

Abstract
Background
Radiotherapy (RT) plays an important role in the multidisciplinary management of Ewing's Sarcoma (ES), especially in unresectable cases.
Aim
Assessment of efficacy of RT in terms of local control in pediatric patients with primary ES of bone.
Materials and methods
Thirty-six patients younger than 17 years old with ES treated with combined RT and chemotherapy with (N = 14) or without (N = 22) prior surgery from 1981 to 2008 were retrospectively reviewed. Since 1995, they were all treated according to the Spanish Society of Pediatric Oncology protocol (55.5% cases). Those patients received vincristine, ifosfamide, doxorubicin and etoposide. The TNM classification was as follows: 17 T1, 18 T2 and 1 T3; 36 N0; 29 M0, 5 M1a and 2 M1b. Analysis was stratified by treatment: definitive RT or pre/postoperative RT.
Results
The 36 patients (21 male; 15 female) had a median age of 10 years (range 2–17 years). Median follow-up of living patients was 105 months. The 2-year local control (LC) rate for all patients was 88%. Five-year LC rates for patients treated with definitive and pre/postoperative RT were 91% and 86%, respectively. Two-year overall survival and disease-free survival rates for all patients were 68% and 66%, respectively. Low phosphatase alkaline levels and local and distant recurrences were significantly predictive of worse prognosis (P = 0.021, P = 0.011, P = 0.007, respectively).
Conclusion
Radiotherapy with and without surgery is a highly effective local treatment option in the multidisciplinary management of ES in pediatric patients.
Keywords: Ewing's Sarcoma, Local control, Radiotherapy
1. Background
The Ewing family of tumors is the second most common primary osseous malignancy in childhood and adolescence.1 Classically, these tumors originate in the bone, although they can also occur in soft tissue and be the result of a translocation between chromosomes 11 and 22.2 The annual incidence of Ewing Sarcoma (ES) in the United States is 2.93 per million children.3 Approximately 50% of patients with Ewing tumors are aged 10–20 years. Another 20–30% of cases are diagnosed in the first decade. Although cases continue to be diagnosed throughout the third decade and beyond, the incidence is much lower. Since 1970, the survival of patients with ES has increased substantially.4 These tumors are aggressive, and multimodality therapy is always required. The selection of appropriate systemic therapy has been guided by both prospective and randomized studies5–9 integrating a multiagent systemic therapy into induction and adjuvant therapeutic approaches. No randomized study has addressed the selection of local tumor control modalities for patients with ES, but during the past 20 years, local management has generally evolved from definitive radiotherapy (RT) to limb salvage surgical approaches.5,6,9,10 The reason for this evolution is multifactorial and includes the suggestion of a greater local control with surgery, improvements in surgical techniques, and a concern about possible secondary malignancies and late effects caused by RT.11–16
2. Aim
The objective of the present study is to evaluate the clinical characteristics and local control of children patients treated with definitive RT for unresectable ES or with pre/postoperative RT for patients with high risk disease (axial primaries or large residual tumors after induction chemotherapy). Another goal of this study is to find whether there is a significant difference in terms of local control between these two groups. Finally, our objective for this retrospective study is to evaluate other survival outcomes and prognostic factors.
3. Materials and methods
3.1. Patients
The institutional review board approved a retrospective chart review which was conducted for all pediatric (≤17 years old) patients diagnosed with ES between 1981 and 2008 at our institution. We identified 79 patients; 2 were excluded for the following reasons: disease progression before planned RT (N = 1) and wrong diagnosis (N = 1). Of the remaining 77 patients, 41 (53%) were treated with chemotherapy and definitive surgery; 14 (18%) were treated with chemotherapy, definitive surgery (wide local or marginal excision), and RT; and 22 (29%) were treated with chemotherapy and definitive RT. Therefore, the 36 patients, who underwent definitive or pre/postoperative RT for local management, were the subject of this report. The patients in this group were treated on several institutional or national prospective studies of ES that included different protocol guidelines. The TNM classification according to the American Joint Committee on Cancer 2002 staging system for bone cancer was as follows: 17 T1, 18 T2 and 1 T3; 36 N0; 29 M0, 5 M1a and 2 M1b.
3.2. Approach to management
Our practice has largely reflected the evolving cooperative group standards for RT. Since 1995, they were all treated according to the Spanish Society of Pediatric Oncology (SEOP) protocol for ES. This protocol divided patients into five categories: (1) resectable location and no distant metastasis, (2) unresectable location and no distant metastasis, (3) resectable location and lung metastasis, (4) unresectable location and lung metastasis, and (5) multicentric tumor or other distant metastasis. The standard treatment scheme was as follows: induction chemotherapy, surgery (resectable cases) with or without adjuvant radiotherapy, consolidation chemotherapy (concurrent with postoperative radiotherapy if indicated), bone marrow transplant, when indicated, and definitive radiation therapy for unresectable cases.
3.3. Surgery
Fourteen patients (66.2%) were treated with surgery. Four (28.6% of patients treated with surgery) had positive margins and ten (71.4%) poor histological tumor response after induction chemotherapy (>10% viable tumor cells). Limb-salvage surgery was preferred over amputation.
3.4. Systemic therapy
The first protocol used at our institution (T-9) consisted of five cycles of Actinomycin D, doxorubicin, cyclophosphamide, vincristine, methotrexate, and bleomycin administered over a forty-five-week period.17 More recently, pediatric patients have been treated according to the Memorial Sloan-Kettering Cancer Center P6 protocol,18 which includes cycles of cyclophosphamide/doxorubicin/vincristine and cycles of ifosfamide/etoposide, or according to the Pediatric Oncology Group (POG) 8346 protocol,7 which calls for either surgery or definitive RT to be delivered in week 12 after initiation of chemotherapy with cyclophosphamide and doxorubicin (Table 1). The most common scheme used was the one based on the SEOP protocol. Cycles of the SEOP protocol included six induction cycles of vincristine (1.5 mg/m2, day 1), doxorubicin (20 mg/m2, days 1–3; alternating with actinomycin D 0.5 mg/m2 before 2001), ifosfamide (2 g/m2 before 2001 and 3 g/m2 after 2001, days 1–3) and etoposide (150 mg/m2, days 1–3). All patients received consolidation chemotherapy. The most common scheme used was vincristine (1.5 mg/m2) day 1, actinomycin (0.75 mg/m2) days 1–2 and cyclophosphamide (1500 mg/m2) day 1, for a total of eight cycles. Since 1995, end-intensification with megatherapy using high-dose chemotherapy (busulfan and melphalan) and stem cell rescue was delivered in 14 patients (38.9%) according to our bone marrow (BM) transplant protocol. Indications for dose-intensive/myeloablative therapy against ES were: (1) patients with non metastatic unresectable tumors; (2) patients with lung/pleural metastasis, if they had disappeared at week 18 after neoadjuvant chemotherapy; (3) patients with multicentric tumor or with BM metastasis if there had been a response ≥50% at week 18 after neoadjuvant chemotherapy.
Table 1.
Ewing's Sarcoma treatment protocols.
| Protocol | N (%) | Date |
|---|---|---|
| T9 | 10 (27.8%) | 1981–1990 |
| POG/P6 | 2/4 (5.6/11.1%) | 1991–1994 |
| Spanish Society of Pediatric Oncology | 20 (55.5%) | 1995–2008 |
Abbreviations: POG = Pediatric Oncology Group.
3.5. Radiation therapy
Specific characteristics of RT treatment, including dose, fractionation, and technique, were determined by reviewing radiation treatment charts and films. Patients treated before 2000 (53% of cases) had treatment planned with two-dimensional non conformal techniques, in which initial fields encompassed the primary tumor plus a 2 cm margin. Patients treated in 2000 or later were treated with three-dimensional conformal techniques targeting the primary lesion at diagnosis plus a 4 cm craneocaudal margin and 2 cm in the other axis, except in cases where this would result in overdosing of an adjacent critical structure (such us epiphysis, spinal cord or ovary).
Definitive radiation therapy was delivered in the following cases: unresectable location or not enough tumor regression to allow the surgery. Postoperative RT was delivered if the histological tumor response was poor (>10% viable tumor cells) and if the margins were positive after surgery. According to the SEOP protocol, postoperative RT was initiated as soon as possible if there was a residual tumor after surgery or concurrently with the second cycle of the adjuvant chemotherapy (approximately three weeks after surgery) if there was a complete resection. Preoperative radiation therapy was indicated in emergency cases (such us medullar compression) or tumor progression after induction chemotherapy. In our series, there was only one case that received a preoperative radiotherapy. Definitive RT was initiated after eight weeks of the bone marrow transplant when indicated. High-risk patients received more aggressive systemic therapy, but RT techniques were not modified. Twenty-two (61.1%) patients were treated with primary RT. In general, definitive RT tumor volumes have been shrinking with the availability of superior imaging in today's era. The RT consisted of radiation to the entire bone or bones containing the original lesion, followed by a boost to the lesion with a 2–4 cm margin.
Dose was prescribed to cover at least 95% of the planning target volume. PreTighter medial margins were occasionally used in the pelvis to spare the bladder and bowel. All RTs were delivered using megavoltage technology, either 60Co (21% of patients, most of them before 1989) or 6–18 MV photons. The dose of RT for all patients ranged from 43.2 Gy to 64.8 Gy. It was performed on a singular basis. Fraction size was 1.8–2 Gy. The median dose was 55 Gy (range 43.2–64.8 Gy) for patients who received definitive RT and 45 Gy (range 30–55.8 Gy) for patients who received pre/postoperative RT. Local failure in definitive RT cases was defined as growth of the residual abnormality or recurrence of the lesion after a complete resolution on imaging and clinically. Sites of distant metastases (lung and bone) were also routinely irradiated. Additional details regarding RT treatment characteristics are outlined in Table 2.
Table 2.
Radiation therapy characteristics.
| Characteristic | No. of patients (%) |
|---|---|
| RT | |
| Definitive | 22 (61) |
| Postoperative | 13 (36) |
| Preoperative | 1 (3) |
| RT type | |
| Two-dimensional | 21 (58) |
| Three-dimensional | 15 (42) |
| RT dose (Gy) | |
| ≤50 | 17 (47) |
| >50 | 19 (53) |
| Fractionation (cGy) | |
| 180 q.d. | 24 (67) |
| 200 q.d. | 12 (33) |
Abbreviations. RT = radiotherapy; q.d. = once daily.
3.6. Outcome measurement and statistics
All statistical computations were performed using SPSS (version 17.0) statistical software. The Kaplan–Meier product-limit method provided estimates of the following end points: overall survival (OS), disease free survival (DFS), local control (LC), and freedom from distant metastasis (FFDM).19 The start of treatment was defined as either the start of radiation or the date of surgery, whichever was earlier. Survival times were calculated as the time from the start of treatment to either the event or the date of last follow-up. The event for the end points of interest was defined as follows: OS equals death from any cause, DFS equals failure (local or distant), LC equals failure at the local site, and FFDM equals distant metastasis. The OS, DFS, LC, and FFDM rates were calculated based on all incidents until censor. The log-rank test statistic allowed detection of statistically significant differences between strata of selected explanatory variables. Variables included in an univariate analyses were age (≤10 years versus >10 years), tumor volume (<150 cm3 versus ≥150 cm3), resectability (resectable versus unresectable), dose intensive/myeloablative therapy (yes versus no), surgery (yes versus no), radiation therapy (pre/postoperative versus definitive), phosphatase alkaline (≤279 IU/l versus >279 IU/l), year of treatment (<1995 versus ≥1995), lag time between symptom onset and diagnosis (<3 months versus ≥3 months), local recurrence (yes versus no) and distant recurrence (yes versus no).
4. Results
4.1. Patients
Between 1981 and 2008, a total of 36 children patients (defined as <18 years) were treated with radiation therapy for ES at our Institution. The median age at diagnosis was 10 years (range 2–17 years). Nineteen patients were 2–10 years old and 17 patients were older than 10 years. All were treated with chemotherapy (55.5%, according to the SEOP protocol for ES), 13 underwent surgery before RT and 1 after RT. All patients had pathologically confirmed ES. Diagnosis was based on biopsy specimens, tumor morphologic characteristics, and immunohistochemical analysis. Staging was based on the patient's history, physical examination, chest X-ray, X-ray of the primary tumor, blood chemistry test results, complete blood count, computed tomography or magnetic resonance imaging of the primary tumor, and computed tomography of the chest, when available. Patients who presented with metastatic disease at the time of diagnosis were not excluded. We divided patients into two treatment groups: those treated with definitive radiotherapy and those who received pre/postoperative radiotherapy. There were 22 patients in the first group and 14 patients in the other one. The most common site of primary tumor was the pelvis, followed by the spine. More than half the patients had tumors larger than 8 cm. Soft-tissue invasions were common (Table 3).
Table 3.
Patient characteristics.
| Characteristic | Definitive radiotherapy (N = 22) | Pre/postoperative radiotherapy (N = 14) |
|---|---|---|
| Gender | ||
| Male | 17 (77.3%) | 4 (28.6%) |
| Female | 5 (22.7%) | 10 (71.4%) |
| Median age (range), y | 9 (3–17) | 10 (2–14) |
| Clinical presentation | ||
| Swelling | 5 (22.7%) | 4 (28.6%) |
| Functional disability | 8 (36.5%) | 0 |
| Pain | 3 (13.6%) | 3 (21.4%) |
| Neurological symptoms | 3 (13.6%) | 1 (7.1%) |
| Others | 3 (13.6%) | 6 (42.9%) |
| Primary tumor site | ||
| Upper extremity | 0 | 1 (7.1%) |
| Lower extremity | 0 | 5 (35.7%) |
| Head | 1 (4.5%) | 3 (21.5%) |
| Spine | 6 (27.3%) | 0 |
| Ribs | 0 | 3 (21.5%) |
| Scapula | 1 (4.5%) | 1 (7.1%) |
| Pelvis | 14 (63.7%) | 1 (7.1%) |
| Tumor size (cm) | ||
| ≤ 8 | 10 (45.5%) | 7 |
| > 8 | 12 (54.5%) | 7 |
| Metastasis at diagnosis | ||
| Lung | 5 (22.7%) | 0 |
| Bone | 1 (4.5%) | 1 (7.1%) |
| Without metastasis | 16 (72.3%) | 13 (92.9%) |
4.2. Treatment outcome
Median follow-up was 105 months (range 10–222 months) for living patients. There were 5 local relapses (four located in the pelvis and one in the orbit); 4 occurred within the first 2 years. Two out of the five local recurrences were successfully treated with salvage surgery. At 2 and 5 years, the overall LC rate was 88% for all patients. Comparing patient treated with definitive or pre/postoperative RT, there was no significant difference (P = 0.972). Five-year LC rates were 91% and 86%, respectively (Fig. 1). There were 22 deaths (one due to sepsis postchemotherapy, four due to progression of the disease during the oncologic treatment [two of them were initially stage IV], three due to local progression [all of them located at the pelvis] and fourteen due to metastatic relapses in the follow up); 11 occurred within the first 2 years and 19 out of 22 occurred within 5 years. The 2 and 5-year OS rate for all patients was 68% (95% CI: 52–83%) and 44% (95% CI: 28–61%), respectively. There was no significant overall survival difference between patients treated with definitive or pre/postoperative RT (P = 0.395). Two-year OS rates were 60% and 78%, respectively (Fig. 2). The 2 and 5-year DFS rate for all patients was 66% (95% CI: 49–82%) and 48% (95% CI: 31–66%), respectively. Two-year DFS rates for patients treated with definitive or pre/postoperative RT were 61% and 70%, respectively (P = 0.474). Distant metastases (14 cases, 38.9%) were the primary mode of disease recurrence. At 2 and 5 years, the overall FFDM rate was 73% (95% CI: 57–89%) and 53% (95% CI: 35–72%), respectively, for all patients. Although the rate was greater in patients treated with pre/postoperative RT, the difference is not statistically significant. Two-year FFDM rates were 75% in the patients treated with pre/postoperative RT and 64% in the patients treated with definitive RT (P = 0.199). There were 14 distant relapses; 9 occurred within the first 2 years and 13 out of 14 occurred within 5 years.
Fig. 1.
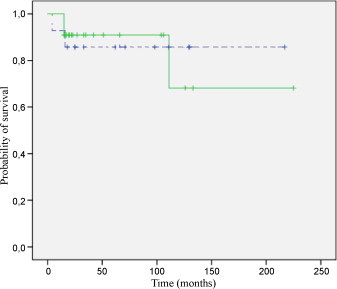
Five-year local control. Solid line, definitive radiotherapy, N = 22; dotted line, pre/postoperative radiotherapy, N = 14.
Fig. 2.
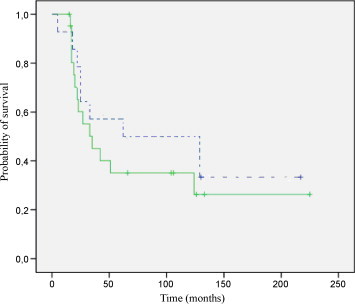
Five-year overall survival. Solid line, definitive radiotherapy, N = 22; dotted line, pre/postoperative radiotherapy, N = 14.
Lungs were the most common site of distant metastasis (6 cases), followed by the bone (one case). Six out of 7 (6 male, one female) metastatic patients had the primary tumor located at the pelvis and were treated with definitive RT after no evidence of metastatic disease was found after induction chemotherapy. Two of them received pulmonary RT. Four were treated with two-dimensional technique and before 1998. Only one out of 6 patients had no tumor recurrence (last follow up: 19 years after diagnosis). The other five plus the one that was treated with pre/postoperative RT died after developing distant metastasis (most of them 2 years after treatment).
In the present study, we compared OS stratified to several factors (Table 4). Phosphatase alkaline was evaluated at diagnosis in 70% of patients. In our laboratory, the lower normal reference limit for phosphatase alkaline is 280 IU/l. We divided these patients in two groups: one >279 IU/l (84%) and the other one ≤279 IU/l (16%). Phosphatase alkaline ≤279 IU/l at diagnosis and local and distant recurrences were significantly predictive of worse prognosis (P = 0.021; P = 0.011; P = 0.007, respectively). We also analyzed the patients’ outcome depending on if they were treated before or after the SEOP protocol, which was initiated in 1995. Fifteen were treated before 1995 while 21 were treated after 1995. Three patients had metastatic disease at diagnosis in the group treated before 1995 and 4 in the other group. Forty percent (6 cases) of patients were treated with definitive RT before 1995 and 76.2% (16 cases) in the other group. Two-year OS rates for all patients were 60% before 1995 and 74% after 1995 (P = 0.356) and two-year LC rates were 80% and 95%, respectively (P = 0.423).
Table 4.
Results of univariate analysis showing risk factors affecting overall mortality.
| Hazard ratio | Confidence interval (95%) | P | |
|---|---|---|---|
| Age (years) | |||
| ≤10 | 1.00 | ||
| >10 | 1.24 | (0.54–2.87) | 0.609 |
| Tumor volume (cm3) | |||
| <150 | 1.00 | ||
| ≥150 | 1.24 | (0.50–3.07) | 0.640 |
| Resectability | |||
| Resectable | 1.00 | ||
| Unresectable | 1.27 | (0.49–3.25) | 0.624 |
| Dose intensive/myeloablative therapy | |||
| Yes | 1.00 | 0.424 | |
| No | 1.45 | (0.59–3.57) | |
| Surgery | |||
| Yes | 1.00 | ||
| No | 1.33 | (0.57–3.08) | 0.508 |
| Radiation therapy | |||
| Pre/postoperative | 1.00 | ||
| Definitive | 1.45 | (0.61–3.47) | 0.403 |
| Phosphatase alkaline (IU/l) | |||
| >279 | 1.00 | ||
| ≤279 | 4.43 | (1.25–15.75) | 0.021 |
| Year of treatment | |||
| ≥1995 | 1.00 | ||
| <1995 | 1.48 | (0.63–3.48) | 0.364 |
| Lag time between symptom onset and diagnosis (months) | |||
| ≥3 | 1.00 | ||
| <3 | 1.65 | (0.71–3.84) | 0.246 |
| Local recurrence | |||
| No | 1.00 | ||
| Yes | 3.76 | (1.35–10.46) | 0.011 |
| Distant recurrence | |||
| No | 1.00 | ||
| Yes | 3.66 | (1.43–9.38) | 0.007 |
5. Discussion
The Ewing family of tumors is a group of highly aggressive neoplasms that occur predominantly in children and young adults.20 Multicenter trials have shown the importance of both aggressive local treatment and adjuvant systemic chemotherapy. The 5-year outcome following multidisciplinary therapy reveals local control rates of 74–93%.8,21–24 Our 2 and 5-year LC rate of 88% is consistent with the literature. With definitive irradiation, the 5-year local regional control rate was 91%. These rates confirm the value of RT for local control. Local control rates were high in both arms: the arm of patients treated with definitive RT and the arm of patients treated with adjuvant RT. It also needs to be reminded that patients treated with definitive RT of our series were those with unresectable locations, most of them in the pelvis. In patients with localized disease at diagnosis, established predictors for poor survival include a large primary tumor and primary tumor of the pelvis.25 Studies report outcomes comparing surgery, RT, or the combination thereof, with little explanation as to selection factors, which dictate local therapy decisions. There never will be, nor should there be a randomized study comparing irradiation versus surgery, because of the inherent bias in the selection of one therapy over another. Resectable lesions are usually small, peripheral in location, and follow a good response to induction chemotherapy. Irradiated lesions are often large, central in location, and follow a poor response to induction chemotherapy. These factors make proper randomization impossible. The most commonly employed local management currently is risk adapted, using local therapy guidelines. Using such an approach, the outcome and function have continuously improved. Radiation therapy standards and accuracy have improved with quality assurance programs. While external beam radiation will continue to be a widely used method, innovative approaches using intraoperative RT or other targeted approaches may also be useful. Technical advances using highly conformal three-dimensional techniques, such as intensity modulated RT and proton therapy in selected cases, will further optimize radiation treatment.
Local and distant recurrences were significantly predictive statistically of worse prognosis. In our study, overall survival and disease free survival rates were almost equal, which suggests the worse prognostic of patients with either local or distant metastasis. The location of the local recurrences observed (such as the pelvis) makes a surgical approach difficult, which has a strong influence on the outcome. On the other hand, we also found that low phosphatase alkaline level at diagnosis was a worse prognostic factor. Since alkaline phosphatase concentrations rise during active bone formation in growth, infants and children normally have levels that may be three times as high as those of adults. Some studies26,27 have shown that age is a prognostic factor for Ewing's Sarcoma, with better prognosis for younger patients. We speculate that patients that have low phosphatase alkaline level at diagnosis are those that are older and, therefore, they could be associated with a worse prognosis. Nevertheless, we consider that this result should be confirmed in studies with more number of patients.
The U.S. Surveillance Epidemiology and End Results database published survival data for Ewing family tumors for patients aged 0–19 years. Five-year OS rates were 42% in 1975–1984 to 58% in 1985–1994 and 60% in 1996–2004. Treatments of our study were delivered from 1981 to 2008 with 2 and 5-year OS rate for all patients of 68% and 44%, respectively. We wanted to assess if the use of modern protocol, such as the SEOP protocol and modern radiotherapy with conformal three-dimensional techniques along with optimal chemotherapy, had an influence on patients’ outcome. Although we did not find statistically significant survival differences between patients treated before or after 1995, we observed a trend of better results in the modern era (two-year OS rates 60% versus 74% and LC rates 80% versus 95%, respectively).
As any study that utilizes an observational database, ours has several limitations. These include: (1) heterogeneity of systemic treatments, (2) small number of patients treated over a period of 30 years during which time advances in imaging have occurred and (3) RT was given at different times during chemotherapy, using different techniques. Despite all these limitations, the results in terms of local control were excellent.
6. Conclusions
We have reported the results of multiagent systemic therapy and RT for patients with ES treated at a single institution during the last decades. Within this group of patients, the LC rates were high in patients treated with definitive radiation and pre/postoperative RT. The findings of this retrospective review highlight the important role of RT in the multidisciplinary management of ES of bone in pediatric patients, typically involving sites difficult to approach with surgical resection. Radiotherapy is also a good option for consolidating the surgical treatment of such patients with high risk disease.
Conflict of interest statement
Authors declare not to have any financial support or relationships that may involve a conflict of interest.
Acknowledgements
The authors wish to acknowledge and thank the following individuals for the contribution to this report: Dr. Alvarez Silan, Dr. Sebastian, and Dr. Robles.
References
- 1.Bernstein M., Kovar H., Paulussen M. Ewing sarcoma family of tumors: Ewing sarcoma of bone and soft tissue and the peripheral primitive neuroectodermal tumors. In: Pizzo P., Poplack D., editors. Principles and practice of pediatric oncology. 5th ed. Lippincott Williams and Wilkins; Philadelphia, PA: 2005. pp. 1002–1032. [Google Scholar]
- 2.de Alava E. Molecular pathology in sarcomas. Clin Transl Oncol. 2007;9(3):130–144. doi: 10.1007/s12094-007-0027-2. [DOI] [PubMed] [Google Scholar]
- 3.Esiashvili N., Goodman M., Marcus R.B., Jr. Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: SEER data. J Pediatr Hematol Oncol. 2008;30(6):425–430. doi: 10.1097/MPH.0b013e31816e22f3. [DOI] [PubMed] [Google Scholar]
- 4.Granowetter L., Womer R., Devidas M. Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children's Oncology Group Study. J Clin Oncol. 2009;27(15):2536–2541. doi: 10.1200/JCO.2008.19.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grier H.E., Krailo M.D., Tarbell N.J. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348:694–701. doi: 10.1056/NEJMoa020890. [DOI] [PubMed] [Google Scholar]
- 6.Paulussen M., Ahrens S., Dunst J. Localized Ewing tumor of bone: final results of the Cooperative Ewing's Sarcoma Study CESS 86. J Clin Oncol. 2001;19:1818–1829. doi: 10.1200/JCO.2001.19.6.1818. [DOI] [PubMed] [Google Scholar]
- 7.Donaldson S.S., Torrey M., Link M.P. A multidisciplinary study investigating radiotherapy in Ewing's sarcoma: end results of POG #8346. Int J Radiat Oncol Biol Phys. 1998;42:125–135. doi: 10.1016/s0360-3016(98)00191-6. [DOI] [PubMed] [Google Scholar]
- 8.Schuck A., Ahrens S., Paulussen M. Local therapy in localized Ewing tumors: results of 1058 patients treated in the CESS 81, CESS 86, and EICESS 92 trials. Int J Radiat Oncol Biol Phys. 2003;55:168–177. doi: 10.1016/s0360-3016(02)03797-5. [DOI] [PubMed] [Google Scholar]
- 9.Rosito P., Mancini A.F., Rondelli R. Italian Cooperative Study for the treatment of children and young adults with localized Ewing sarcoma of bone: a preliminary report of 6 years of experience. Cancer. 1999;86:421–428. doi: 10.1002/(sici)1097-0142(19990801)86:3<421::aid-cncr10>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 10.Raney R.B., Asmar L., Newton W.A., Jr. Ewing's sarcoma of soft tissues in childhood: a report from the Intergroup Rhabdomyosarcoma Study, 1972 to 1991. J Clin Oncol. 1997;15:574–582. doi: 10.1200/JCO.1997.15.2.574. [DOI] [PubMed] [Google Scholar]
- 11.Dunst J., Ahrens S., Paulussen M. Second malignancies after treatment for Ewing's sarcoma: a report of the CESS studies. Int J Radiat Oncol Biol Phys. 1998;42:379–384. doi: 10.1016/s0360-3016(98)00228-4. [DOI] [PubMed] [Google Scholar]
- 12.Kuttesch J.F., Jr., Wexler L.H., Marcus R.B. Second malignancies after Ewing's sarcoma: radiation dose-dependency of secondary sarcomas. J Clin Oncol. 1996;14:2818–2825. doi: 10.1200/JCO.1996.14.10.2818. [DOI] [PubMed] [Google Scholar]
- 13.Bertucio C.S., Wara W.M., Matthay K.K. Functional and clinical outcomes of limb-sparing therapy for pediatric extremity sarcomas. Int J Radiat Oncol Biol Phys. 2001;49:763–769. doi: 10.1016/s0360-3016(00)01415-2. [DOI] [PubMed] [Google Scholar]
- 14.Marinus J., Niël C.G., de Bie R.A. Measuring radiation fibrosis: the interobserver reliability of two methods of determining the degree of radiation fibrosis. Int J Radiat Oncol Biol Phys. 2000;47:1209–1217. doi: 10.1016/s0360-3016(00)00528-9. [DOI] [PubMed] [Google Scholar]
- 15.Okunieff P., Wang X., Rubin P. Radiation-induced changes in bone perfusion and angiogenesis. Int J Radiat Oncol Biol Phys. 1998;42:885–889. doi: 10.1016/s0360-3016(98)00339-3. [DOI] [PubMed] [Google Scholar]
- 16.Schvartzman E., Chantada G., Fandiño A. Results of a stage-based protocol for the treatment of retinoblastoma. J Clin Oncol. 1996;14:1532–1536. doi: 10.1200/JCO.1996.14.5.1532. [DOI] [PubMed] [Google Scholar]
- 17.Rosen G., Caparros B., Nirenberg A. Ewing's sarcoma: ten-year experience with adjuvant chemotherapy. Cancer. 1981;47:2204–2213. doi: 10.1002/1097-0142(19810501)47:9<2204::aid-cncr2820470916>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 18.Kushner B.H., Meyers P.A., Gerald W.L. Very high-dose short-term chemotherapy for poor-risk peripheral primitive neuroectodermal tumors, including Ewing's sarcoma, in children and young adults. J Clin Oncol. 1995;13:2796–2804. doi: 10.1200/JCO.1995.13.11.2796. [DOI] [PubMed] [Google Scholar]
- 19.Kaplan E.L., Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 20.Marcus R.B., Jr., Berrey B.H., Graham-Pole J. The treatment of Ewing's sarcoma of bone at the University of Florida: 1969 to 1998. Clin Orthop Relat Res. 2002;397:290–297. doi: 10.1097/00003086-200204000-00033. [DOI] [PubMed] [Google Scholar]
- 21.Nesbit M.E., Jr., Gehan E.A., Burgert E.O., Jr. Multimodal therapy for the management of primary, nonmetastatic Ewing's sarcoma of bone: a long-term follow-up of the First Intergroup study. J Clin Oncol. 1990;8:1664–1674. doi: 10.1200/JCO.1990.8.10.1664. [DOI] [PubMed] [Google Scholar]
- 22.Burgert E.O., Jr., Nesbit M.E., Garnsey L.A. Multimodal therapy for the management of nonpelvic, localized Ewing's sarcoma of bone: intergroup study IESS-II. J Clin Oncol. 1990;8:1514–1524. doi: 10.1200/JCO.1990.8.9.1514. [DOI] [PubMed] [Google Scholar]
- 23.Dunst J., Jürgens H., Sauer R. Radiation therapy in Ewing's sarcoma: an update of the CESS 86 trial. Int J Radiat Oncol Biol Phys. 1992;32:919–930. doi: 10.1016/0360-3016(95)00016-r. [DOI] [PubMed] [Google Scholar]
- 24.Craft A., Cotterill S., Malcolm A. Ifosfamide-containing chemotherapy in Ewing's sarcoma: The Second United Kingdom Children's Cancer Study Group and the Medical Research Council Ewing's Tumor Study. J Clin Oncol. 1998;16:3628–3633. doi: 10.1200/JCO.1998.16.11.3628. [DOI] [PubMed] [Google Scholar]
- 25.Fizazi K., Dohollou N., Blay J.Y. Ewing's family of tumors in adults: multivariate analysis of survival and long-term results of multimodality therapy in 182 patients. J Clin Oncol. 1998;16:3736–3743. doi: 10.1200/JCO.1998.16.12.3736. [DOI] [PubMed] [Google Scholar]
- 26.Ullmann C., Beck J.D., Holter W. Long-term results following multidisciplinary treatment of localized Ewing's sarcoma in children and adolescents. Strahlenther Onkol. 2008;184(3):137–144. doi: 10.1007/s00066-008-1838-y. [DOI] [PubMed] [Google Scholar]
- 27.Yonemori K., Yamaguchi U., Kaneko M.J. Prediction of response and prognostic factors for Ewing family of tumors in a low incidence population. Cancer Res Clin Oncol. 2008;134(3):389–395. doi: 10.1007/s00432-007-0295-9. [DOI] [PubMed] [Google Scholar]