

Abstract
Mixed lineage kinases (MLKs) are members of the mitogen-activated protein kinase kinase kinase (MAP3K) family and are reported to activate MAP kinase pathways. There have been at least 9 members of the MLK family identified to date, although the physiological functions of all the family members are yet unknown. However, MLKs in general have been implicated in neurodegenerative diseases, including Parkinson and Alzheimer diseases. Recent reports suggest that some of the MLK members could play a role in cancer via modulating cell migration, invasion, cell cycle, and apoptosis. This review article will first describe the biology of MLK members and then discuss the current progress that relates to their functions in cancer.
Keywords: MLKs, JNK, cancer, MAPKs, phosphorylation
Introduction
Programmed cell death (i.e., apoptosis) is necessary for normal development, and a fine balance exists between cell death and survival processes to maintain normal homeostasis.1,2 Therefore, unrestrained cell death is expected to promote degenerative diseases, like Parkinson disease (PD) and Alzheimer disease (AD), whereas antagonism, combined with uncontrolled cell growth, could lead to tumorigenesis (i.e., cancer). The balance between cell death and survival is regulated by numerous proapoptotic and prosurvival proteins, and several of these factors and their role in cancer are described in the literature.2,3
The family members of mixed lineage kinase (MLK) are understudied, and their detailed functions are still obscure. However, the recent advances made in the field suggest a reasonable function of MLKs as proapoptotic kinases that can promote neurodegenerative diseases. This close involvement of the MLK group of kinases in neurodegenerative diseases has led to the use of pan-MLK inhibitor CEP-1347 in clinical trials for PD.4 The recent advancements made in this field also suggest that some of the MLK members could play a role in tumorigenesis, suggesting that these specific MLK members could serve as potential therapeutic targets to prevent metastasis and tumor growth.5-7
MLKs
MLKs are members of a large family of mitogen-activated protein kinase kinase kinase (MAP3K) and are reported to activate MAPKs including c-Jun N-terminal kinase (JNK), p38, and extracellular signal–regulated kinases (ERKs).8 Common to all the MLK members is the catalytic domain that contains signature sequences of both serine/threonine (Ser/Thr) and tyrosine (Tyr) kinases, which led to their name as MLKs. It is established that MLKs are functional Ser/Thr kinases,9 but there is no report that suggests that any of the MLK members function as a Tyr kinase. It is possible that some or all MLK members are functional Tyr kinases towards very specific substrate(s), which are yet to be identified. Another possibility is that the Tyr kinase activities of MLKs are regulated by specific stimuli, and identification of such specific stimuli will be important to define their Tyr kinase activities. The other common domain that is present in all MLK members is a leucine zipper domain. Based on their protein domain structures, the 9 MLK family members are divided into 3 subfamilies (Fig. 1). The members of the first subfamily are called MLKs and contain 4 members, MLK1 to MLK4, with MLK4 having 2 isoforms, MLK4α and MLK4β. The second subfamily, called dual leucine zipper–bearing kinases (DLKs), contains 2 members, DLK and LZK. The third subfamily is known as zipper sterile-α motif kinases (ZAKs), and there are 2 members in this subgroup, ZAKα and ZAKβ.
Figure 1.

Classification of MLKs and their conserved domains. Classification of the MLK family in 3 subgroups showing the conserved domains: SH3, kinase, leucine zipper, CRIB, and SAM.
The members of the MLK subfamily are unique because they contain the amino-terminal Src homology 3 (SH3) domain that is absent in the 2 other subfamilies. The SH3 domain is a protein-protein–interacting motif and is known to recognize polyproline-rich sequences within the target proteins.10 In addition to the SH3 domain, the MLK subfamily members also contain the Cdc42/Rac-interactive binding (CRIB) motif necessary to interact with small GTPases (Fig. 1). It is reported that some members of this family do interact with active Cdc42 and Rac1.11,12
The DLK subfamily members are characterized by a kinase domain followed by 2 leucine zipper motifs, interrupted by a 31–amino acid spacer. The DLK members do not contain either SH3 or CRIB domains; however, they contain a proline-rich region in the carboxy-terminal, but its regulatory role is not yet reported.
Members of the third subfamily, ZAKs, are characterized by the presence of a sterile-α motif (SAM). The SAM domain is found in many signaling proteins, including adapter proteins, GTPase-activating proteins, and receptor tyrosine kinases.13 The approximately 70–amino acid SAM domain can mediate homodimerization or heterodimerization. The ZAKs are also known as MLK-like mitogen-activated protein triple kinases (MLTKs). ZAKβ (MLTKβ) is the splice variant and has a much smaller C-terminal domain than ZAKα and as a result lacks the SAM domain. The ZAK proteins are very loosely related to other MLKs and only share the catalytic domain and leucine zipper (Fig. 1).
MLKs and MAPK Signaling
The conserved subdomains of the MLK group of kinases are very similar to MAP3K members, such as MEKKs and Raf Ser/Thr kinases, and therefore, it was speculated that MLKs are members of the MAP3K family and might activate MAP kinase pathways. Early experiments to elucidate the role of different members of MLKs in MAPK pathways were performed with overexpressed proteins in mammalian cells. Therefore, some of these data obtained from overexpression studies are confusing. It was shown that MLK3 functions as a MAP3K and activates JNK potently (Fig. 2) and p38 quite modestly.9 Subsequently, it was shown that MLK1, MLK2, and DLK also activate JNK.14-17 Our group and others showed that MLK3 activated JNK via direct phosphorylation of JNK upstream, MKK4/SEK1,9,18 whereas others showed that MLK3 chiefly activated JNK via another MEK member in the JNK pathway, MKK7.19 Interestingly, DLK was also shown to activate JNK mainly via MKK7 phosphorylation.20 Furthermore, it was shown that in response to specific agonists of MLK3, TNFα, and ceramides, JNK was exclusively targeted, suggesting the notion that MLK3 and perhaps other MLK-mediated MAPK activations could be agonist specific.
Figure 2.
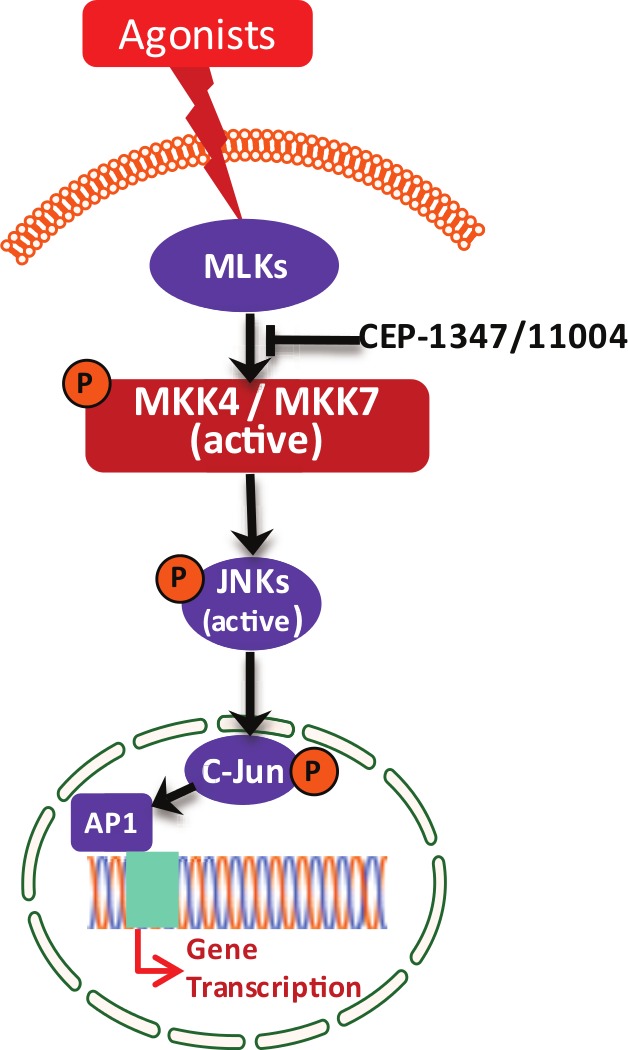
Activation of JNK by MLK3/MLKs. MLK3 is activated by its agonist TNFα and ceramide. The activation of MLK3 phosphorylates MEK members MKK4 and MKK7. The activated MKK4 and MKK7 phosphorylate and activate JNK that ultimately regulates AP-1–mediated gene expression. The pan-MLK inhibitors CEP-1347 and CEP-11004 are reported to block MLK-mediated downstream signaling.
The role of MLK family members in extracellular signal–regulated activation of MAPKs, ERK1/2, is controversial. It was reported that overexpression of MLK3 activates ERKs via MEK1 phosphorylation, and ERK activation by MLK3 can be blocked by the MEK1 inhibitor PD98059 or by expressing kinase-dead MEK1.21 However, it was demonstrated by our group that MLK3 indeed phosphorylates and activates MEK1, but this does not result in ERK activation.22 This confusion, whether MLKs activate all MAPK pathways or are specific to some, was further complicated when MLK3 was shown to activate ERKs via B-Raf activation in a kinase-independent manner.23 It was reported that MLK3 is required to maintain the integrity of the B-Raf–Raf1 complex, and this multiprotein complex is essential for ERK activation.23 Therefore, a clearer picture of MLKs’ role in ERK-MAPK activation will be evident upon identification of specific agonists and studies with knockdown of MLK members instead of overexpression.
Identification of Agonists of MLKs and Mechanisms of Activation
The identification of agonist(s) of MLK members was mired due to the lack of reliable antibodies that can immunoprecipitate endogenous MLKs. The other problem that was encountered during agonist identification with recombinant MLKs from mammalian cells was the constitutive activation of overexpressed MLKs in mammalian cells. The third problem that delayed this process was the lack of specific MLK knockout models. The fourth and most significant hurdle that hindered agonists’ identification was the presence of multiple MLK subtypes in mammalian cells.8 The first 2 agonists of MLK3 were identified initially in Drosophila cells due to the presence of a single isoform of MLK in Drosophila, named Drosophila MLK (dMLK) or slipper (Slpr).24,25 In Drosophila S2 cells, knockdown of endogenous dMLK prevented ceramide- and TNFα-induced JNK but not p38 or ERK activations.26 Similarly, in mammalian cells, incubation with ceramide and TNFα induced MLK3 kinase activity, suggesting that these 2 are natural agonists of MLK3.26 These experiments clearly indicated that MLK3 is an activator of JNK and that MLK3 itself is activated by bioactive lipid ceramides and the proinflammatory cytokine TNFα. Later on, Brancho et al.27 also showed that TNFα-mediated JNK activation was down-regulated in MEFs from MLK3 knockout animals, corroborating that TNFα is indeed an agonist of MLK3. Recently, it has been shown that MLK3 is also activated by other cytokines, such as IL-1β in pancreatic β-cells28 and CXCL12 in breast cancer cells.29 However, these reports did not provide any direct evidence to show whether these cytokines regulate the kinase activity of MLK3. Unfortunately, the agonists of other MLK members are still not known. It is also not known whether ceramide or the other cytokines can regulate the kinase activity of MLKs other than MLK3. Failure to identify the specific agonists for other MLKs could similarly be attributed to the lack of reliable antibodies that can immunoprecipitate endogenous MLK members for direct kinase assay. Other information that is lacking and that has contributed to this is the identity of specific substrates of other MLKs. So far, only 1 physiological substrate of MLK3 is known in which it was shown that MLK3 directly phosphorylates SEK1/MKK4.9 Therefore, the identification of a specific substrate for each MLK member and the development of isoform-specific MLK antibodies will be needed to identify the specific agonist(s) and understand their regulation.
Regulation of MLKs
All MLKs differ at their C-terminal regulatory domain, and therefore, it is quite conceivable that each MLK member will be regulated differentially and could play a distinct physiological role. The regulation of kinase activity for most of the MLK members is also largely unexplored. Much of MLK regulation has been reported with 1 member of this family, MLK3. The proinflammatory cytokine TNFα (now an established agonist of MLK3) exerts its downstream signaling by recruiting adaptor proteins, including TRAFs. In fact, our recent work has shown that TRAF2-MLK3 interaction is essential for TNFα-induced MLK3 activation and downstream signaling.30 However, it is not clear how this mere interaction promotes MLK3 activation because TRAF2 does not contain any kinase activity. Several speculations can be put forward to explain this scenario: 1) the interaction between TRAF2 and MLK3 might lead to the recruitment of a (yet to be identified) kinase that promotes MLK3 activation, or 2) TRAF2-MLK3 interaction might cause structural changes in MLK3 that lead to increased kinase activity. The other agonist, ceramide, is known to be generated in response to several cytokines, including TNFα, and therefore, it is also possible that TNFα activates MLK3 via ceramide generation, which needs to be established. Thus, the obvious next question that remains unanswered is how ceramide activates MLK3. Is this through direct binding or through regulating ceramide-induced specific kinases or phosphatases? The involvement of any ceramide-induced kinase or phosphatase in MLK3 activation has never been reported, nor is it known whether ceramide directly binds to MLK3. Our group has been working to establish the mechanism of ceramide-induced activation of MLK3 kinase activity.
Our studies have shown that MLK3 activity can also be regulated by other upstream kinases. We reported that glycogen synthase kinase 3β (GSK3β) activates MLK3 kinase activity upon growth factor deprivation.31 Upon growth factor deprivation, GSK3β is activated, and active GSK3β then phosphorylates 2 serine residues (i.e., Ser 789 and 793) in the C-terminus of MLK3 that ultimately induces the kinase activity of MLK3 (Fig. 3) and cell death.31 The other kinase that has been implicated in the negative regulation of MLK3 kinase activity is Akt1.32 It is reported that in response to growth factor signaling, MLK3 kinase activity was down- regulated. It was identified that Akt1 phosphorylates MLK3 on Ser 674 in the C-terminus domain, which leads to the down-regulation of MLK3 kinase activity and increased cell survival (Fig. 3).
Figure 3.

MLK3-mediated signaling. The details of MLK3 regulation/signaling are described within the text.
All 9 members of the MLK family contain some form of leucine zipper. It is reported that MLK3 leucine zipper induces homodimerization of MLK3 and promotes the autophosphorylation/activation of the downstream MKK4-JNK pathway.33 Similarly, the leucine zipper in DLK has also been shown to cause the homodimerization and activation of JNK.16 Whether homodimerization or heterodimerization of MLKs is required for their activation is also not without controversy. It is reported that the dimerization-defunct form of MLK3 binds with Cdc42 and is sufficient for MLK3 autophosphorylation/activation. However, downstream JNK activation was compromised with the leucine zipper mutant.34 These results are quite intriguing because they suggest that perhaps dimerization of MLKs is not a mechanism of its own activation; rather, binding of Cdc42 probably helps the translocation of the monomeric kinase to the plasma membrane for activation by other kinases or perhaps by its agonist, ceramides. Thus, dimerization is not a requirement for autophosphorylation-mediated activation but presumably increases the chance of MLK-MLK interaction.
The MLK subfamily members, MLK1 to MLK4, which contain the SH3 domain, also contain a central CRIB domain. The proteins Cdc42 and Rac1 are Rho family GTPases and bind to their effectors in activated GTP-bound forms. MLK3 contains a well-conserved CRIB consensus sequence and has been reported to bind activated Cdc42 and Rac1.11 Through co-expression of the active form of Cdc42 along with MLK3, it is shown that MLK3 was marginally activated and potentiated JNK activation.11 However, in the same report, active Cdc42 was unable to increase the kinase activity of recombinant MLK3 under in vitro conditions.11 These results again suggested that binding of Cdc42 merely is not enough for MLK3 activation; rather, Cdc42 binding possibly causes the translocation of MLK3 to the plasma membrane, where MLK3 meets its activator/agonists. It was also shown that co-expression of Cdc42 with MLK3 induced the phosphorylation of MLK3 at 2 sites.11 Although intriguing, it is however unclear how Cdc42 induces MLK3 phosphorylation since it lacks any kinase activity. These results also indirectly support the same hypothesis that Cdc42 binding to MLK3 probably is required for its translocation to the plasma membrane, where it can be phosphorylated by other yet-to-be-identified kinase(s). The other mechanism by which Cdc42/Rac1 possibly can activate MLK3 kinase activity could be by disrupting the autoinhibitory interaction between the SH3 domain and proline-rich domain within the MLK3 sequence. On this line, it was shown that Cdc42 indeed disrupts the intramolecular interaction and causes the activation of MLK3.35 However, these results still do not rule out the possibility that Cdc42 binding and thereby disruption of the SH3 intramolecular interaction leads to MLK3 translocation to the membrane for its full activation. Since Cdc42, like other Rho family members, is posttranslationally prenylated, there is every likelihood that Cdc42-mediated activation of MLK3 is mediated via a membrane-targeting property of GTPases and not by mere binding to the CRIB domain.
In addition to the CRIB domain, all members of the MLK subfamily also contain an N-terminal SH3 domain. The SH3 domain is a 60 amino acid–containing protein motif that binds to proline-rich polypeptides in the consensus sequence PXXXP.10 The SH3 domain of MLK3 has been shown to bind hematopoietic progenitor kinase 1 (HPK1); however, the physiological significance of this interaction is not known.36 It has been shown that the SH3 domain in MLK3 binds to the SH3 binding site located between the leucine zipper and CRIB domains.35 A point mutation within the SH3 domain increased the kinase activity of MLK3.35 These results demonstrate that MLK3 itself autoregulates its kinase activity via intramolecular interactions by the SH3 domain. A similar mechanism of autoregulation is also observed in the Src family in which the intramolecular interaction between the SH3 domain and tyrosine residue in the C-terminus regulates Src kinase activity.37 The critical proline residue in the SH3 domain of MLK3 is also conserved in other MLK subfamily members, thus suggesting the possibility that MLK1, MLK2, and MLK4 might also be autoregulated via intramolecular interactions. Since the SH3 binding sequences are located in between the CRIB and leucine zipper domains, one can speculate that one mechanism by which kinase activities of MLKs are maintained in the basal level could be due to the intramolecular interaction between the SH3 and polyproline sites that prevent Cdc42 or Rac binding, which is needed for MLK translocation and full activation at the membrane. These are open questions and need experimental proof.
It has also been suggested that MLKs’ downstream JNK regulates the kinase activities of MLKs by the feedback phosphorylation mechanism.38 In these studies, the authors showed that JNK phosphorylation of MLK3 promotes its activation,38 whereas inhibition of JNK causes hypophosphorylation, leading to the inactivation and translocation of MLK3 in triton-insoluble fractions.38 These observations are supported by another study, which showed that MLK2 can be phosphorylated by JNK in the C-terminus, although a specific phosphorylation site was not identified.39
Functions of MLK Members and Cancer Connection
Most of the MLK family members are potent activators of the JNK pathway, and activation of JNK has been implicated chiefly in cell death. Therefore, the first cellular function of MLKs that came to light was its role in promoting cell death.17,31,32 Activation of JNK not only plays a role as a regulator of cell death, but it has also been implicated in cell migration and invasion.40,41 The first hint regarding a potential link of MLKs with cancer came out when MLK3 overexpression was shown to transform NIH3T3 cells via MEK1 activation,21 suggesting that some members of the MLK family could play a role in cancer progression. Quite recently, several important findings have implicated MLKs in cell death, cell migration, and the cell cycle, suggesting that these so-called proapoptotic kinases could also paradoxically promote oncogenesis.
MLKs’ role in cell migration/invasion
The first evidence that a MLK family member could play a role in cell migration came from a study with gastric cancer cells in which gastrin was shown to promote gastric cell migration via JNK activation.42 In this interesting study, the authors also showed that pretreatment of gastric cancer cells, either with the pan-MLK inhibitor CEP-1347 or JNK inhibitor SP600125, blocked gastrin-induced cell migration.42 Gastrin also regulated MMP7 promoter activity via the MLK3-JNK pathway, suggesting an involvement of this axis in cell migration and invasion.42 Corroborating MLK3’s role in gastric cancer cell migration, it is reported that MLK3 plays a similar role in breast cancer cell migration via MLK3–JNK–AP-1 signaling.7 Expression of active MLK3 was sufficient to induce mammary epithelial cell migration/invasion via AP-1–regulated expression of genes involved in invasion.7 Quite recently, Cronan et al.5 also identified MLK3 as one of the primary MAP3K members whose knockdown in the highly invasive breast cancer cell line MDA-MB-231 prevented tumor growth and metastasis. In this study, they also showed that loss of MLK3 expression increased mitotic infidelity and apoptosis in vitro.5 Involvement of MLK3 in cell migration and invasion has been confirmed in another recent study that suggests that MLK3 promotes the invasion of ovarian cancer cells.43 In this study, the authors demonstrated that MLK3 silencing inhibits the expression of MMP-1, -2, -9, and -12.43 Quite recently, it has been shown that MLK3 promotes cell migration via phosphorylation of the focal adhesion scaffold protein, paxillin.29 Whether paxillin is directly phosphorylated by MLK3 or via some other unknown kinase is, however, not indicated in this report. Collectively, all these reports quite convincingly point to a role of MLK3 in cancer cell migration and invasion. Whether other MLKs also have similar roles in cell migration and invasion is still unknown. Knowing the functions of other MLKs in cell migration and invasion will be important to further investigate whether MLK isoform–specific inhibitors or a pan-MLK inhibitor (CEP-1347) could possibly be used to inhibit the metastasis of cancer cells.
MLKs’ role in the cell cycle
In differentiated neuronal cells, activation of MLKs has been shown to promote cell death17; however, it has been speculated that MLKs could play a proliferative role in dividing cells. The first evidence that MLK3 plays a role in cell cycle progression came from its high homology with the noncatalytic domain of enigmatic fungal kinase, never in mitosis A (NIMA) kinase.44 The NIMA kinase has been reported to be indispensable for G2-M cell cycle transition.45 The noncatalytic domain is critical for NIMA function, and based on this analogy, it was shown that knockdown of MLK3 in HeLa cells prevents G2-M transition.44 It was also shown in this study that MLK3 kinase activity was increased during G2-M transition; however, JNK activity was unaltered,44 suggesting that MLK3’s function in cell cycle regulation is independent of JNK activation (probably via some unknown pathway). Another report similarly showed that MLK3 and perhaps other MLKs play a role in cell cycle progression. By using the pan-MLK inhibitor CEP-11004, these authors showed that the G2-M transition in HeLa cells was blocked and was partially reversed by overexpressing exogenous MLK3.46 CEP-11004 also inhibited histone H3 phosphorylation during prophase and prior to nuclear envelope breakdown and formation of aberrant mitotic spindles.46 These 2 studies collectively showed that MLK3 and perhaps other MLKs play an important role in cell cycle progression. However, these studies did not provide any molecular mechanism of MLK3/MLK-mediated cell cycle regulation. Our group recently showed that MLK3 phosphorylates the oncogene β-catenin, which leads to stabilization of the β-catenin protein.6 Interestingly, this stabilized β-catenin protein did not increase β-catenin/TCF-mediated transcription, which was rather inhibited due to the recruitment of KLF4 in the complex.6 Finally, stabilization of β-catenin by MLK3 blocked G2-M transition,6 suggesting that MLK3-induced cell cycle events are complex and likely governed by downstream proteins. Quite recently, we have also shown a mechanism by which MLK3 could promote the cell cycle. We showed that MLK3 phosphorylates a prolyl isomerase, Pin1, on the Ser 138 site.47 This phosphorylation was sufficient for Pin1 translocation to the nucleus, which ultimately promoted G2-M transition. Supporting the cell cycle regulation of the MLK3-Pin1 axis, the phosphorylation of the Pin1–Ser 138 protein was enriched in breast tumors, indicating that this pathway plays an important role in cell cycle and cell proliferation pathways.47 This cell cycle function of MLKs also suggests that MLK inhibitors could serve as a potential therapeutic towards controlling the growth of transformed cells.
MLKs’ role in cell death
Initial functional analyses of MLKs suggested that almost all the MLK members play an important role in promoting cell death.4,17 All these studies were done in neuronal cells, and therefore, the inhibitor of MLKs, CEP-1347, was involved in clinical trials for PD.4 The cell death–related functions of MLKs in nonneuronal cells, specifically in transformed cells, are not well known. Our recent studies with estrogen receptor–positive (ER+) breast cancer cells showed that the proapoptotic function of MLK3 was blocked by estrogen.48 We showed that the kinase activity of MLK3 was down-regulated by estrogen via activation of the PI3K-AKT pathway.48 In this report, we also showed that cytotoxic drug, Taxol-induced (Taxol Source, Ivax Laboratories, Florida) cell death requires MLK3 kinase activity and that estrogen antagonizes Taxol-induced cell death via MLK3/MLKs inhibition. This important study sheds light on the therapeutic potential of MLK3 activators in ER+ breast cancer treatment. Coincidently, MLK3 kinase activity was also inhibited in ER+ human breast tumors, suggesting that blockage of ER signaling in ER+ breast cancer would promote cell death via MLK3 activation.
Future Directions
The MLK-JNK axis has emerged as a viable therapeutic target of neurodegenerative diseases; however, recent reports clearly show that this same axis could have opposite and protumorigenic effects in epithelial cancer cells. The use of JNK inhibitors has been proposed in various diseases, including in metastasis of cancer cells. Despite a known involvement of JNK in cancer pathways, it is anticipated that the use of a JNK inhibitor clinically might be challenging due to the presence of multiple downstream targets of JNKs, with numerous functions. Thus, it is quite logical to propose that inhibitors of JNK upstream kinases, such as MLKs, could serve a viable alternative to target particular functions of JNK, such as metastasis and cell growth in epithelial cells. Nevertheless, we are in the initial stages of understanding the physiological functions of MLK members and exploring the pharmacological and therapeutic potentials of MLK inhibitors, specifically for cancer. The initial findings, especially with 1 member of the MLK family, MLK3, open up new avenues of research that need to be broadened in the future. Some of the future directions are outlined below:
We need to determine the detailed function of all members of the MLK family. At this time, the availability of some MLK isoform–specific knockout animal models will help to determine the function of various MLKs and also assist to investigate any crosstalk among the members of this novel family of kinases.
Once the functions of all the MLK members are outlined, and it is established that they are functionally nonredundant (most likely), then it will be important to develop isoform-specific inhibitors that could block specific MLK activity and its downstream pathological functions. Various groups have been working to develop MLK isoform–specific inhibitors, which will help to determine the function of MLKs in various diseases, including cancer.
An initial step that will help in elucidating the biological functions of various MLK members will be to identify specific agonists of all MLKs. Since MLKs differ in their regulatory C-terminal tail, it is quite conceivable that each member of this family will be regulated by a specific agonist.
Another critical step towards understanding the biology of MLKs will be the identification of specific substrates of all the MLK members. The identification of specific substrates will also help in the identification of agonists of other MLKs and help us to determine their physiological as well as pathological functions.
With the recent advances made to understand the functions of MLKs in cancer, and with the availability of new tools and reagents, it is expected that this area will expand and yield valuable information about the therapeutic potential of MLK inhibitors in cancer.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no conflict of interest with respect to research, authorship, and/or publication of this article.
Funding: The author(s) received the following financial support for the research, authorship, and/or publication of this article: This work was supported by Veterans Administration Research funds to Ajay Rana (BX000312) and Basabi Rana (BX000571). Part of this work was also supported by grants from the National Institutes of Health to Ajay Rana (GM55835), Basabi Rana (CA121221), and Anuman- tha Kanthasamy (NS074443 and ES10586) and by a grant from the Department of Defense to Basabi Rana (W81XWH-10-1-0195/PC093099).
References
- 1. Bialik S, Zalckvar E, Ber Y, Rubinstein AD, Kimchi A. Systems biology analysis of programmed cell death. Trends Biochem Sci. 2010;35:556-64 [DOI] [PubMed] [Google Scholar]
- 2. Garrido C, Kroemer G. Life’s smile, death’s grin: vital functions of apoptosis-executing proteins. Curr Opin Cell Biol. 2004;16:639-46 [DOI] [PubMed] [Google Scholar]
- 3. Rufini A, Melino G. Cell death pathology: the war against cancer. Biochem Biophys Res Commun. 2011;414:445-50 [DOI] [PubMed] [Google Scholar]
- 4. Wang LH, Besirli CG, Johnson EM., Jr. Mixed-lineage kinases: a target for the prevention of neurodegeneration. Annu Rev Pharmacol Toxicol. 2004;44:451-74 [DOI] [PubMed] [Google Scholar]
- 5. Cronan MR, Nakamura K, Johnson NL, et al. Defining MAP3 kinases required for MDA-MB-231 cell tumor growth and metastasis. Oncogene. 2012;31:3889-900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thylur RP, Senthivinayagam S, Campbell EM, et al. Mixed lineage kinase 3 modulates beta-catenin signaling in cancer cells. J Biol Chem. 2011;286:37470-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen J, Miller EM, Gallo KA. MLK3 is critical for breast cancer cell migration and promotes a malignant phenotype in mammary epithelial cells. Oncogene. 2010;29:4399-411 [DOI] [PubMed] [Google Scholar]
- 8. Gallo KA, Johnson GL. Signalling: mixed- lineage kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol. 2002;3:663-72 [DOI] [PubMed] [Google Scholar]
- 9. Rana A, Gallo K, Godowski P, et al. The mixed lineage kinase SPRK phosphorylates and activates the stress-activated protein kinase activator, SEK-1. J Biol Chem. 1996;271:19025-8 [DOI] [PubMed] [Google Scholar]
- 10. Mayer BJ, Baltimore D. Signalling through SH2 and SH3 domains. Trends Cell Biol. 1993;3:8-13 [DOI] [PubMed] [Google Scholar]
- 11. Bock BC, Vacratsis PO, Qamirani E, Gallo KA. Cdc42-induced activation of the mixed-lineage kinase SPRK in vivo: requirement of the Cdc42/Rac interactive binding motif and changes in phosphorylation. J Biol Chem. 2000;275:14231-41 [DOI] [PubMed] [Google Scholar]
- 12. Teramoto H, Coso OA, Miyata H, Igishi T, Miki T, Gutkind JS. Signaling from the small GTP-binding proteins Rac1 and Cdc42 to the c-Jun N-terminal kinase/stress-activated protein kinase pathway: a role for mixed lineage kinase 3/protein-tyrosine kinase 1, a novel member of the mixed lineage kinase family. J Biol Chem. 1996;271:27225-8 [DOI] [PubMed] [Google Scholar]
- 13. Qiao F, Bowie JU. The many faces of SAM. Sci STKE. 2005;2005:re7. [DOI] [PubMed] [Google Scholar]
- 14. Poitras L, Bisson N, Islam N, Moss T. A tissue restricted role for the Xenopus Jun N-terminal kinase kinase kinase MLK2 in cement gland and pronephric tubule differentiation. Dev Biol. 2003;254:200-14 [DOI] [PubMed] [Google Scholar]
- 15. Poitras L, Jean S, Islam N, Moss T. PAK interacts with NCK and MLK2 to regulate the activation of Jun N-terminal kinase. FEBS Lett. 2003;543:129-35 [DOI] [PubMed] [Google Scholar]
- 16. Nihalani D, Merritt S, Holzman LB. Identification of structural and functional domains in mixed lineage kinase dual leucine zipper-bearing kinase required for complex formation and stress-activated protein kinase activation. J Biol Chem. 2000;275:7273-9 [DOI] [PubMed] [Google Scholar]
- 17. Xu Z, Maroney AC, Dobrzanski P, Kukekov NV, Greene LA. The MLK family mediates c-Jun N-terminal kinase activation in neuronal apoptosis. Mol Cell Biol. 2001;21:4713-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tibbles LA, Ing YL, Kiefer F, et al. MLK-3 activates the SAPK/JNK and p38/RK pathways via SEK1 and MKK3/6. EMBO J. 1996;15:7026- 35 [PMC free article] [PubMed] [Google Scholar]
- 19. Yasuda J, Whitmarsh AJ, Cavanagh J, Sharma M, Davis RJ. The JIP group of mitogen-activated protein kinase scaffold proteins. Mol Cell Biol. 1999;19:7245-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Merritt SE, Mata M, Nihalani D, Zhu C, Hu X, Holzman LB. The mixed lineage kinase DLK utilizes MKK7 and not MKK4 as substrate. J Biol Chem. 1999;274:10195-202 [DOI] [PubMed] [Google Scholar]
- 21. Hartkamp J, Troppmair J, Rapp UR. The JNK/SAPK activator mixed lineage kinase 3 (MLK3) transforms NIH 3T3 cells in a MEK-dependent fashion. Cancer Res. 1999;59:2195-202 [PubMed] [Google Scholar]
- 22. Shen YH, Godlewski J, Zhu J, et al. Cross-talk between JNK/SAPK and ERK/MAPK pathways: sustained activation of JNK blocks ERK activation by mitogenic factors. J Biol Chem. 2003;278:26715-21 [DOI] [PubMed] [Google Scholar]
- 23. Chadee DN, Xu D, Hung G, et al. Mixed-lineage kinase 3 regulates B-Raf through maintenance of the B-Raf/Raf-1 complex and inhibition by the NF2 tumor suppressor protein. Proc Natl Acad Sci U S A. 2006;103:4463-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sathyanarayana P, Barthwal MK, Lane ME, et al. Drosophila mixed lineage kinase/slipper: a missing biochemical link in Drosophila JNK signaling. Biochim Biophys Acta. 2003;1640:77-84 [DOI] [PubMed] [Google Scholar]
- 25. Stronach B, Perrimon N. Activation of the JNK pathway during dorsal closure in Drosophila requires the mixed lineage kinase, slipper. Genes Dev. 2002;16:377-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sathyanarayana P, Barthwal MK, Kundu CN, et al. Activation of the Drosophila MLK by ceramide reveals TNF-alpha and ceramide as agonists of mammalian MLK3. Mol Cell. 2002;10:1527-33 [DOI] [PubMed] [Google Scholar]
- 27. Brancho D, Ventura JJ, Jaeschke A, Doran B, Flavell RA, Davis RJ. Role of MLK3 in the regulation of mitogen-activated protein kinase signaling cascades. Mol Cell Biol. 2005;25:3670-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Humphrey RK, Newcomb CJ, Yu SM, et al. Mixed lineage kinase-3 stabilizes and functionally cooperates with TRIBBLES-3 to compromise mitochondrial integrity in cytokine-induced death of pancreatic beta cells. J Biol Chem. 2010;285:22426-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen J, Gallo KA. MLK3 regulates paxillin phosphorylation in chemokine-mediated breast cancer cell migration and invasion to drive metastasis. Cancer Res. 2012;72:4130-40 [DOI] [PubMed] [Google Scholar]
- 30. Sondarva G, Kundu CN, Mehrotra S, et al. TRAF2-MLK3 interaction is essential for TNF-alpha-induced MLK3 activation. Cell Res. 2010;20:89-98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mishra R, Barthwal MK, Sondarva G, et al. Glycogen synthase kinase-3beta induces neuronal cell death via direct phosphorylation of mixed lineage kinase 3. J Biol Chem. 2007;282:30393-405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barthwal MK, Sathyanarayana P, Kundu CN, et al. Negative regulation of mixed lineage kinase 3 by protein kinase B/AKT leads to cell survival. J Biol Chem. 2003;278:3897-902 [DOI] [PubMed] [Google Scholar]
- 33. Leung IW, Lassam N. Dimerization via tandem leucine zippers is essential for the activation of the mitogen-activated protein kinase kinase kinase, MLK-3. J Biol Chem. 1998;273:32408-15 [DOI] [PubMed] [Google Scholar]
- 34. Vacratsis PO, Gallo KA. Zipper-mediated oligomerization of the mixed lineage kinase SPRK/MLK-3 is not required for its activation by the GTPase cdc 42 but is necessary for its activation of the JNK pathway: monomeric SPRK L410P does not catalyze the activating phosphorylation of Thr258 of murine mitogen-activated protein kinase kinase 4. J Biol Chem. 2000;275:27893-900 [DOI] [PubMed] [Google Scholar]
- 35. Zhang H, Gallo KA. Autoinhibition of mixed lineage kinase 3 through its Src homology 3 domain. J Biol Chem. 2001;276:45598-603 [DOI] [PubMed] [Google Scholar]
- 36. Kiefer F, Tibbles LA, Anafi M, et al. HPK1, a hematopoietic protein kinase activating the SAPK/JNK pathway. EMBO J. 1996;15:7013-25 [PMC free article] [PubMed] [Google Scholar]
- 37. Boggon TJ, Eck MJ. Structure and regulation of Src family kinases. Oncogene. 2004;23:7918-27 [DOI] [PubMed] [Google Scholar]
- 38. Schachter KA, Du Y, Lin A, Gallo KA. Dynamic positive feedback phosphorylation of mixed lineage kinase 3 by JNK reversibly regulates its distribution to triton-soluble domains. J Biol Chem. 2006;281:19134-44 [DOI] [PubMed] [Google Scholar]
- 39. Phelan DR, Price G, Liu YF, Dorow DS. Activated JNK phosphorylates the C-terminal domain of MLK2 that is required for MLK2-induced apoptosis. J Biol Chem. 2001;276:10801-10 [DOI] [PubMed] [Google Scholar]
- 40. Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Genet Dev. 2002;12:14-21 [DOI] [PubMed] [Google Scholar]
- 41. Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537-49 [DOI] [PubMed] [Google Scholar]
- 42. Mishra P, Senthivinayagam S, Rangasamy V, Sondarva G, Rana B. Mixed lineage kinase-3/JNK1 axis promotes migration of human gastric cancer cells following gastrin stimulation. Mol Endocrinol. 2010;24:598-607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhan Y, Abi Saab WF, Modi N, Stewart AM, Liu J, Chadee DN. Mixed lineage kinase 3 is required for matrix metalloproteinase expression and invasion in ovarian cancer cells. Exp Cell Res. 2012;318:1641-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Swenson KI, Winkler KE, Means AR. A new identity for MLK3 as an NIMA-related, cell cycle-regulated kinase that is localized near centrosomes and influences microtubule organization. Mol Biol Cell. 2003;14:156-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Osmani AH, McGuire SL, Osmani SA. Parallel activation of the NIMA and p34cdc2 cell cycle-regulated protein kinases is required to initiate mitosis in A. nidulans. Cell. 1991;67:283-91 [DOI] [PubMed] [Google Scholar]
- 46. Cha H, Dangi S, Machamer CE, Shapiro P. Inhibition of mixed-lineage kinase (MLK) activity during G2-phase disrupts microtubule formation and mitotic progression in HeLa cells. Cell Signal. 2006;18:93-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rangasamy V, Mishra R, Sondarva G, et al. Mixed-lineage kinase 3 phosphorylates prolyl-isomerase Pin1 to regulate its nuclear translocation and cellular function. Proc Natl Acad Sci U S A. 2012;109:8149-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rangasamy V, Mishra R, Mehrotra S, et al. Estrogen suppresses MLK3-mediated apoptosis sensitivity in ER+ breast cancer cells. Cancer Res. 2010;70:1731-40 [DOI] [PMC free article] [PubMed] [Google Scholar]