting its effects on an array of different cell types, including central neurons. AngII intra-neuronal signaling is mediated, at least in part, by reactive oxygen species, particularly superoxide (O2•−). Recently, it has been discovered that mitochondria are a major subcellular source of AngII-induced O2•−. We have previously reported that over-expression of manganese superoxide dismutase (MnSOD), a mitochondrial matrix-localized O2•− scavenging enzyme, inhibits AngII intra-neuronal signaling. Interestingly, over-expression of copper/zinc superoxide dismutase (CuZnSOD), which is believed to be primarily localized to the cytoplasm, similarly inhibits AngII intra-neuronal signaling and provides protection against AngII-mediated neurogenic hypertension. Herein, we tested the hypothesis that CuZnSOD over-expression in central neurons localizes to mitochondria and inhibits AngII intra-neuronal signaling by scavenging mitochondrial O2•−. Using a neuronal cell culture model (CATH.a neurons), we demonstrate that both endogenous and adenovirus-mediated over-expressed CuZnSOD (AdCuZnSOD) are present in mitochondria. Furthermore, we show that over-expression of CuZnSOD attenuates the AngII-mediated increase in mitochondrial O2•− levels and the AngII-induced inhibition of neuronal potassium current. Taken together, these data clearly show that over-expressed CuZnSOD in neurons localizes in mitochondria, scavenges AngII-induced mitochondrial O2•−, and inhibits AngII intra-neuronal signaling.
Abbreviations: AngII, angiotensin II; RAS, renin–angiotensin system; MnSOD, manganese superoxide dismutase; CuZnSOD, copper/zinc superoxide dismutase; AT1R, angiotensin type 1 receptor; NOX, NADPH oxidase; MIMS, mitochondrial inter-membrane space; Ikv, neuronal potassium current; ROS, reactive oxygen species
Keywords: Mitochondria, Angiotensin II, CuZnSOD, Superoxide, Neurons, Potassium current
Graphical abstract
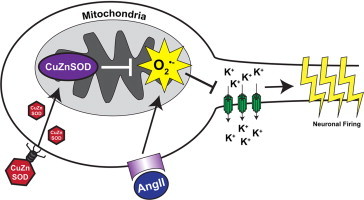
Highlights
-
•
Endogenous CuZnSOD is localized to mitochondria of AngII-sensitive neurons.
-
•
Adenovirus-mediated over-expressed CuZnSOD is localized to neuron mitochondria.
-
•
AngII-induced mitochondrial O2•− flux is attenuated by CuZnSOD over-expression.
-
•
Over-expressed CuZnSOD reduces AngII-mediated inhibition of neuronal K+ current.
Introduction
The brain renin–angiotensin system (RAS) plays an essential role in the pathogenesis of hypertension [1]. Angiotensin II (AngII) is the main effector peptide of the RAS, and elicits its pressor response primarily through angiotensin type 1 receptors (AT1R) [2]. AT1R stimulation initiates a signaling cascade that activates NADPH oxidases (NOX) and increases intra-cellular levels of superoxide (O2•−) [3–5]. While the importance of O2•− in AngII intra-neuronal signaling has been repeatedly and convincingly shown, the exact mechanism of how O2•− acts as an effector molecule and the subcellular localization of its production in AngII signaling is not fully understood.
Superoxide is formed in numerous subcellular compartments, but recent evidence indicates a major location of AngII-induced O2•− production is the mitochondria. For example, mitochondrial O2•− has been shown to mediate both the pressor response and sympathoexcitation induced by AngII in the rostral ventrolateral medulla of the brain [6,7]. Additionally, we have demonstrated that AngII-mediated mitochondrial O2•− leads to the inhibition of neuronal potassium (K+) current [8]. Recently, we reported that one of the NOX isoforms (i.e. NOX4) is located in neuron mitochondria, and contributes to the increase in mitochondrial O2•− flux after AngII stimulation [9]. Due to the significant role mitochondrial O2•− plays in AngII-mediated signaling, increased scavenging of this reactive oxygen species (ROS) could prove beneficial in the treatment of AngII-mediated hypertension.
The superoxide dismutase (SOD) family of enzymes possesses the unique ability to convert O2•− to hydrogen peroxide (H2O2) and oxygen [10]. In mammalian systems, two intra-cellular isoforms of SOD exist: copper/zinc (CuZnSOD) and manganese (MnSOD). MnSOD is exclusively located within the mitochondrial matrix, while CuZnSOD is thought to be primarily located in the cytoplasm of cells [10]. Previous studies have demonstrated that adenovirus-mediated over-expression of either CuZnSOD or MnSOD in the brain decreases blood pressure in hypertensive animal models [5,8,11,12], but how these differentially-located enzymes elicit the same effect is unknown.
Previous studies have shown CuZnSOD to be expressed not only in the cytoplasm, but also in the mitochondrial inter-membrane space (MIMS) of certain cell types including neurons [13–17]. However, the function of mitochondrial-localized CuZnSOD as it relates to AngII intra-neuronal signaling has not been fully elucidated. In addition, it remains unclear if adenoviral-mediated over-expression of CuZnSOD results in increased expression of active CuZnSOD in mitochondria. Here, we present data demonstrating that both endogenously and adenovirus-mediated over-expressed CuZnSOD are associated with the mitochondria of central neurons. Furthermore, we reveal that over-expression of CuZnSOD in neurons attenuates mitochondrial-derived O2•− and inhibition of neuronal potassium current (Ikv) in response to AngII. Overall, this study shows the presence of active CuZnSOD in neuron mitochondria following adenovirus-mediated gene transfer, and furthers the importance of mitochondrial O2•− in AngII intra-neuronal signaling.
Methods and materials
Cell culture
Mouse catecholaminergic neurons (CATH.a cell line, ATCC #11179), were cultured in the RPMI 1640 medium (supplemented with 8% normal horse serum, 4% fetal bovine serum, and 1% penicillin–streptomycin) and maintained in 5% CO2 at 37 °C. Prior to experimentation, neurons were differentiated utilizing N6,2′-O-dibutyryladenosine 3′,5′-cyclic monophosphate sodium salt (1 mM, Sigma, St. Louis, MO, USA) for 6–8 days [8,18].
Adenovirus transduction
For over-expression studies, replication-deficient recombinant adenovirus (Ad5-CMV) encoding human CuZnSOD (AdCuZnSOD) or control vector (AdEmpty) was obtained from ViraQuest Inc. (North Liberty, IA). On day 3 of differentiation, CATH.a neurons were transduced with 50 multiplicity of infection (MOI) of respective virus for 24 h in serum-free media. Complete media was replaced for an additional 4 days post-infection prior to experimentation [19].
Immunofluorescence and confocal microscopy
Differentiated and transduced CATH.a neurons were incubated with 250 nM MitoTracker Red (Invitrogen, Molecular Probes, Carlsbad, CA) for 20 min, as previously described [8], to localize mitochondria. Following this, cells were fixed in 4% paraformaldehyde and subjected to CuZnSOD immunofluorescence staining. Neurons were incubated with blocking buffer (phosphate buffer supplemented with 10% normal horse serum and 0.3% Triton X-100) for 1 h at room temperature, then incubated with primary CuZnSOD antibody (1:500 dilution, The Binding Site Limited, Birmingham, UK) overnight at 4 °C. Following washout of the primary antibody, neurons were incubated with FITC-conjugated secondary antibody (1:500 dilution, The Binding Site Limited, Birmingham, UK) for 2 h at room temperature. Fluorescent images were acquired with a Zeiss 510 Meta Confocal Laser Scanning Microscope.
Mitochondrial isolation
Mitochondria were isolated as previously described [8,20]. Briefly, CATH.a neurons were homogenized in ice-cold buffer A (225 mM mannitol, 65 mM sucrose, 10 mM HEPES, and 1 mM EGTA) using a glass Dounce homogenizer. The homogenates were centrifuged at 500g for 6 min at 4 °C to eliminate cellular debris. The supernatant was collected and centrifuged at 10,000g for 10 min at 4 °C to obtain mitochondria-enriched pellet. This mitochondria-enriched pellet was resuspended with ice-cold buffer B (225 mM mannitol, 65 mM sucrose, and 10 mM HEPES), and washed twice by centrifugation. The final mitochondrial fraction was subjected to standard Western blot analysis.
Western blot analysis
Immunoblotting was performed on whole cell lysates and isolated mitochondrial fractions. Briefly, samples were separated on 4–20% gradient pre-casted denaturing gels, followed by a transfer to nitrocellulose membranes. After blocking, membranes were incubated with primary antibody (CuZnSOD – 1:1000 dilution, Santa Cruz Biotechnology, CA; MnSOD – 1:2000 dilution, Upstate Biotech/Millipore, Billerica, MA; cytochrome c oxidase subunit IV, COXIV – 1:1000 dilution, Abcam, Cambridge, MA; lactate dehydrogenase, LDH – 1:1000 dilution, Abcam, Cambridge, MA; calnexin – 1:1000 dilution, Abcam, Cambridge, MA) overnight at 4 °C. Following washout of primary antibody, membranes were incubated with secondary antibody (1:10,000, Thermo Scientific, Rockford, IL) for 1 h at room temperature. After addition of chemiluminescence substrate (Pierce Enhanced Detection System, Thermo Scientific, Rockford, IL), images were acquired by a UVP Bioimaging System.
SOD activity assay
CuZnSOD and MnSOD activity in whole cell lysates and mitochondrial fractions from CATH.a neurons was determined by a semi-quantitative in-gel activity assay as previously reported [21]. Briefly, 60 µg of protein was separated by electrophoresis on a 12.5% native gel, which was then stained with 2.4 mM nitroblue tetrazolium, 28 µM riboflavin, and 28 mM N,N,N,N-tetramethylethylenediamine for 20 min in dark. Following washout of the staining solution with distilled water, the gel was illuminated under a fluorescent light until achromatic bands appeared. SOD enzymatic activity is indicated by the intensity of achromatic bands.
Mitochondrial superoxide analysis
CATH.a neurons were incubated with MitoSOX Red (Invitrogen, Molecular Probes, Carlsbad, CA), a O2•− sensitive fluorogenic probe, and MitoTracker Green (Invitrogen, Molecular Probes, Carlsbad, CA), a mitochondrial marker, as previously described [8,22]. Briefly, non-transduced control, AdEmpty, and AdCuZnSOD-transduced CATH.a neurons were loaded with 1 µM of MitoSOX Red (excitation: 405 nm and emission: 505–550 nm) for 20 min and 50 nM of MitoTracker Green (excitation: 488 nm and emission: 575–615 nm) for 30 min. Fluorescence images were acquired with a Zeiss 510 Meta Confocal Laser Scanning Microscope before and after addition of 100 nM AngII for 30 min. Individual neurons within an image were identified as a region of interest (ROI) and fluorescence intensity from each ROI was quantified using the Zeiss LSM 510 analysis software. AngII-induced changes in MitoSOX Red fluorescence are reported as fold-change from baseline (pre-AngII) fluorescence.
Electrophysiological recordings
Neuronal K+ currents (Ikv) were recorded from CATH.a neurons using an Axopatch 200B amplifier (Axon Instruments) in the standard whole cell configuration of the patch-clamp technique, as we previously described [19,23]. Current traces were sampled at 10 kHz and filtered at 5 kHz. Holding potential was −80 mV. Current–voltage (I–V) relations were elicited by test potential over the range of −80 to +80 mV with 200-ms duration in 20-mV increments. Using this protocol, we were able to measure peak K+ current (Ipeak), which includes the transient outward K+ current, and the steady-state current (Isteady-state) at the end of the 200-ms pulse. Resulting data were acquired and analyzed with Clampfit 9.2 software (Axon Instruments). The effect of AngII on Ipeak and Isteady-state was tested by superfusing CATH.a neurons with AngII (100 nM) for 5 min and repeating the voltage pulse regimen. Recordings were performed at 22–24 °C.
Statistical analysis
Data are presented as mean±standard error of the mean (SEM) and were analyzed by Student's t-test for two-group comparisons or by ANOVA followed by the Newman–Keuls correction for multiple comparisons. GraphPad Prism 5.0 statistical and graphing software was used. Differences were considered significant at p<0.05.
Results
Endogenously and exogenously expressed CuZnSOD are found in neuron mitochondria
Previous reports have shown CuZnSOD expression in the inter-membrane space of mitochondria in various cell types including neurons [13–17]. To validate that CuZnSOD is present in the mitochondria of AngII-sensitive neurons, mouse catecholaminergic neuronal cells (CATH.a neurons), which have been used extensively to investigate AngII intra-neuronal signaling mechanisms, were studied. Representative confocal microscopy images reveal co-localization of endogenous mouse CuZnSOD with the mitochondrial-targeted fluorescent probe MitoTracker Red (Fig. 1, top row). Furthermore, over-expression of human CuZnSOD via adenoviral-mediated gene transfer drastically enhances this co-localization (Fig. 1, bottom row). Taken together, these data infer the association of CuZnSOD with mitochondria in AngII-sensitive CATH.a neurons.
Fig. 1.
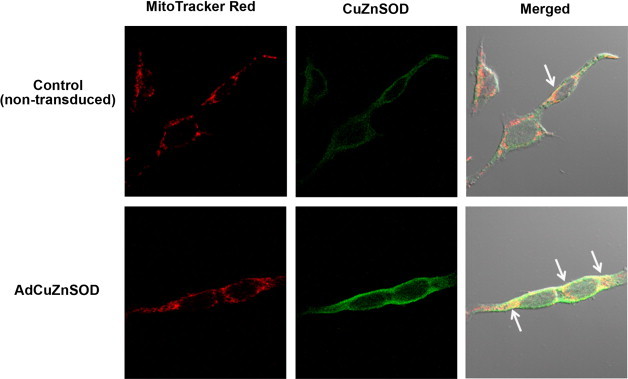
CuZnSOD protein expression in neuron mitochondria. Representative confocal microscopy images showing MitoTracker Red (red fluorescence; left), CuZnSOD (green fluorescence; middle), and merged images (yellow with DIC; right) in control non-transduced CATH.a neurons (top row) or neurons transduced with AdCuZnSOD (50 MOI). Arrows in merged image indicate co-localization of MitoTracker Red and CuZnSOD.
CuZnSOD protein and activity are found in enriched neuron mitochondrial fractions
To further authenticate the localization and function of CuZnSOD in mitochondria of neurons, mitochondrial-enriched sub-cellular fractions were isolated from CATH.a neurons. Western blot analysis confirms the purity of the mitochondrial preparation as only MnSOD and cytochrome C oxidase subunit IV (COXIV), two well-characterized mitochondrial-localized proteins, were enriched in the mitochondrial fraction (Fig. 2A). In contrast, cytosolic (i.e. lactate dehydrogenase [LDH]) and endoplasmic reticulum (i.e. calnexin) proteins demonstrated negligible immunoreactivity in the mitochondrial fraction, implying a highly pure representation of only mitochondria in the preparation (Fig. 2A). Endogenous mouse CuZnSOD (mCuZnSOD) was shown to be located in both the whole cell lysate and mitochondrial fraction as determined by Western blot analysis. This distribution was carried through to adenovirus-mediated over-expressed human CuZnSOD (hCuZnSOD) as well (Fig. 2A). Moreover, endogenous and exogenous (i.e. human) CuZnSOD enzymes demonstrated activity in both the whole cell lysate and mitochondrial fraction as indicated by the in-gel SOD activity assay (Fig. 2B). Overall, these data strongly indicate the localization of catalytically active CuZnSOD in mitochondria of neurons and that over-expressed CuZnSOD protein via adenovirus transduction is robustly expressed and active in neuron mitochondria.
Fig. 2.
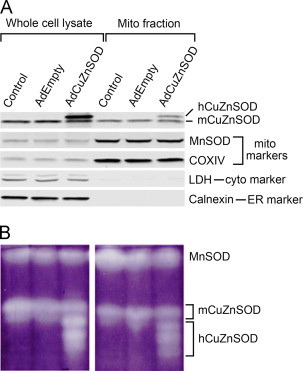
Adenovirus-mediated over-expression of CuZnSOD in CATH.a neurons results in expression of active CuZnSOD in mitochondria. (A) Representative Western blot analysis showing CuZnSOD protein expression in whole cell lysates and mitochondrial fractions collected from control non-transduced, AdEmpty-transduced, or AdCuZnSOD-transduced CATH.a neurons. Adenovirus-expressed human CuZnSOD (hCuZnSOD) is distinguishable from endogenous mouse CuZnSOD (mCuZnSOD). Robust immunoreactivity of mitochondrial markers (MnSOD, COXIV) and lack of immunoreactivity of cytoplasmic (lactate dehydrogenase, LDH) and endoplasmic reticulum (calnexin) markers in the mitochondrial fraction indicate purity of mitochondrial preparation. (B) Representative in-gel SOD activity assay showing activity of MnSOD, endogenous mouse CuZnSOD, and adenovirus-mediated expressed human CuZnSOD in whole cell lysates and mitochondrial fractions collected from non-transduced, AdEmpty-transduced, or AdCuZnSOD-transduced CATH.a neurons.
Over-expression of CuZnSOD attenuates AngII-induced increase in mitochondrial O2•−
We previously reported that AngII significantly increases mitochondrial O2•− in CATH.a neurons, and this escalation is attenuated by over-expressing the mitochondrial-localized MnSOD [8]. Considering our new data (Figs. 1 and 2) showing that CuZnSOD is localized in part to the mitochondria, we aimed to investigate if over-expression of CuZnSOD could also diminish AngII-mediated mitochondrial O2•− flux. To query this, live CATH.a neurons transduced with either AdCuZnSOD or control adenovirus (AdEmpty) were stained with both the mitochondrially-targeted O2•−-sensitive dye, MitoSOX Red, and a fluorescent mitochondrial marker, MitoTracker Green. As expected, in AdEmpty-transduced neurons AngII stimulated an increase (approximately 5.5-fold) in MitoSOX Red fluorescence that co-localized with the MitoTracker Green fluorescence, suggesting an increase in mitochondrial O2•− production (Fig. 3). In contrast, AngII-stimulated neurons transduced with AdCuZnSOD displayed significantly less MitoSOX Red fluorescence (Fig. 3). These data indicate that increased activity of CuZnSOD in neuron mitochondria following adenoviral transduction can attenuate the AngII-mediated increase in mitochondrial O2•− levels.
Fig. 3.
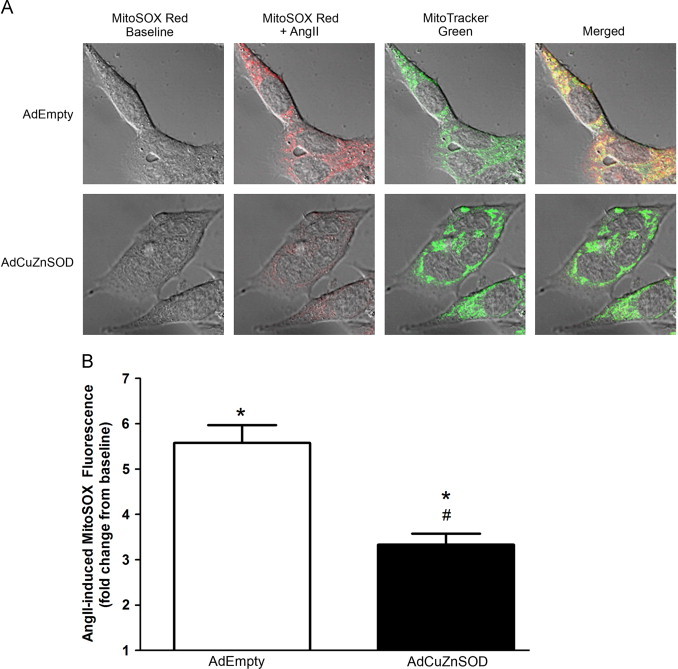
Over-expressed CuZnSOD attenuates AngII-induced increases in mitochondrial O2•− levels. (A) High magnification confocal microscopy images showing baseline MitoSOX Red oxidation (red fluorescence; far left), MitoSOX Red oxidation after 20 min of 100 nM AngII stimulation (red fluorescence; middle left), MitoTracker Green (green fluorescence; middle right), and merged images (yellow with DIC; far right) in CATH.a neurons following adenoviral transduction (50 MOI) of either CuZnSOD (AdCuZnSOD) or vector control (AdEmpty). (B) Quantification of MitoSOX Red fluorescence intensity from confocal images using Zeiss Confocal LSM analysis software. Fluorescence intensity was normalized to baseline MitoSOX red fluorescence in each neuron (n=32 neurons for AdEmpty; n=46 neurons for AdCuZnSOD). *p<0.05 versus baseline fluorescence; #p<0.05 versus AdEmpty.
CuZnSOD over-expression attenuates AngII-induced inhibition of neuronal potassium current (Ikv)
To investigate the functional significance of over-expressing CuZnSOD in neuron mitochondria, whole-cell patch-clamping to elucidate AngII-induced changes in Ikv was performed on AdCuZnSOD-transduced CATH.a neurons. As previously reported [8], AngII significantly decreased outward Ipeak and Isteady-state in non-transduced control neurons and neurons transduced with AdEmpty (Fig. 4). In contrast, this attenuation in Ikv was significantly inhibited in CATH.a neurons over-expressing CuZnSOD (Fig. 4). Taken together, these data indicate that increased expression of active CuZnSOD in neuron mitochondria via adenovirus-mediated gene transfer is sufficient to offset AngII-mediated intra-neuronal signaling and downstream physiological endpoints.
Fig. 4.
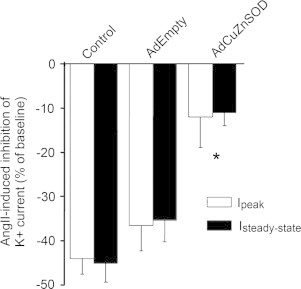
AngII-mediated inhibition of Ikv is attenuated by the over-expression of CuZnSOD. Summary data showing AngII-induced inhibition of Ipeak and Isteady-state in control non-transduced, AdEmpty-transduced, or AdCuZnSOD-transduced CATH.a neurons (n=7–8 neurons per group). Data presented as percent change of Ipeak and Isteady-state from baseline current (Ibaseline−IAngII/Ibaseline). *p<0.05 versus control and AdEmpty.
Discussion
In the present study, we confirm the observation of endogenous CuZnSOD localization to the mitochondria of neurons. Furthermore, we found that over-expression of CuZnSOD via adenoviral transduction significantly increases the levels of active CuZnSOD in neuron mitochondria. This increased expression of CuZnSOD in mitochondria attenuated AngII-mediated increases in mitochondrial O2•− and the AngII-induced inhibition of Ikv. We have previously reported similar findings by over-expressing MnSOD [8], which extends the importance of mitochondrial O2•− in AngII-mediated signaling. While CuZnSOD activity is canonically thought to be cytoplasmic in origin, our data presented here suggest a functional role for the antioxidant enzyme at a specific subcellular organelle. Most importantly, our findings challenge previous interpretations of AdCuZnSOD inhibiting AngII intra-cellular signaling because of its ability to scavenge only cytoplasmic O2•−, and suggest that CuZnSOD over-expression may inhibit AngII signaling because of its ability to scavenge mitochondrial-localized O2•−.
Reactive oxygen species (ROS) are critical mediators of cellular signaling, and one specific ROS, O2•−, has been demonstrated as a main effector molecule in AngII intra-neuronal signaling [4,5,24,25]. One established mechanism of increased O2•− in neurons in response to AngII is NADPH oxidase activation [26,27]. We have recently shown that NADPH oxidase 4 (NOX4) is located in neuron mitochondria, and is a source of O2•− in response to AngII [9]. While NOX4 may be a source of AngII-mediated O2•− in neuron mitochondria, other sources of mitochondrial O2•− have also been proposed. A secondary source of mitochondrial O2•− may derive from the opening of mitochondrial KATP channels (mitoKATP). mitoKATP have shown to be activated in the presence of O2•−, which would occur during time of increased NOX activation and could lead to aberrant mitochondrial function due to electron leakage from complexes I and III [28]. Another source of mitochondrial O2•− can be from the mitochondrial permeability transition (MPT). Similar to the mitoKATP channels, the MPT has been shown to be sensitive to ROS as well as calcium [29]. The opening of the MPT pore alters mitochondrial metabolism and function, and thus creates a viscous cycle of ROS generation following a preliminary ROS stimulus, which may be initiated by AngII and the NOX family of enzymes. Interestingly, while numerous sources of mitochondrial O2•− have been identified as potential manufacturers of the AngII-mediated ROS, very little is known about the O2•− spatial location within the mitochondria. O2•− produced on the matrix side of the mitochondria may exert very different effects from O2•− produced in the inter-membrane space. Understanding of the spatial O2•− production in mitochondria may be critical in AngII-mediated intra-neuronal signaling, and thus further studies are warranted to fully elucidate the specific origins of the O2•− in response to AngII.
CuZnSOD was first identified in the MIMS by Irwin Fridovich in 1973 [17], but how CuZnSOD is transported into the MIMS has remained elusive. CuZnSOD requires disulfide formation as well as copper insertion that are performed by the copper chaperone for SOD1 (CCS) [30,31]. Interestingly, CuZnSOD has been shown to only be able to cross the outer membrane of the mitochondria in its non-metallated and reduced state [32], which means that maturation of CuZnSOD must occur within the mitochondria. The most current proposed mechanism of CuZnSOD translocation is dependent upon the Erv1/Mia40 disulfide relay system, where CCS facilitates the folding, copper insertion, and trapping of CuZnSOD in the MIMS [33]. Recently, Kawamata and Manfredi identified that oxygen tension also affects the import of CuZnSOD to the mitochondria. They demonstrated that 20% oxygen inhibited while 6% oxygen augmented the import of CuZnSOD to the mitochondria [34]. One explanation for this phenomenon could be that in low oxygen tensions, electrons may leak from the electron transport chain at a higher rate due to a lack of molecular oxygen as a final acceptor, which causes increases in mitochondrial O2•− production. The increase in CuZnSOD in the MIMS may be a preventative measure in the counteracting of any potential damage or signaling that could arise from this change in oxygen tension. Overall, while the mechanisms for CuZnSOD translocation to the mitochondria are not fully understood, the amount of energy and cellular machinery involved in transporting CuZnSOD into the mitochondria suggests this is an important and vital cellular process. In the current study, we demonstrate that over-expression of CuZnSOD has the ability to attenuate AngII-mediated increases in mitochondrial O2•− levels. We did not examine if endogenous CuZnSOD mitochondrial import was increased during AngII stimulation, as a potential regulatory mechanism for the increased mitochondrial O2•− production. Additionally, our experiments were performed at 21% oxygen, which may limit the amount of CuZnSOD (both exogenous and endogenous) imported to the mitochondria. In summary, the data presented here demonstrate a novel function for over-expressed CuZnSOD in the attenuation of AngII-mediated mitochondrial O2•− flux in neurons, but more work is necessary to fully elucidate the mechanism of this process.
In conclusion, our present study has demonstrated that over-expression of CuZnSOD via adenovirus-mediated gene transfer in neurons significantly increases both mitochondrial CuZnSOD protein level and enzyme activity. Additionally, the over-expressed CuZnSOD is able to attenuate the increased mitochondrial O2•− levels observed after AngII stimulation, which prevents AngII-induced inhibition of Ikv. These data suggest that mitochondrial-produced O2•− plays a significant role in AngII-mediated neuronal signaling, and that over-expression of CuZnSOD has the ability to diminish these effects. Furthermore, these data challenge the interpretation of previous studies that CuZnSOD over-expression via adenoviral transduction inhibits AngII intra-cellular signaling because it scavenges only cytoplasmic O2•−. Our findings suggest that inhibition of AngII signaling via CuZnSOD over-expression may be due to its mitochondrial localization and ability to scavenge mitochondrial O2•−.
Acknowledgments
This study was supported by a National Institutes of Health Grant R01-HL103942-01 (M.C. Zimmerman) and by an American Heart Association Scientist Development Grant 0930204N (M.C. Zimmerman). A.J. Case is supported by an American Heart Association Postdoctoral Fellowship (13POST17060015). We thank Janice A. Taylor and James R. Talaska of the Confocal Laser Scanning Microscope Core Facility at the University of Nebraska Medical Center for providing assistance with confocal microscopy and the Nebraska Research Initiative and the Eppley Cancer Center for their support of the Core Facility.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Andersson B., Eriksson S., Rundgren M. Angiotensin and the brain. Acta Physiol. Scand. 1995;155:117–125. doi: 10.1111/j.1748-1716.1995.tb09956.x. [DOI] [PubMed] [Google Scholar]
- 2.Allen A.M., Moeller I., Jenkins T.A., Zhuo J., Aldred J.P., Chai S.Y. Mendelsohn FAO. Angiotensin receptors in the nervous system. Brain Res. Bull. 1998;47:17–28. doi: 10.1016/s0361-9230(98)00039-2. [DOI] [PubMed] [Google Scholar]
- 3.Zimmerman M.C. Angiotensin II and angiotensin-1-7 redox signaling in the central nervous system. Curr. Opin. Pharmacol. 2011;11:138–143. doi: 10.1016/j.coph.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zimmerman M.C., Davisson R.L. Redox signaling in central neural regulation of cardiovascular function. Prog. Biophys. Mol. Biol. 2004;84:125–149. doi: 10.1016/j.pbiomolbio.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 5.Zimmerman M.C., Lazartigues E., Lang J.A., Sinnayah P., Ahmad I.M., Spitz D.R., Davisson R.L. Superoxide mediates the actions of angiotensin II in the central nervous system. Circ. Res. 2002;91:1038–1045. doi: 10.1161/01.res.0000043501.47934.fa. [DOI] [PubMed] [Google Scholar]
- 6.Chan S.H., Wu K.L., Chang A.Y., Tai M.H., Chan J.Y. Oxidative impairment of mitochondrial electron transport chain complexes in rostral ventrolateral medulla contributes to neurogenic hypertension. Hypertension. 2009;53:217–227. doi: 10.1161/HYPERTENSIONAHA.108.116905. [DOI] [PubMed] [Google Scholar]
- 7.Nozoe M., Hirooka Y., Koga Y., Araki S., Konno S., Kishi T., Ide T., Sunagawa K. Mitochondria-derived reactive oxygen species mediate sympathoexcitation induced by angiotensin II in the rostral ventrolateral medulla. J. Hypertens. 2008;26:2176–2184. doi: 10.1097/HJH.0b013e32830dd5d3. [DOI] [PubMed] [Google Scholar]
- 8.Yin J.X., Yang R.F., Li S., Renshaw A.O., Li Y.L., Schultz H.D., Zimmerman M.C. Mitochondria-produced superoxide mediates angiotensin II-induced inhibition of neuronal potassium current. Am. J. Physiol.: Cell Physiol. 2010;298:C857–C865. doi: 10.1152/ajpcell.00313.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Case A.J., Li S., Basu U., Tian J., Zimmerman M.C. Mitochondrial-localized nadph oxidase 4 is a source of superoxide in angiotensin II-stimulated neurons. Am. J. Physiol.: Heart Circ. Physiol. 2013;305:H19–H28. doi: 10.1152/ajpheart.00974.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fukai T., Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011;15:1583–1606. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosenbaugh E.G., Roat J.W., Gao L., Yang R.F., Manickam D.S., Yin J.X., Schultz H.D., Bronich T.K., Batrakova E.V., Kabanov A.V., Zucker I.H., Zimmerman M.C. The attenuation of central angiotensin II-dependent pressor response and intra-neuronal signaling by intracarotid injection of nanoformulated copper/zinc superoxide dismutase. Biomaterials. 2010;31:5218–5226. doi: 10.1016/j.biomaterials.2010.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan S.H., Tai M.H., Li C.Y., Chan J.Y. Reduction in molecular synthesis or enzyme activity of superoxide dismutases and catalase contributes to oxidative stress and neurogenic hypertension in spontaneously hypertensive rats. Free Radic. Biol. Med. 2006;40:2028–2039. doi: 10.1016/j.freeradbiomed.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 13.He C., Murthy S., McCormick M.L., Spitz D.R., Ryan A.J., Carter A.B. Mitochondrial Cu,Zn-superoxide dismutase mediates pulmonary fibrosis by augmenting H2O2 generation. J. Biol. Chem. 2011;286:15597–15607. doi: 10.1074/jbc.M110.187377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Igoudjil A., Magrane J., Fischer L.R., Kim H.J., Hervias I., Dumont M., Cortez C., Glass J.D., Starkov A.A., Manfredi G. In vivo pathogenic role of mutant SOD1 localized in the mitochondrial intermembrane space. J. Neurosci.: Off. J. Soc. Neurosci. 2011;31:15826–15837. doi: 10.1523/JNEUROSCI.1965-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inarrea P., Moini H., Rettori D., Han D., Martinez J., Garcia I., Fernandez-Vizarra E., Iturralde M., Cadenas E. Redox activation of mitochondrial intermembrane space Cu,Zn-superoxide dismutase. Biochem. J. 2005;387:203–209. doi: 10.1042/BJ20041683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okado-Matsumoto A., Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J. Biol. Chem. 2001;276:38388–38393. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- 17.Weisiger R.A., Fridovich I. Mitochondrial superoxide simutase. Site of synthesis and intramitochondrial localization. J. Biol. Chem. 1973;248:4793–4796. [PubMed] [Google Scholar]
- 18.Du J.Q., Sun C.W., Tang J.S. Effect of angiotensin II type 1 receptor on delayed rectifier potassium current in catecholaminergic cath.a cells. Acta Pharmacol. Sin. 2004;25:1145–1150. [PubMed] [Google Scholar]
- 19.Yin J.X., Yang R.F., Li S., Renshaw A.O., Li Y.L., Schultz H.D., Zimmerman M.C. Mitochondria-produced superoxide mediates angiotensin II-induced inhibition of neuronal potassium current. Am. J. Physiol.: Cell Physiol. 2010;298:C857–865. doi: 10.1152/ajpcell.00313.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rehncrona S., Mela L., Siesjo B.K. Recovery of brain mitochondrial function in the rat after complete and incomplete cerebral ischemia. Stroke. 1979;10:437–446. doi: 10.1161/01.str.10.4.437. [DOI] [PubMed] [Google Scholar]
- 21.Beauchamp C., Fridovich I. Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Anal. Biochem. 1971;44:276–287. doi: 10.1016/0003-2697(71)90370-8. [DOI] [PubMed] [Google Scholar]
- 22.Zimmerman M.C., Oberley L.W., Flanagan S.W. Mutant SOD1-induced neuronal toxicity is mediated by increased mitochondrial superoxide levels. J. Neurochem. 2007;102:609–618. doi: 10.1111/j.1471-4159.2007.04502.x. [DOI] [PubMed] [Google Scholar]
- 23.Yang R.F., Yin J.X., Li Y.L., Zimmerman M.C., Schultz H.D. Angiotensin-(1-7) increases neuronal potassium current via a nitric oxide-dependent mechanism. Am. J. Physiol.: Cell Physiol. 2011;300:C58–C64. doi: 10.1152/ajpcell.00369.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zimmerman M.C., Lazartigues E., Sharma R.V., Davisson R.L. Hypertension caused by angiotensin ii infusion involves increased superoxide production in the central nervous system. Circ. Res. 2004;95:210–216. doi: 10.1161/01.RES.0000135483.12297.e4. [DOI] [PubMed] [Google Scholar]
- 25.Zimmerman M.C., Sharma R.V., Davisson R.L. Superoxide mediates angiotensin II-induced influx of extracellular calcium in neural cells. Hypertension. 2005;45:717–723. doi: 10.1161/01.HYP.0000153463.22621.5e. [DOI] [PubMed] [Google Scholar]
- 26.Sun C., Sellers K.W., Sumners C., Raizada M.K. Nad(p)h oxidase inhibition attenuates neuronal chronotropic actions of angiotensin II. Circ. Res. 2005;96:659–666. doi: 10.1161/01.RES.0000161257.02571.4b. [DOI] [PubMed] [Google Scholar]
- 27.Zimmerman M.C., Dunlay R.P., Lazartigues E., Zhang Y., Sharma R.V., Engelhardt J.F., Davisson R.L. Requirement for rac1-dependent nadph oxidase in the cardiovascular and dipsogenic actions of angiotensin II in the brain. Circ. Res. 2004;95:532–539. doi: 10.1161/01.RES.0000139957.22530.b9. [DOI] [PubMed] [Google Scholar]
- 28.Zhang D.X., Chen Y.F., Campbell W.B., Zou A.P., Gross G.J., Li P.L. Characteristics and superoxide-induced activation of reconstituted myocardial mitochondrial ATP-sensitive potassium channels. Circ. Res. 2001;89:1177–1183. doi: 10.1161/hh2401.101752. [DOI] [PubMed] [Google Scholar]
- 29.Kowaltowski A.J., Castilho R.F., Vercesi A.E. Mitochondrial permeability transition and oxidative stress. FEBS Lett. 2001;495:12–15. doi: 10.1016/s0014-5793(01)02316-x. [DOI] [PubMed] [Google Scholar]
- 30.Culotta V.C., Klomp L.W., Strain J., Casareno R.L., Krems B., Gitlin J.D. The copper chaperone for superoxide dismutase. J. Biol. Chem. 1997;272:23469–23472. doi: 10.1074/jbc.272.38.23469. [DOI] [PubMed] [Google Scholar]
- 31.Furukawa Y., Torres A.S., O'Halloran T.V. Oxygen-induced maturation of SOD1: a key role for disulfide formation by the copper chaperone ccs. EMBO J. 2004;23:2872–2881. doi: 10.1038/sj.emboj.7600276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Field L.S., Furukawa Y., O'Halloran T.V., Culotta V.C. Factors controlling the uptake of yeast copper/zinc superoxide dismutase into mitochondria. J. Biol. Chem. 2003;278:28052–28059. doi: 10.1074/jbc.M304296200. [DOI] [PubMed] [Google Scholar]
- 33.Mesecke N., Terziyska N., Kozany C., Baumann F., Neupert W., Hell K., Herrmann J.M. A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell. 2005;121:1059–1069. doi: 10.1016/j.cell.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 34.Kawamata H., Manfredi G. Different regulation of wild-type and mutant cu,zn superoxide dismutase localization in mammalian mitochondria. Hum. Mol. Genet. 2008;17:3303–3317. doi: 10.1093/hmg/ddn226. [DOI] [PMC free article] [PubMed] [Google Scholar]