

Abstract
Deregulation of c-Jun NH2-terminal kinase (JNK) signaling occurs frequently in a variety of human cancers, yet the exact role(s) of JNK deregulation in cancer cell biology remains to be fully elucidated. Our recent demonstration that the activity of JNK is required not only for self-renewal of glioma stem cells but also for their tumor initiation has, however, identified a new role for JNK in the control of the stemness and tumor-initiating capacity of cancer cells. Significantly, transient JNK inhibition was sufficient to cause sustained loss of the tumor-initiating capacity of glioma stem cells, suggesting that the phenotype of “lost tumor-initiating capacity” may be as stable as the differentiated state and that the tumor-initiating capacity might therefore be under the control of JNK through an epigenetic mechanism that also governs stemness and differentiation. Here, in this article, we review the role and mechanism of JNK in the control of this “stemness-associated tumor-initiating capacity” (STATIC), a new hypothetical concept we introduce in this review article. Since the idea of STATIC is essentially applicable to both cancer types that do and do not follow the cancer stem cell hypothesis, we also give consideration to the possible involvement of JNK-mediated control of STATIC in a wide range of human cancers in which JNK is aberrantly activated. Theoretically, successful targeting of STATIC through JNK could contribute to long-term control of cancer. Issues to be considered before clinical application of therapies targeting this JNK-STATIC axis are also discussed.
Keywords: cancer stem cell, epigenetic control, glioblastoma, JNK, stemness, tumor-initiating capacity
Introduction
The c-Jun NH2-terminal kinases (JNKs) are a subgroup of mitogen-activated protein kinases that has long been implicated in cancer development. Although accumulating evidence suggests that JNK may contribute to the development of cancer as a signal transducer of cellular proliferation, differentiation, survival, and migration, the exact role(s) of JNK in cancer still remains to be fully delineated.1-4 Only recently, we and others have shown that JNK is required for self-renewal and the tumor-initiating capacity of human glioma stem cells, giving rise to the novel possibility that the maintenance of cancer stem/initiating cells may be one of the critical roles of JNK in cancer development.5,6 Most importantly, we have demonstrated for the first time using preclinical animal tumor models that therapeutic targeting of JNK is a safe and effective measure to selectively eliminate the tumor-initiating stem cell population of glioblastoma cells in vivo.5 Given the presumed role of cancer stem/initiating cells in therapy resistance and the recurrence of cancer,7,8 our study suggests that JNK could be a potential target molecule to control cancer stem/initiating cells and thus overcome therapy resistance and prevent recurrence, which leads to long-term survival and ultimately the cure of cancer patients. Here, in this review article, we discuss the possible role and mechanism of JNK in the regulation of cancer stem/initiating cells as well as those issues that need to be addressed before we can develop and institute the clinical application of JNK-targeted therapies for the curative treatment of cancer. In addition, since the plausibility of the cancer stem cell concept per se is now being challenged and put to question,9-11 we also try to introduce a novel perspective through which to view the cancer stem cell concept in a different way in order to overcome and reconcile the current controversies over the concept.
Tumor-Initiating Capacity and Self-Renewal: 2 Disparate yet Closely Associated Characteristics of Cancer Stem/Initiating Cells That May Be under the Control of JNK
The cancer stem cell hypothesis posits that tumors are heterogeneous, being composed of a rare subpopulation of tumor cells termed cancer stem cells and the remaining cells accounting for the vast majority of the tumor cells.12-16 The hypothetical cancer stem cells, but not the remaining tumor cells (nonstem cancer cells), possess the capacity to initiate a tumor that reproduces the heterogeneity and characteristics of the original tumor when transplanted in vivo, and at the same time, the cancer stem cells are “immature” tumor cells that retain the self-renewal capacity and potential to differentiate into multiple lineages similarly to normal tissue stem cells. The hypothesis also holds that there is hierarchy between the cancer stem cells and the other cells: cancer stem cells are capable of giving rise to nonstem cancer cells but not vice versa. This core concept of the cancer stem cell hypothesis has been formulated over several decades or even centuries.15,16 Since the 19th century, the histological heterogeneity of tumors has been recognized. Then, a series of tumor cell transplantation studies demonstrated that tumors contain a small number of tumor cells capable of initiating tumors that retain the morphological heterogeneity of the original tumors.17-20 This was followed by the landmark observations made by Pierce and others,21,22 which further embodied and formed the basis of the current cancer stem cell concept. What Pierce and colleagues21,22 found was that, just like their normal tissue counterparts, immature tumor cells are the actively proliferating cells within a tumor and are capable of tumor initiation, and they also give rise to more differentiated progenies that do not proliferate actively and are no longer capable of tumor formation. Also, based on such similarity between tumor tissue homeostasis and that of normal tissues, Pierce and Speers22 described “tumors as caricatures of the process of tissue renewal.” Subsequently, the validity of this hypothetical concept was underscored by the initial identification of a cancer stem cell in leukemia that fitted so nicely into the concept of the cancer stem cell hypothesis,23 and ever since, a large body of evidence has demonstrated that some cancers do follow this cancer stem cell hypothesis. However, on the other hand, mounting evidence now suggests that there are other cancers that apparently do not.9 Significantly, cancers may not necessarily be heterogeneous and/or may have a large subpopulation of tumor-initiating cells, which means that it is indeed difficult and may even be dangerous to generalize the hypothesis as it is.9 If so, then, does it imply that cancer stem cell research is of no value to those cancer types that do not follow the hypothesis? Can we not view the cancer stem cell concept from a different perspective and formulate a new concept that may be more comprehensively applicable to a wide range of cancer types that do and do not follow the cancer stem cell hypothesis?
Here, we introduce a novel hypothetical concept of “stemness-associated tumor-initiating capacity” (STATIC), an evolutionary version of the cancer stem cell hypothesis. Significantly, since Pierce’s observations, researchers have consistently observed that self-renewing, undifferentiated tumor cells have a higher capacity to initiate a tumor compared to their differentiated counterparts.15,24 This is, in a sense, quite surprising because the control of self-renewal/differentiation and that of tumor initiation are both intricate cellular functions apparently disparate from each other except that both may be associated with the capacity to undergo limitless cellular replication. The close association between these apparently distinct cellular mechanisms, however, points to the possibility that they share a common regulatory module. Also, the regulatory module most likely involves epigenetic mechanisms since the control of self-renewal and differentiation is known to be epigenetically controlled.25,26 In addition, the mechanism controlling self-renewal/differentiation should originally be subject to control by cell-extrinsic factors,27 so that cell fate decision may be coordinated to maintain the homeostasis of the whole tissue (e.g., to keep the stem cell population small relative to the entire population). Put together, these ideas are schematically presented in Figure 1A. Figure 1A illustrates that 1) the tumor-initiating capacity is under the control of a common mechanism that maintains the self-renewal capacity/undifferentiated state (= stemness), 2) the mechanism is most likely epigenetic, and 3) the mechanism is subject not only to cell-intrinsic but also to cell-extrinsic factors. According to this hypothetical model (STATIC), the subpopulation of immature tumor-initiating cells may not necessarily be small in cancers in which the extrinsic factor–mediated control mechanism is disrupted (Fig. 1B). In cancers in which the epigenetic control of differentiation is lost (Fig. 1C), tumor-initiating cells may no longer be sorted by markers associated with a cellular stem/differentiation status. It is also possible that self-renewing tumor cells may not necessarily be tumor-initiating cells in some cancers (Fig. 1D). Thus, while being applicable to cancers that do not follow the cancer stem cell hypothesis, on one hand, this hypothetical model, on the other hand, inherits the advantages of the original cancer stem cell hypothesis and has even more:
Figure 1.
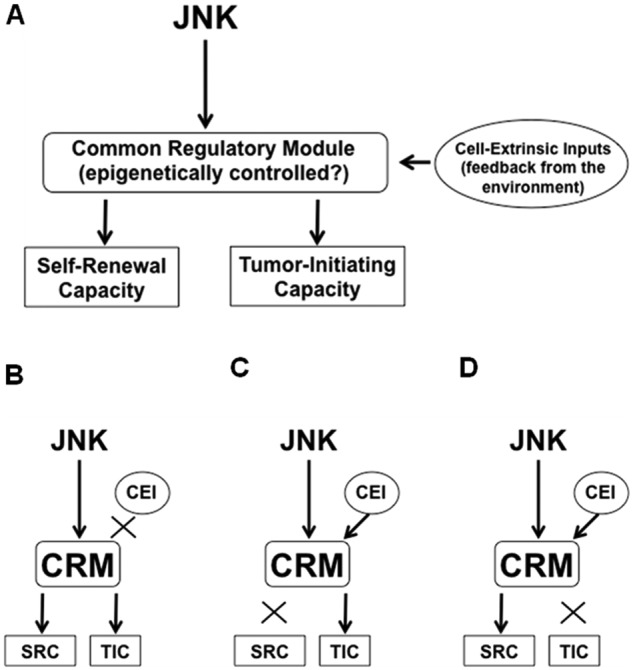
Stemness (= capacity to self-renew as immature, undif- ferentiated cells) and tumor-initiating capacity may be 2 disparate yet closely associated properties of cancer cells that are controlled by a common regulatory module and are also subject to modulation by cell-extrinsic inputs. (A) A hypothetical model for the control of the stemness and tumor-initiating capacity of cancer stem cells. In this hypothetical model, we assume that the cancer stem cell population is kept small within the tumor through feedback inputs from the environment. (B) Once the feedback control mechanism is disrupted, the cancer stem cell population may not be kept small, and consequently, the tumor could lose heterogeneity. (C) Tumor-initiating cells may not necessarily have stem-like properties. Once the mechanism for self-renewal loses the upstream control, cancer cells that do not look like a stem cell may nevertheless have the tumor-initiating capacity regulated in a similar manner to cancer stem cells. (D) Cancer cells that look like a stem cell may not necessarily be tumor-initiating cells. Once the mechanism for tumor initiation loses the upstream control, the small population of immature, stem-like cancer cells may no longer be capable of tumor initiation. Put another way, the self-renewal capacity of the cancer cells may not necessarily reflect their tumor-initiating capacity in this type of cell. CRM = common regulatory module; SRC = self-renewing capacity; TIC = tumor-initiating capacity; CEI = cell-extrinsic input.
The identification of genes/molecules controlling tumor initiation is time consuming and laborious because it inevitably involves animal xenograft studies. However, both the cancer stem cell hypothesis and our new one predict that we can greatly facilitate the identification process by focusing on surrogate markers of tumor initiation. For instance, by using stem cells derived from cancers that conform to the cancer stem cell hypothesis as a model, exploration of candidate molecules can be done simply by in vitro screening of molecules controlling the cellular stem/differentiation status (= surrogate marker of tumor-initiating capacity).
According to the hypotheses, transient targeting of the identified genes/molecules is supposed to provide a sustained inhibitory effect on the tumor-initiating capacity of tumor cells because the condition of “lost tumor-initiating capacity” is expected to be as epigenetically stable as the differentiated state. In case the loss of tumor-initiating capacity is “irreversible,” the therapeutic intervention could have a “curative” effect.
Once identified, it would be feasible and relevant to explore the role of the genes/molecules involved in the regulation of STATIC even in cancers that do not conform to the cancer stem cell hypothesis, as may be explained by Figure 1B and 1C.
Based on a prototypical idea of this hypothetical STATIC model, we set out to search for molecules involved in the control of the tumor-initiation capacity of glioblastoma cells. Consequently, we discovered that JNK is among the key molecules regulating STATIC of glioblastoma cells.5
Role of JNK in the Control of STATIC of Glioblastoma Cells
JNK is more activated in glioma stem cells than in their differentiated counterparts
In our recently reported study,5 we searched for molecules differentially expressed and/or activated in self-renewing glioma stem cells and in those that have undergone serum-induced differentiation, with the intention to identify molecules involved in the control of STATIC of glioblastoma cells. Examination using 6 glioma stem cell lines established directly from patient glioblastoma tissues or from conventional glioblastoma cell lines revealed that the JNK pathway is consistently more activated in self-renewing glioma stem cells than in their differentiated counterparts, suggesting that JNK may be involved in the maintenance of the undifferentiated stem cell state (i.e., stemness) of glioblastoma cells.
Activation of JNK in human glioblastoma
So far, a series of studies examining the expression and activation (= expression of the phosphorylated form) of JNK in human glioblastoma tissues by immunoblot analysis have demonstrated that JNKs are expressed and activated in the majority of glioblastoma cases.28-30 Strong expression of phosphorylated JNK in the majority (>90%) of glioblastoma cases has been confirmed independently by an immunohistochemical study, which also showed that JNK activation is associated with the histological grade of glioma and is virtually nil in the normal brain.31 A subcutaneous xenograft experiment using serum-cultured U87 glioblastoma cells constitutively expressing a dominant-negative form of JNK showed that the tumor growth of glioblastoma is retarded when JNK activity is inhibited.30 Together, the reports consistently demonstrated that JNK is activated in glioblastoma tissues and also suggested that the activation of JNK may have a role in promoting the tumor growth of glioblastoma. Nevertheless, the exact role(s) of JNK in glioblastoma biology still remained to be fully elucidated.
JNK is essential for the maintenance of the stemness of glioma stem cells
To determine, therefore, whether the maintenance of stemness is among the critical roles of JNK in glioblastoma cells, we examined, in a recently reported study,5 the impact of JNK inhibition on the stem cell properties of glioma stem cells. Both pharmacological and genetic inactivation of JNK in glioma stem cells in vitro resulted in inhibition of the stem cell–like properties of glioma stem cells, such as the expression of stem cell markers and the capacity to self-renew as spheres, and induced their differentiation. Thus, the activity of JNK is essential for the maintenance of the stemness of glioma stem cells, which was also reproduced by an independent study.6 Notably, both studies demonstrated that knockdown of either JNK1 or JNK2 is sufficient to inhibit the stemness of glioma stem cells, suggesting that JNK1 and JNK2, instead of being redundant to each other, may function in a coordinated manner as previously reported32 in the control of the stemness of glioma stem cells. The demonstration of the consistently essential role of JNK in a variety of glioma stem cells derived directly from glioblastoma tissues and from conventional glioblastoma cell lines in independent studies5,6 strongly supports the idea that JNK may be a fundamental regulator of the stemness of glioblastoma cells.
JNK is essential for the maintenance of the tumor-initiating capacity of glioma stem cells
We then examined whether JNK, which has just been shown to control the stemness of glioma stem cells, is also involved in the maintenance of the tumor-initiating capacity in a manner associated with the stemness of glioma stem cells. Orthotopic xenograft experiments demonstrated that transient JNK inactivation in vitro results in sustained inhibition of brain tumor formation by glioma stem cells. Whereas control glioma stem cells invariably formed brain tumors and killed the host animals within 100 days, those undergoing transient JNK inactivation failed to form lethal brain tumors within the life span of the host animals (>300 days) in most cases, clearly indicating that the number of cells capable of tumor initiation was reduced by transient inactivation of JNK below the threshold for tumor formation. In support of our finding, it has been independently shown that shRNA-mediated inhibition of JNK2 results in delayed brain tumor formation by glioma stem cells.6 Since JNK2 knockdown failed to “prevent” brain tumor formation in that study, the data alone may not discriminate whether the JNK2 knockdown simply delayed the tumor growth or actually reduced the number of tumor-initiating cells, although insufficiently, to prevent tumor formation. However, combined with our results, the data strongly suggest that the latter is the case.
Therapeutic targeting of JNK to control STATIC of glioma stem cells in vivo
Given that cancer stem cell research is an applied medical science in contrast to normal stem cell research, which is essentially a basic one, it was of utmost importance for us to demonstrate the medical significance of our findings. To test, therefore, the clinical relevance of JNK as a therapeutic target, we examined whether in vivo inhibition of JNK successfully controls STATIC of glioma stem cells. To this end, the effect of transient systemic administration of SP600125, a JNK inhibitor, was studied using the serial xenograft transplantation assay, the most reliable “gold standard” in the human cancer stem cell assay field.16 The results indicated that systemic JNK inhibitor treatment of pre-established primary tumors successfully prevents the formation of secondary brain tumors during the extended observation period, which were invariably formed when primary tumors were control treated. Systemic JNK inhibitor treatment for a defined period of only 10 days was also sufficient to inhibit brain tumor formation by glioma stem cells implanted in the brain parenchyma and to confer a survival advantage that would be equivalent to the reduction of tumor-initiating cells by 1 to 2 orders of magnitude. Finally, we confirmed that the above treatment protocol has no long-term (= for the life span of nude mice) adverse effects on the general health status of the treated animals and does not cause overt deficits in their cognitive function. Together, our study demonstrated for the first time that therapeutic targeting of JNK safely and effectively inhibits STATIC of glioblastoma cells at least in preclinical animal models.5
Mechanism of JNK-Mediated Regulation of the Stemness and Tumor-Initiating Capacity of Glioblastoma Cells
While it is now clear from what has been described so far that JNK plays a pivotal role in the control of the stemness and tumor-initiating capacity of glioblastoma cells, much remains to be shown as to how JNK is activated in glioblastoma cells and how JNK controls the stemness and tumor-initiating capacity of glioblastoma cells. We therefore discuss possible mechanisms in this section (Fig. 2).
Figure 2.
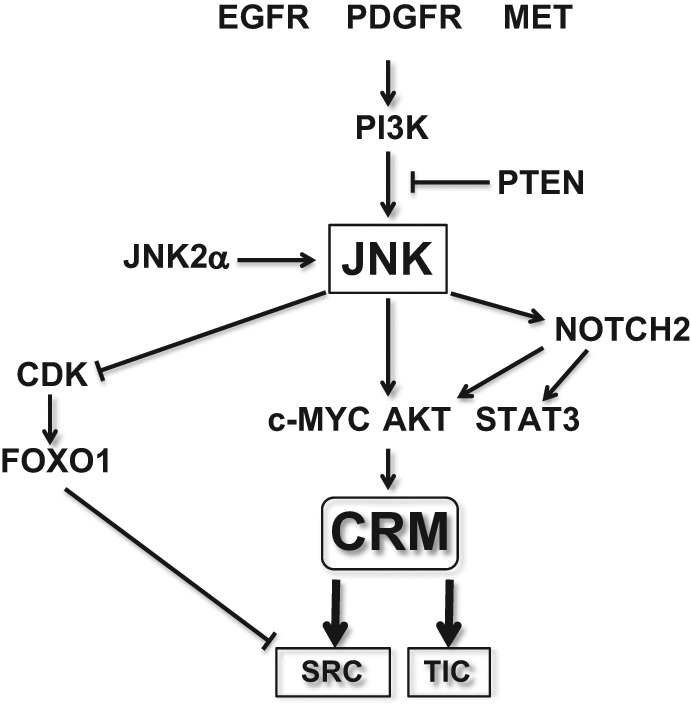
A hypothetical model for JNK-mediated regulation of the stemness and associated tumor-initiating capacity in glioma stem cells. The possible mechanistic components that are assumed to control or to be under the control of JNK in glioma stem cells are schematically depicted. See text for details. Also note that not all the functional interactions depicted in the figure have been demonstrated in glioma stem cells. CRM = common regulatory module; SRC = self-renewing capacity; TIC = tumor-initiating capacity.
How is JNK activated in glioblastoma cells?
EGFR gene amplification and/or intragenic deletion of the EGFR gene (EGFR vIII) are among the most common genetic aberrations occurring in glioblastoma.33 Because overexpression of wild-type or the vIII mutant of EGFR causes JNK activation in vitro,31,34,35 EGFR may be responsible for JNK activation in glioblastoma cells. In support of this idea, an immunohistochemical analysis of glioblastoma tissues revealed that EGFR expression shows some positive correlation with JNK activation.31 Nevertheless, the same study also demonstrated that JNK activation is detected in more than 80% of EGFR-“negative” glioblastomas,31 suggesting that there exist EGFR-independent JNK-activating mechanisms in glioblastoma. In this regard, activating aberrations of the PDGFRA and MET receptor tyrosine kinase genes, which occur in small but significant populations of glioblastomas,36 may be among the possible mechanisms for EGFR-independent JNK activation. Indeed, both PDGFRα and MET are known to activate JNK,37-39 and PDGF-B, a ligand for PDGFR, and MET have been implicated in the maintenance of glioma stem cells.40-42 Intriguingly, MET expression was mutually exclusive with that of EGFR in glioma stem cells.42 MET, therefore, may account for the JNK activation in EGFR-negative glioblastoma cells. On the other hand, a recent study reported that PDGFRβ, rather than PDGFRα, is preferentially expressed with a functional role in glioma stem cells.43 Given that PDGFRβ reportedly activates JNK,44 PDGFRβ-mediated JNK activation could contribute to the maintenance of glioma stem cells.
Then, how do these receptor tyrosine kinases control JNK and the stemness of glioma stem cells? Recent evidence suggests that PI3K is required for the maintenance of the self-renewal capacity and stem cell marker expression of glioma stem cells45,46 and that the JNK signaling pathway is a downstream target of PTEN/PI3K.47 Just in line with these findings, PI3K-dependent activation of JNK has been implicated in the maintenance of the stemness of glioma stem cells.6 Since EGFR, PDGFR, and MET activate intracellular pathways mediated by PI3K,48 PI3K may possibly play a role as a molecular link between these receptor tyrosine kinases and JNK, although this idea has not yet been experimentally validated. It would also be of interest to speculate that the significant impact of the PTEN status on the stem cell phenotypes of glioma stem cells49 is mediated through PI3K-dependent activation of JNK.
In addition to the mechanism of JNK activation by upstream signals, an autoactivation mechanism of JNK has been reported.29,30,50 JNK2 isoforms, in particular JNK2α2, autophosphorylate and activate themselves without requiring the participation of upstream kinases.50,51 Significantly, it has been suggested that such autoactivated JNK isoforms are expressed in a great majority of glioblastoma cases.29,30 Apparently, the expression of the autoactivated JNK isoforms is of therapeutic importance since attempts to control JNK by targeting upstream molecules would be futile if such isoforms account for the majority of the JNK activity in glioblastoma cells.
Another important issue that may deserve consideration in this section is whether the JNK activity level serves as a key determinant in the cell fate (i.e., stem or nonstem) decision of glioma stem cells in patient glioblastomas. Although we observed that JNK activity is decreased in association with the serum-induced differentiation of glioma stem cells in vitro,5 this does not necessarily imply that JNK activity is coupled with the stem cell/differentiation status of tumor cells in patient glioblastomas. Indeed, most of the tumor cells in glioblastoma tissues express detectable levels of activated JNK.31 This finding is not mutually exclusive with the idea of JNK being the determinant of the cell fate decision because there may exist a certain threshold level of JNK activity that divides tumor cells into stem and nonstem populations. However, it is also possible that JNK is similarly activated in both stem and nonstem tumor cell populations in patient glioblastomas. In the latter case, molecules other than JNK are considered to play a pivotal role in the cell fate decision in vivo.
How does JNK control the stemness and tumor-initiating capacity of glioblastoma cells?
We and others have shown that JNK maintains the stemness and tumor-initiating capacity of glioma stem cells through its kinase activity.5,6 Among known direct substrates of JNK, AKT and c-MYC have been implicated in the control of the stemness and tumor-initiating capacity of glioma stem cells.52-54 Since JNK phosphorylation of Thr450 of AKT reportedly activates its kinase activity55 and phosphorylation of Ser62 and Ser71 of c-MYC is required for its biological function,56 JNK may control the characteristic properties of glioma stem cells through such phosphorylation-mediated regulation of AKT and/or c-MYC. Of note, in addition to their role in the control of the stem cell state of glioma stem cells, AKT and c-MYC may also contribute to the maintenance of glioma stem cells by promoting their survival.52,53 Another JNK substrate of interest is STAT3. Accumulating evidence suggests that dysregulation of STAT3 has a role not only in glioblastoma cells in general but also in their stem cell population, and as such, STAT3 is currently regarded as a potential target of glioblastoma therapy.57 Although the roles of JNK in STAT3 regulation are not simple and appear to be multifaceted, JNK may possibly maintain the stemness and/or tumor-initiating capacity of glioblastoma cells through phosphorylation-mediated activation of STAT3 since direct phosphorylation of STAT3 by JNK on its serine residue (Ser727) has been shown to promote STAT3-mediated gene regulation.58-60
We found in our study that FOXO1 is activated upon JNK inhibition and that its activation is required for JNK inhibition–induced differentiation of glioma stem cells.5 Although FOXO1 may not be a direct substrate for JNK and the mechanism of JNK-mediated inhibition of FOXO1 remains largely obscure at this moment, a recent report suggested a role for CDK in the JNK control of FOXO1.61 Importantly, knockdown of FOXO1 prevented the differentiation but not loss of stem cell marker expression and sphere-forming capacity caused by JNK inhibition, suggesting that FOXO1 function may not be required in the earlier phase of differentiation but is required for full differentiation.5 Nevertheless, prevention of premature activation of FOXO1 appears to be one of the mechanisms by which JNK maintains glioma stem cells. Meanwhile, NOTCH2 has also been implicated in JNK-mediated control of glioma stem cells, based on the observation that NOTCH2 expression is closely associated with the JNK activity and hence the stem cell state of glioma stem cells.6 Given the demonstrated role of NOTCH in the control of glioma stem cells that likely involves AKT and STAT3,62 NOTCH2 may also have a role in the JNK control of glioma stem cells through AKT and STAT3.
Issues to Be Considered before Clinical Application of the JNK-Targeting Therapy to Glioblastoma Treatment
How long does the therapeutic effect of transient JNK inhibition last?
Our study demonstrated that the tumor- initiating capacity of glioma stem cells is closely coupled with their stem cell state and that transient JNK inhibition results in sustained loss of their tumor-initiating capacity. These findings indicate that, as discussed earlier, the tumor-initiating capacity of glioma stem cells is indeed stemness associated and is therefore most likely under an epigenetic control. Thus, one could safely expect that once we successfully deprive glioblastoma cells of their tumor-initiating capacity, the condition (loss of tumor-initiating capacity) would then be as stable as the differentiated state and last for a long time period. However, although epigenetic factors are usually stable, they are not necessarily static and can be modulated by the environment.25 In this regard, use of mouse xenograft models apparently has its own limitation in examining how stable the condition of “lost tumor-initiating capacity” is since the stability cannot be assessed beyond the life span of nude mice (~2 years). It is therefore expected that future clinical trials will provide valuable information regarding this issue through the demonstration of whether and to what extent targeting of STATIC is of therapeutic benefit. On the other hand, what should we do if the condition of “lost tumor-initiating capacity” turns out to be not so stable as to prevent tumor recurrence during the rest of the patients’ lives? In such cases, it would be of benefit to try to irreversibly eliminate glioma stem cells with cytotoxic therapies while the cells remain in the nonstem condition after the JNK-targeting therapy since the stem cell state has been associated with higher therapeutic resistance than the nonstem cell state and vice versa.24 Combination of the JNK-targeting therapy followed by cytotoxic therapies may therefore be considered as a rational approach to the treatment of glioblastoma. Here, it might deserve attention that the order and timing of the therapies could have a profound impact on treatment efficacy.63 Quite imaginably, simultaneous JNK-targeting therapy could, for instance, compromise the therapeutic effect of co-administered cytotoxic agents if the agents should induce tumor cell death in a JNK-dependent manner. We therefore propose that cancer stem cell–targeting therapies should in principle “precede” conventional cytotoxic/cytostatic therapies to avoid unexpected interactions between the therapies, which needs to be taken into consideration when designing rational clinical trials. Similarly, use of cancer stem cell–targeting therapies as consolidation and/or maintenance therapies would be justified if they were to be delivered “after the completion of” cytotoxic/cytostatic therapies.
Which should be spared: neural stem cells or the brain function?
For clinical application of therapeutic agents targeting glioma stem cells, it is essential that they do not cause deleterious adverse events at therapeutic doses. Given that glioma stem cells, and possibly other cancer stem cells as well, may share properties with normal tissue stem cells including neural stem cells, the possible effects of cancer stem cell–targeting therapies on neural stem cells should deserve particular consideration. However, rather surprisingly, there has been no definitive proof that adult neurogenesis has an essential role in learning and memory, with most of the studies investigating the role of adult neurogenesis in learning and memory proving to be negative.64,65 There are a few that report positive results; however, the results are inconsistent even among the positive reports.65 Furthermore, the reported differences are in general quite subtle despite substantial ablation of neurogenesis, and the possibility of the ablation methods affecting beyond neurogenesis has not been excluded.65 Most convincingly, the role of adult neurogenesis in learning and memory was called into question by the demonstration that cyclin D2–null mice, in which adult neurogenesis is almost totally lost, performed as well as the wild-type mice in any behavioral tasks known to be hippocampal dependent.65 Although these findings may not totally exclude the possibility that adult neurogenesis has some role in cognitive functions, in particular when the brain is damaged, it is clear from the findings that glioma/cancer stem cell–targeting agents should be assessed not in terms of whether or not they affect neural stem cells and/or neurogenesis but in terms of whether they do affect the brain function per se. In other words, we should be deliberate enough not to remove candidate therapeutic agents from the list simply because they have some effect on neural stem cells and/or neurogenesis since such agents could nonetheless be safe and effective measures with which to treat glioblastomas or other cancers.
In this line, we investigated in our study the effect of systemic JNK inhibitor treatment on the brain function of nude mice.5 Our JNK inhibitor treatment protocol using SP600125 had no discernible effect on mice when they were examined by the Y-maze test, which is currently regarded as a sensitive indicator of brain lesions, including hippocampal lesions, rather than a specific indicator of memory function alone.66-68 Together with the reported neuroprotective activity of SP600125,69,70 the JNK inhibitor may be suitable for clinical application also in view of its effects on the nervous system.
Role for JNK in the Control of STATIC in Human Cancers: Is STATIC a Paradigm Limited to Glioblastoma?
We have demonstrated for the first time that JNK is involved in the control of STATIC, using glioma stem cells as an experimental model.5 Undoubtedly, it is of interest and importance whether such a role for JNK could be generalized to other human cancer types.
Documented role for JNK in the tumor initiation process of human cancers other than glioblastoma
Relevant literature indicates that JNK is deregulated not only in glioblastomas but also in other human cancers.4 Furthermore, the essential role of JNK has been documented at least in some of them using animal models corresponding to human cancers. For instance, in an animal model of chemically induced hepatocellular carcinoma, loss of the JNK1 gene resulted in a sharp decline not only in the size of the formed tumors but also in the incidence of tumor formation per se,71 suggesting that JNK1 is required for chemical initiation of the liver tumors. Similarly, to determine the role of JNK deregulation in human gastric cancers, the effect of JNK1 inactivation was explored in an animal model of chemically induced gastric cancer. Again, both tumor incidence and size were significantly reduced in JNK1-null mice compared to wild-type mice.72 JNK signaling is also known to be activated in human colon cancer tissues, which may have a role in colon carcinogenesis as evidenced by the observation that oral administration of SP600125 was sufficient to reduce the number and size of intestinal tumors spontaneously formed in mice genetically engineered to express a mutant allele of Apc.73 Human non–small cell lung cancer has also been associated with JNK deregulation in part due to the expression of the JNK2α isoform, which is constitutively activated through an autoactivation mechanism.60,74 Intriguingly, experiments using an animal model of Ras-initiated lung cancer clearly demonstrated that Ras-induced lung tumor formation is dependent on JNK.75
JNK control of STATIC may possibly go beyond brain tumors
Collectively, the studies in the literature indicate that JNK has a pivotal role in the initiation of animal models of human cancers in which JNK is deregulated. Of note, in those studies, the mechanism of JNK-dependent tumor initiation has been accounted for mostly by JNK-dependent proliferation of tumor cells, and the possible role of JNK as a regulator of cancer stem cells and/or STATIC has not been discussed. However, as clearly demonstrated by the instance of glioblastoma whereby JNK does have a crucial role in cancer stem cells5,6 in addition to its role in bulk tumor cells,30 it is quite conceivable that JNK also has a cancer stem cell–associated role also in those human cancers with JNK deregulation. In line with this idea, we have recently found, using human cancer cells of nonbrain tumor origin, that JNK is required for what is most likely to be STATIC of the human cancer cells (manuscript in preparation). Together, these lines of evidence strongly suggest that the paradigm may not be unique to glioblastoma and may be shared at least by some other human cancers.
Concluding Remarks
The JNK signaling pathway is aberrantly activated in various human cancers, and the growing body of evidence suggests that it has a definitive role in, albeit not limited to, tumor initiation.1-4 Combined with the recent demonstration that JNK is involved in the control of STATIC of glioblastoma cells, the evidence is now in support of the idea that a similar mechanism may be operative in other human cancers.5,6,76 Apparently, testing of this idea is warranted in future studies: if the mechanism is shared by many human cancers, JNK targeting would realize the long-term control of such cancers, irrespective of whether they follow the cancer stem cell model or not. Currently, the concept of “STATIC” is only operationally defined. Future elucidation of how JNK controls the molecular mechanism underlying STATIC will not only help redefine the concept in molecular terms but also help identify novel molecular targets for the long-term, hopefully curative, control of human cancers.
Acknowledgments
The authors are indebted to Dr. Kimishige Ishizaka for invaluable advice on writing this review article. They also thank members of their laboratory and their collaborators for productive and stimulating discussion and Dr. Tomoko Kagawa for her continuous support and encouragement.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received the following financial support for the research, authorship, and/or publication of this article: This work was supported by Grants-in-Aid for Scientific Research, Challenging Exploratory Research, and Young Scientists from the Ministry of Education, Culture, Sports, Science and Technology of Japan; a Grant-in-Aid from the Global COE Program of the Japan Society for the Promotion of Science; the National Cancer Center Research and Development Fund (23-A-20); and a grant from the Japan Brain Foundation.
References
- 1. Kennedy NJ, Davis RJ. Role of JNK in tumor development. Cell Cycle. 2003;2:199-201 [PubMed] [Google Scholar]
- 2. Manning AM, Davis RJ. Targeting JNK for therapeutic benefit: from junk to gold? Nat Rev Drug Discov. 2003;2:554-65 [DOI] [PubMed] [Google Scholar]
- 3. Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Cell Biol. 2007;19:142-9 [DOI] [PubMed] [Google Scholar]
- 4. Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537-49 [DOI] [PubMed] [Google Scholar]
- 5. Matsuda K, Sato A, Okada M, et al. Targeting JNK for therapeutic depletion of stem-like glioblastoma cells. Sci Rep. 2012;2:516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yoon CH, Kim MJ, Kim RK, et al. c-Jun N-terminal kinase has a pivotal role in the maintenance of self-renewal and tumorigenicity in glioma stem-like cells. Oncogene. 2012;31:4655-66 [DOI] [PubMed] [Google Scholar]
- 7. Yu Y, Ramena G, Elble RC. The role of cancer stem cells in relapse of solid tumors. Front Biosci. 2012;4:1528-41 [DOI] [PubMed] [Google Scholar]
- 8. Malik B, Nie D. Cancer stem cells and resistance to chemo and radio therapy. Front Biosci. 2012;4:2142-9 [DOI] [PubMed] [Google Scholar]
- 9. Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009;138:822-9 [DOI] [PubMed] [Google Scholar]
- 10. Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012;21:283-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10:717-28 [DOI] [PubMed] [Google Scholar]
- 12. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105-11 [DOI] [PubMed] [Google Scholar]
- 13. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755-68 [DOI] [PubMed] [Google Scholar]
- 14. Dick JE. Stem cell concepts renew cancer research. Blood. 2008;112:4793-807 [DOI] [PubMed] [Google Scholar]
- 15. Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17:313-9 [DOI] [PubMed] [Google Scholar]
- 16. Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012;12:133-43 [DOI] [PubMed] [Google Scholar]
- 17. Furth J, Kahn M. The transmission of leukemia of mice with a single cell. Am J Cancer. 1937;31:276-82 [Google Scholar]
- 18. Makino S. Further evidence favoring the concept of the stem cell in ascites tumors of rats. Ann NY Acad Sci. 1956;63:818-30 [DOI] [PubMed] [Google Scholar]
- 19. Hewitt HB. Studies of the dissemination and quantitative transplantation of a lymphocytic leukaemia of CBA mice. Br J Cancer. 1958;12:378-401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bruce WR, Van Der Gaag H. A quantitative assay for the number of murine lymphoma cells capable of proliferation in vivo. Nature. 1963;199:79-80 [DOI] [PubMed] [Google Scholar]
- 21. Pierce GB, Wallace C. Differentiation of malignant to benign cells. Cancer Res. 1971;31:127-34 [PubMed] [Google Scholar]
- 22. Pierce GB, Speers WC. Tumors as caricatures of the process of tissue renewal: prospects for therapy by directing differentiation. Cancer Res. 1988;48:1996-2004 [PubMed] [Google Scholar]
- 23. Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645-8 [DOI] [PubMed] [Google Scholar]
- 24. Alison MR, Lin WR, Lim SM, Nicholson LJ. Cancer stem cells: in the line of fire. Cancer Treat Rev. 2012;38:589-98 [DOI] [PubMed] [Google Scholar]
- 25. Berdasco M, Esteller M. Aberrant epigenetic landscape in cancer: how cellular identity goes awry. Dev Cell. 2010;19:698-711 [DOI] [PubMed] [Google Scholar]
- 26. Berdasco M, Esteller M. DNA methylation in stem cell renewal and multipotency. Stem Cell Res Ther. 2011;2:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lu B, Jan L, Jan YN. Control of cell divisions in the nervous system: symmetry and asymmetry. Annu Rev Neurosci. 2000;23:531-56 [DOI] [PubMed] [Google Scholar]
- 28. Antonyak MA, Kenyon LC, Godwin AK, et al. Elevated JNK activation contributes to the pathogenesis of human brain tumors. Oncogene. 2002;21:5038-46 [DOI] [PubMed] [Google Scholar]
- 29. Tsuiki H, Tnani M, Okamoto I, et al. Constitutively active forms of c-Jun NH2-terminal kinase are expressed in primary glial tumors. Cancer Res. 2003;63:250-5 [PubMed] [Google Scholar]
- 30. Cui J, Han SY, Wang C, et al. c-Jun NH(2)-terminal kinase 2alpha2 promotes the tumorigenicity of human glioblastoma cells. Cancer Res. 2006;66:10024-31 [DOI] [PubMed] [Google Scholar]
- 31. Li JY, Wang H, May S, et al. Constitutive activation of c-Jun N-terminal kinase correlates with histologic grade and EGFR expression in diffuse gliomas. J Neurooncol. 2008;88:11-7 [DOI] [PubMed] [Google Scholar]
- 32. Zhang D, Song L, Li J, Wu K, Huang C. Coordination of JNK1 and JNK2 is critical for GADD45alpha induction and its mediated cell apoptosis in arsenite responses. J Biol Chem. 2006;281:34113-23 [DOI] [PubMed] [Google Scholar]
- 33. Furnari FB, Fenton T, Bachoo RM, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683-710 [DOI] [PubMed] [Google Scholar]
- 34. Antonyak MA, Moscatello DK, Wong AJ. Constitutive activation of c-Jun N-terminal kinase by a mutant epidermal growth factor receptor. J Biol Chem. 1998;273:2817-22 [DOI] [PubMed] [Google Scholar]
- 35. Bonavia R, Inda MM, Vandenberg S, et al. EGFRvIII promotes glioma angiogenesis and growth through the NF-κB, interleukin-8 pathway. Oncogene. 2012;31:4054-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rodrigues GA, Park M, Schlessinger J. Activation of the JNK pathway is essential for transformation by the Met oncogene. EMBO J. 1997;16:2634-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yu J, Deuel TF, Kim HR. Platelet-derived growth factor (PDGF) receptor-alpha activates c-Jun NH2-terminal kinase-1 and antagonizes PDGF receptor-beta-induced phenotypic transformation. J Biol Chem. 2000;275:19076-82 [DOI] [PubMed] [Google Scholar]
- 39. Yu J, Liu XW, Kim HR. Platelet-derived growth factor (PDGF) receptor-alpha-activated c-Jun NH2-terminal kinase-1 is critical for PDGF-induced p21WAF1/CIP1 promoter activity independent of p53. J Biol Chem. 2003;278:49582-8 [DOI] [PubMed] [Google Scholar]
- 40. Jiang Y, Boije M, Westermark B, Uhrbom L. PDGF-B can sustain self-renewal and tumorigenicity of experimental glioma-derived cancer-initiating cells by preventing oligodendrocyte differentiation. Neoplasia. 2011;13:492-503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Joo KM, Jin J, Kim E, et al. MET signaling regulates glioblastoma stem cells. Cancer Res. 2012;72:3828-38 [DOI] [PubMed] [Google Scholar]
- 42. De Bacco F, Casanova E, Medico E, et al. The MET oncogene is a functional marker of a glioblastoma stem cell subtype. Cancer Res. 2012;72:4537-50 [DOI] [PubMed] [Google Scholar]
- 43. Kim Y, Kim E, Wu Q, et al. Platelet-derived growth factor receptors differentially inform intertumoral and intratumoral heterogeneity. Genes Dev. 2012;26:1247-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Panzhinskiy E, Zawada WM, Stenmark KR, Das M. Hypoxia induces unique proliferative response in adventitial fibroblasts by activating PDGFβ receptor-JNK1 signalling. Cardiovasc Res. 2012;95:356-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sunayama J, Sato A, Matsuda K, et al. Dual blocking of mTor and PI3K elicits a prodifferentiation effect on glioblastoma stem-like cells. Neuro Oncol. 2010;12:1205-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sunayama J, Matsuda K, Sato A, et al. Crosstalk between the PI3K/mTOR and MEK/ERK pathways involved in the maintenance of self-renewal and tumorigenicity of glioblastoma stem-like cells. Stem Cells. 2010;28:1930-9 [DOI] [PubMed] [Google Scholar]
- 47. Vivanco I, Palaskas N, Tran C, et al. Identification of the JNK signaling pathway as a functional target of the tumor suppressor PTEN. Cancer Cell. 2007;11:555-69 [DOI] [PubMed] [Google Scholar]
- 48. Moritz A, Li Y, Guo A, et al. Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci Signal. 2010;3:ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen R, Nishimura MC, Bumbaca SM, et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell. 2010;17:362-75 [DOI] [PubMed] [Google Scholar]
- 50. Cui J, Holgado-Madruga M, Su W, Tsuiki H, Wedegaertner P, Wong AJ. Identification of a specific domain responsible for JNK2alpha2 autophosphorylation. J Biol Chem. 2005;280:9913-20 [DOI] [PubMed] [Google Scholar]
- 51. Nitta RT, Chu AH, Wong AJ. Constitutive activity of JNK2 alpha2 is dependent on a unique mechanism of MAPK activation. J Biol Chem. 2008;283:34935-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Eyler CE, Foo WC, LaFiura KM, McLendon RE, Hjelmeland AB, Rich JN. Brain cancer stem cells display preferential sensitivity to Akt inhibition. Stem Cells. 2008;26:3027-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang J, Wang H, Li Z, et al. c-Myc is required for maintenance of glioma cancer stem cells. PLoS One. 2008;3:e3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zheng H, Ying H, Yan H, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008;455:1129-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shao Z, Bhattacharya K, Hsich E, et al. c-Jun N-terminal kinases mediate reactivation of Akt and cardiomyocyte survival after hypoxic injury in vitro and in vivo. Circ Res. 2006;98:111-8 [DOI] [PubMed] [Google Scholar]
- 56. Noguchi K, Kitanaka C, Yamana H, Kokubu A, Mochizuki T, Kuchino Y. Regulation of c-Myc through phosphorylation at Ser-62 and Ser-71 by c-Jun N-terminal kinase. J Biol Chem. 1999;274:32580-7 [DOI] [PubMed] [Google Scholar]
- 57. Liu Y, Li C, Lin J. STAT3 as a therapeutic target for glioblastoma. Anticancer Agents Med Chem. 2010;10:512-9 [DOI] [PubMed] [Google Scholar]
- 58. Lim CP, Cao X. Serine phosphorylation and negative regulation of Stat3 by JNK. J Biol Chem. 1999;274:31055-61 [DOI] [PubMed] [Google Scholar]
- 59. Turkson J, Bowman T, Adnane J, et al. Requirement for Ras/Rac1-mediated p38 and c-Jun N-terminal kinase signaling in Stat3 transcriptional activity induced by the Src oncoprotein. Mol Cell Biol. 1999;19:7519-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nitta RT, Del Vecchio CA, Chu AH, Mitra SS, Godwin AK, Wong AJ. The role of the c-Jun N-terminal kinase 2-α-isoform in non-small cell lung carcinoma tumorigenesis. Oncogene. 2011;30:234-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xu P, Das M, Reilly J, Davis RJ. JNK regulates FoxO-dependent autophagy in neurons. Genes Dev. 2011;25:310-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fan X, Khaki L, Zhu TS, et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells. 2010;28:5-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee MJ, Ye AS, Gardino AK, et al. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell. 2012;149:780-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shors TJ. From stem cells to grandmother cells: how neurogenesis relates to learning and memory. Cell Stem Cell. 2008;3:253-8 [DOI] [PubMed] [Google Scholar]
- 65. Jaholkowski P, Kiryk A, Jedynak P, et al. New hippocampal neurons are not obligatory for memory formation: cyclin D2 knockout mice with no adult brain neurogenesis show learning. Learn Mem. 2009;16:439-51 [DOI] [PubMed] [Google Scholar]
- 66. Reisel D, Bannerman DM, Schmitt WB, et al. Spatial memory dissociations in mice lacking GluR1. Nat Neurosci. 2002;5:868-73 [DOI] [PubMed] [Google Scholar]
- 67. Lalonde R. The neurobiological basis of spontaneous alternation. Neurosci Biobehav Rev. 2002;26:91-104 [DOI] [PubMed] [Google Scholar]
- 68. Hughes RN. The value of spontaneous alternation behavior (SAB) as a test of retention in pharmacological investigations of memory. Neurosci Biobehav Rev. 2004;28:497-505 [DOI] [PubMed] [Google Scholar]
- 69. Sharma N, Deshmukh R, Bedi KL. SP600125, a competitive inhibitor of JNK attenuates streptozotocin induced neurocognitive deficit and oxidative stress in rats. Pharmacol Biochem Behav. 2010;96:386-94 [DOI] [PubMed] [Google Scholar]
- 70. Ramin M, Azizi P, Motamedi F, Haghparast A, Khodagholi F. Inhibition of JNK phosphorylation reverses memory deficit induced by β-amyloid (1-42) associated with decrease of apoptotic factors. Behav Brain Res. 2011;217:424-31 [DOI] [PubMed] [Google Scholar]
- 71. Sakurai T, Maeda S, Chang L, Karin M. Loss of hepatic NF-kappa B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc Natl Acad Sci U S A. 2006;103:10544-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shibata W, Maeda S, Hikiba Y, et al. c-Jun NH2-terminal kinase 1 is a critical regulator for the development of gastric cancer in mice. Cancer Res. 2008;68:5031-9 [DOI] [PubMed] [Google Scholar]
- 73. Fujishita T, Aoki M, Taketo MM. JNK signaling promotes intestinal tumorigenesis through activation of mTOR complex 1 in Apc(Δ716) mice. Gastroenterology. 2011;140:1556-63 [DOI] [PubMed] [Google Scholar]
- 74. Khatlani TS, Wislez M, Sun M, et al. c-Jun N-terminal kinase is activated in non-small-cell lung cancer and promotes neoplastic transformation in human bronchial epithelial cells. Oncogene. 2007;26:2658-66 [DOI] [PubMed] [Google Scholar]
- 75. Cellurale C, Sabio G, Kennedy NJ, et al. Requirement of c-Jun NH(2)-terminal kinase for Ras-initiated tumor formation. Mol Cell Biol. 2011;31:1565-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chen F. JNK-induced apoptosis, compensatory growth, and cancer stem cells. Cancer Res. 2012;72:379-86 [DOI] [PMC free article] [PubMed] [Google Scholar]