

Abstract
Objective:
To determine whether the association between multiple sclerosis (MS) and Leber hereditary optic neuropathy (LHON) (known as “Harding disease”) is a chance finding, or the 2 disorders are mechanistically linked.
Methods:
We performed a United Kingdom–wide prospective cohort study of prevalent cases of MS with LHON mitochondrial DNA (mtDNA) mutations. The new cases were compared with published cases, enabling a comprehensive clinical description. We also performed a meta-analysis of studies screening patients with MS for LHON mtDNA mutations to find evidence of a genetic association.
Results:
Twelve new patients were identified from 11 pedigrees, and 44 cases were identified in the literature. The combined cohort had the following characteristics: multiple episodes of visual loss, predominance for women, and lengthy time interval before the fellow eye is affected (average 1.66 years), which is very atypical of LHON; conversely, most patients presented without eye pain and had a poor visual prognosis, which is unusual for optic neuritis associated with MS. The number of UK cases of LHON-MS fell well within the range predicted by the chance occurrence of MS and the mtDNA mutations known to cause LHON. There was no association between LHON mtDNA mutations and MS in a meta-analysis of the published data.
Conclusions:
Although the co-occurrence of MS and LHON mtDNA mutations is likely to be due to chance, the resulting disorder has a distinct phenotype, implicating a mechanistic interaction. Patients with LHON-MS have a more aggressive course, and prognostication and treatment should be guarded.
Leber hereditary optic neuropathy (LHON) is a maternally inherited form of blindness characterized by the preferential loss of the retinal ganglion cells and subsequent optic nerve degeneration. The visual prognosis is poor, and the majority of patients remain registered legally blind for the rest of their lives with a substantial impact on their quality of life.1
Shortly after the identification of mitochondrial DNA (mtDNA) mutations causing LHON, case series described the m.11778A>G mtDNA mutation in patients with features of both LHON and multiple sclerosis (MS).2 These patients were all women, and 6 were considered to have clinically definite MS, while the other 2 had visual loss with white matter abnormalities on MRI suggestive of demyelination. Further reports and case series were published of MS associated with all 3 primary LHON mutations (m.11778A>G, m.3460A>G, and m.14484T>C),3,4 and this particular disease association became known as “Harding disease” (here termed LHON-MS).
Given that LHON is a rare disorder, affecting approximately 1 in 30,000 to 50,000 in England and other northern European populations,5−7 the growing number of patients described with LHON-MS has been considered to be greater than expected by chance.8 This led to speculation of an interaction between the mitochondrial dysfunction in LHON and the emerging immunologic mechanism in MS.
To resolve this issue, we conducted the first prospective study of patients with LHON mutations and an MS-like illness (LHON-MS cases) and compared these findings with cases systematically ascertained from the published literature. The published cases were identified from both clinical case reports and case series, and molecular genetic screening of MS cohorts. Our aims were to 1) define the clinical phenotype in a large cohort of patients with an overlapping LHON-MS phenotype, 2) compare this phenotype with “classic” LHON and MS, and 3) understand potential underlying disease mechanisms, particularly whether the association occurs purely by chance.
METHODS
Survey of prevalent cases.
Prevalent cases in the United Kingdom were identified through the British Neurological Surveillance Unit (BNSU) coordinated by the Association of British Neurologists. Between 2005 and 2010, the BNSU contacted more than 600 Association of British Neurologists members every month, requesting details of new or existing suspected cases of LHON and MS. Members notifying the BNSU were contacted by the study team to obtain consent and collect 1) clinical data describing the sex, age at onset, and clinical course, and 2) the results of relevant investigations. These included brain MRI; CSF cell count, protein content, and oligoclonal band analysis; evoked potential results analysis; and mtDNA analysis. Cases were included if they met current diagnostic criteria for MS9 and harbored an mtDNA mutation known to cause LHON, or harbored a LHON mutation and had clinical investigations compatible with MS.9 Patients were considered to qualify as being registered blind if their best-corrected visual acuity was 3/60 (Snellen) or worse.10
Systematic review of published cases of LHON and an MS-like illness.
A systematic review of the medical literature, with no date limits, for articles written in English or French was conducted using the following search terms in MEDLINE on May 28, 2013: (leber or leber's or lhon or mitochondrial disease) and (MS or transverse myelitis or neuromyelitis optica or devic or devic's or nmo or white matter lesions or demyelination or harding or harding's). The search was conducted independently by 2 of the authors (G.P. and A.B.). Data collection was conducted independently by 2 of the authors (G.P. and A.B.) and in the case of any disagreement, the relevant articles were re-reviewed to reach a consensus. We defined cases of LHON-MS as those with a primary LHON mutation and a clinical description compatible with MS with appropriate diagnostic test results, or patients described as having “clinically definite” MS based on contemporaneous diagnostic criteria. Patients not meeting these criteria were excluded. Data extracted included mutation status, sex, age at onset, first symptom (visual or neurologic), type of visual loss (unilateral, sequential, or bilateral simultaneous), interval before involvement of fellow eye, number of episodes of visual loss, response to corticosteroids, presence of ocular pain, subtype of MS (relapsing-remitting MS [RRMS] or primary progressive MS [PPMS]), and presence of oligoclonal bands and diagnostic MRI findings. When indicated, study authors were contacted to provide additional clinical information.
Meta-analysis of mtDNA studies in MS.
A systematic review of the medical literature written in English or French with no date limits was conducted using the following search terms in OvidSP MEDLINE on May 20, 2013: (multiple sclerosis and [mitochondrial dna or mtdna] and mutation). We restricted our analysis to known pathogenic mutations of mtDNA detected using either allele-specific assays or mtDNA sequencing. We excluded reports describing associations with common polymorphic mtDNA alleles (>1% minor allele frequency) or mtDNA haplogroups. Data collected included the number of positive MS patients for each of the 3 primary LHON mutations, the number of MS patients and controls screened, the entry criteria of the MS patients, and race/ethnicity of participants.
Standard protocol approvals, registrations, and patient consents.
Institutional research ethics board approval was obtained, and written informed consent was secured from all patients participating in the study.
RESULTS
Survey of prevalent cases.
The BNSU received 27 notifications of cases not previously reported, of whom 12 originating from 11 pedigrees met our criteria for LHON-MS (table 1). One case (patient 9) who had MS without visual symptoms or signs was identified with the m.11778G>A mutation. All other patients had a clinical picture of MS with severe involvement of the anterior visual pathway. Fifteen patients were excluded: i) 10 patients in whom we were not able to substantiate the molecular genetic diagnosis, ii) 3 patients who did not meet criteria for MS, and iii) 2 patients for whom no further clinical data were obtainable.
Table 1.
Case series of 12 new patients with LHON-MS
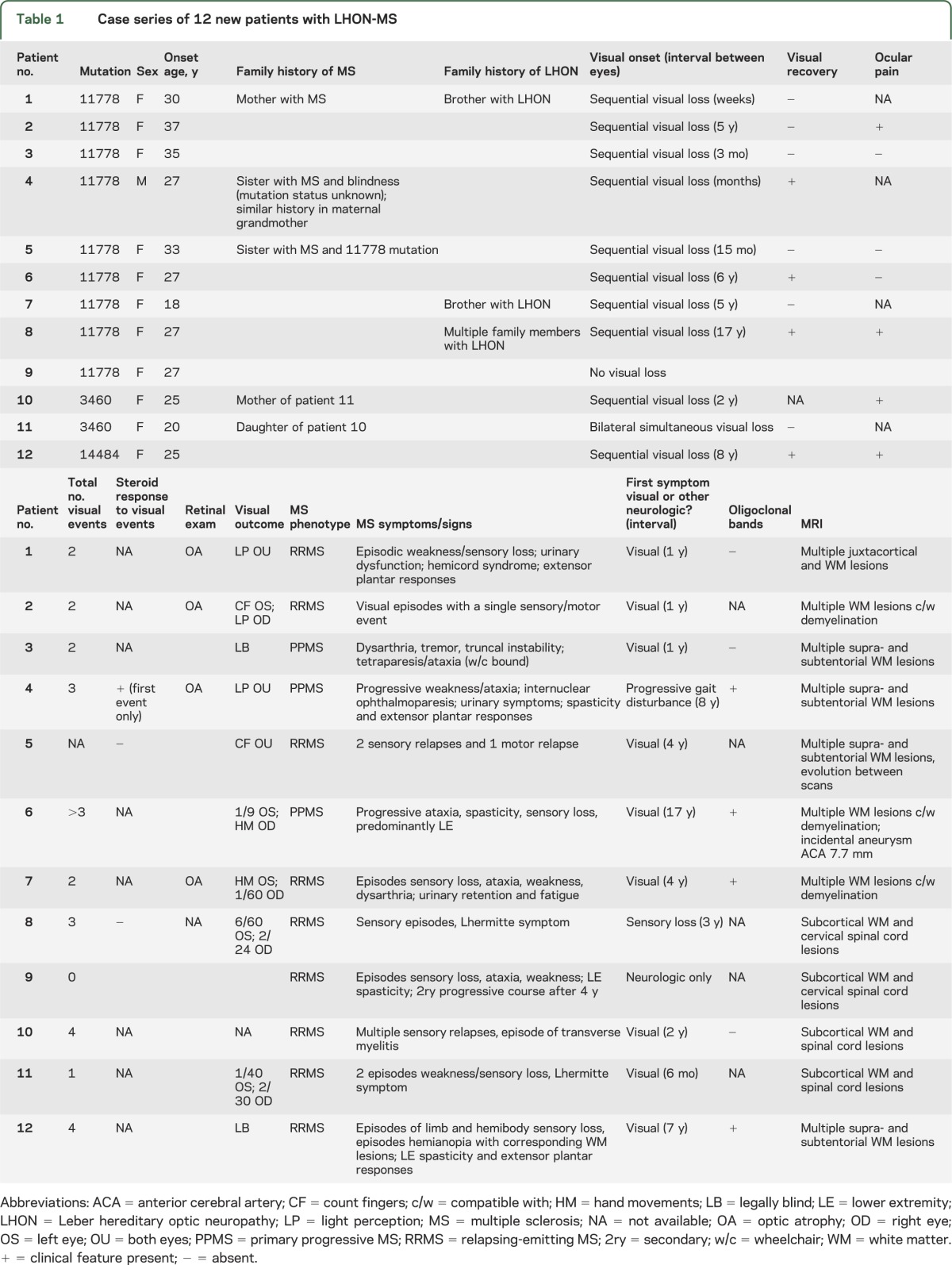
Of the 12 patients identified in this BNSU survey, 9 harbored the m.11778A>G mutation, 2 the m.3460A>G mutation, and 1 the m.14484A>G mutation. Eleven of the patients were women. All presented in early to mid adulthood (average age 25.6 years, range 18–37 years), with clinical and paraclinical evidence of MS (table 1). Nine of 12 patients had RRMS, and in 3 the course was PPMS.
Of the 11 patients having visual loss, 6 had visual acuity meeting criteria for blindness registration. Four of the patients had ocular pain at the onset of visual symptoms. One patient had bilateral visual loss at presentation, and the other 9 had sequential visual loss with intervals of up to 17 years before both eyes were affected (the interval was more than 1 year in 7 patients). Five of the patients had more than 2 episodes of visual loss over their disease course. Four of the patients had some degree of visual recovery after the onset of symptoms, and patient 4 experienced slight initial improvement with corticosteroid therapy. A representative MRI from patient 8 is shown in figure 1, with fundal images and optical coherence tomography (figure 2).
Figure 1. Head and cervical spine MRI findings in patient 8.
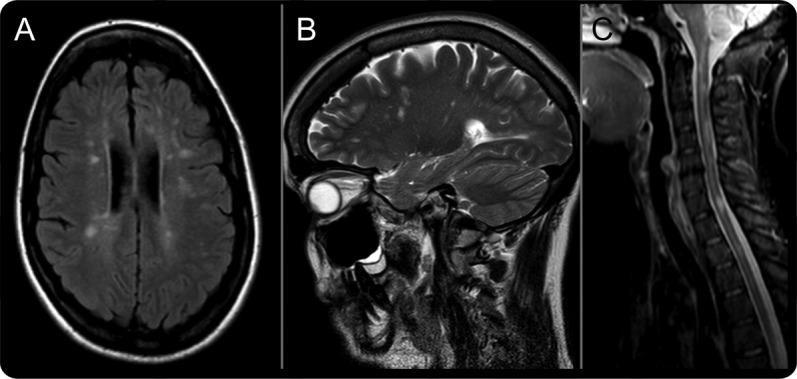
(A) Transverse axial T2 fluid-attenuated inversion recovery image of the head demonstrates multiple ovoid high-signal lesions in the periventricular white matter and juxtacortically. (B) Sagittal T2 image reveals lesions with an orientation resembling Dawson fingers. (C) Sagittal T2 image of the cervical spine demonstrates multiple high-signal lesions that are each less than one spinal segment. Taken together, these findings meet MRI diagnostic criteria for multiple sclerosis.
Figure 2. Visual examination and optical coherence tomography findings in patient 8.

(A) Bilateral optic atrophy is demonstrated on fundoscopic examination (RE = right eye, LE = left eye). (B) Optical coherence tomography (OCT) measurements were obtained with the high-resolution spectral-domain Cirrus platform (Carl Zeiss Meditec, Dublin, CA). The average retinal nerve fiber layer (RNFL) thickness was 70 μm in the right eye (OD) and 66 μm in the left eye (OS). (C) The analysis software automatically selects the appropriate normative range for the patient, and the peripapillary RNFL measurements (dark traces) are represented within color-coded distribution centiles: i) red <1%, ii) yellow 1%–5%, and iii) green 5%–95%. The OCT measurements confirm significant RNFL thinning in keeping with the optic disc pallor. TSNIT = temporal-superior-nasal-inferior-temporal.
Systematic review of published cases.
The search strategy identified 122 articles. After reading the abstracts, 39 titles were selected for review of the full-text articles. The bibliography of these articles revealed 8 additional publications, which were also included in the final review. From these 47 articles, we identified 44 patients who met our criteria for inclusion. These patients and their clinical details, with references, are summarized in table e-1 on the Neurology® Web site at www.neurology.org. Eleven patients were excluded from the analysis on the basis that they did not have sufficient evidence for the diagnosis of MS, despite the presence of LHON mutations and paraclinical investigations suggestive of MS (table e-2). We excluded 29 patients who had been reported from 13 publications, and have provided descriptions and reasons for exclusion in table e-3 (the most common reason for exclusion, in 22 cases, was the absence of a documented primary LHON mutation).
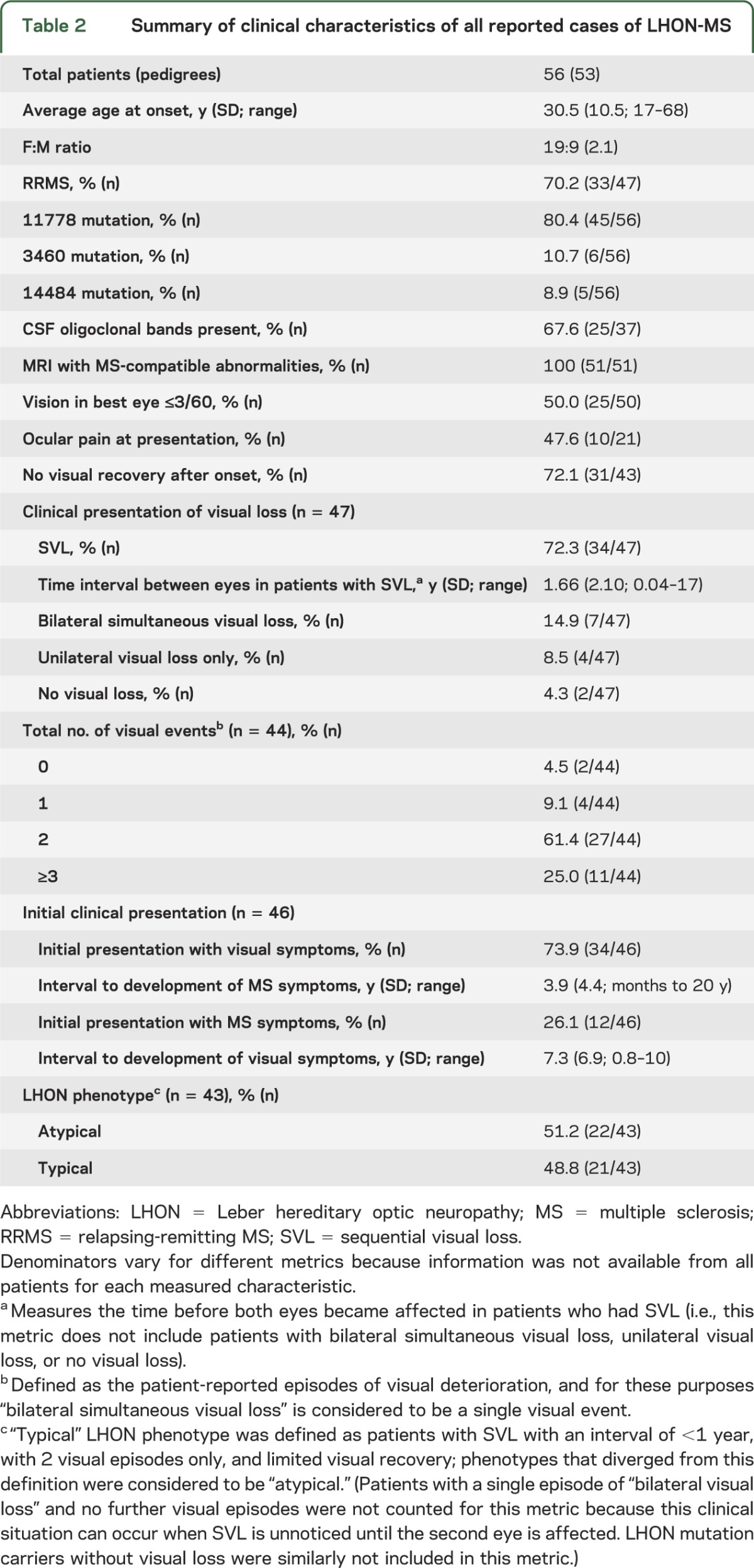
There were no statistically significant differences between the 12 new cases and the 44 patients identified from the published literature; therefore, we combined the 2 datasets in order to summarize the salient features of all 56 patients. We present various metrics in aggregate in table 2. Most patients presented from early to mid adulthood with a predominance of affected women (2.1:1). Most had RRMS, the m.11778 G>A mutation, and a poor visual prognosis. Many of the patients had features atypical for LHON, such as the presence of ocular pain at presentation (47.6%), a lengthy time interval before involvement of the fellow eye (average of 1.66 years, and up to 17 years in one case), and more than 2 episodes of visual loss (25%).
Table 2.
Summary of clinical characteristics of all reported cases of LHON-MS
Meta-analysis of mtDNA studies in MS.
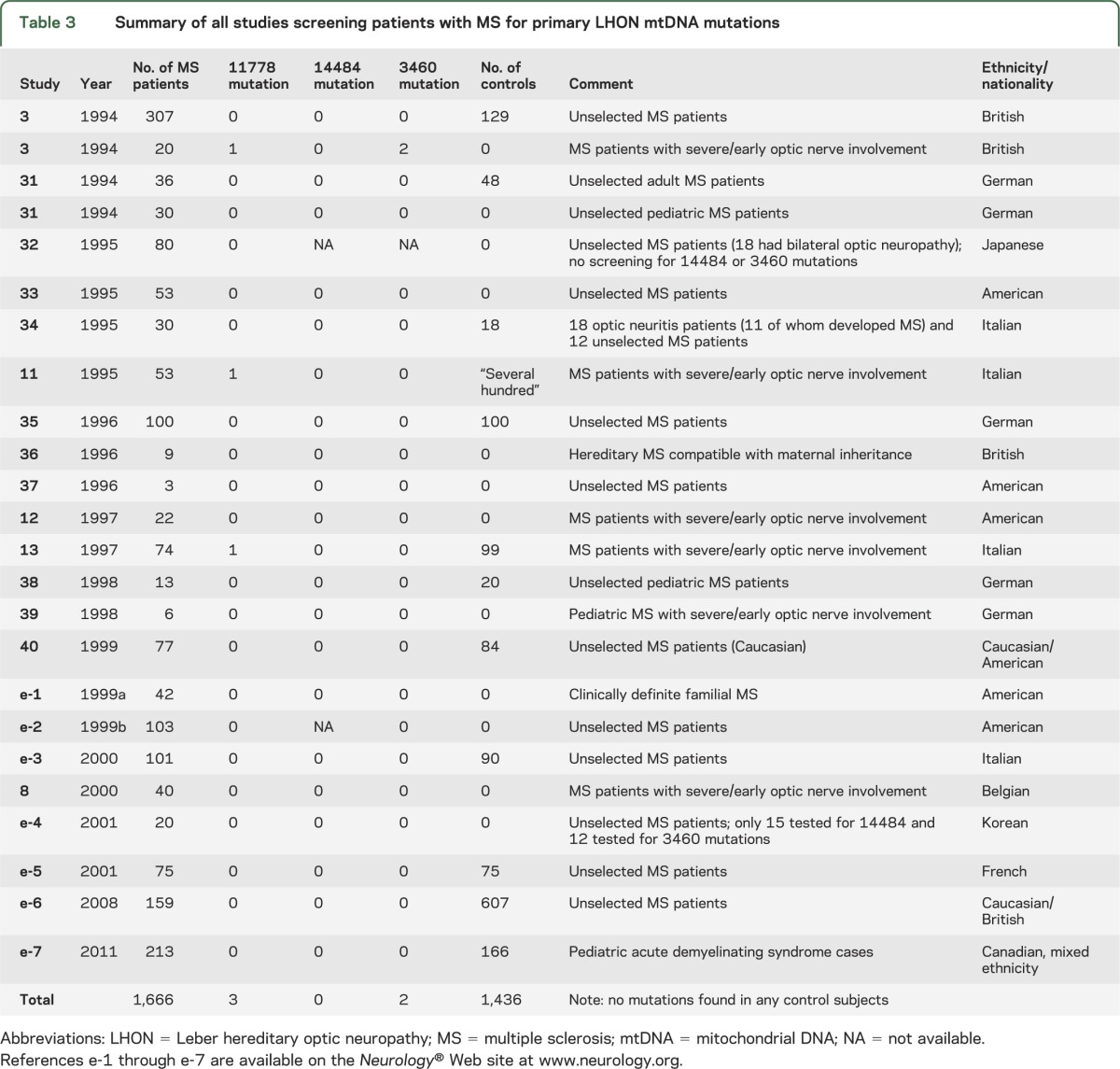
The search strategy identified 59 articles, 18 of which were relevant and thus included in this study. Reference searching of these 18 articles yielded 4 further articles for inclusion. We summarize the results from these 22 articles reporting the frequency of pathogenic mtDNA mutations in MS cases in table 3. Of the 1,666 patients with MS screened, 5 patients with primary LHON mutations were identified (these patients are also individually described in table e-1). All 5 patients with mutations had severe and early optic nerve involvement (a subgroup containing only 209 patients from 5 studies,3,8,11−13 although the lack of control groups in most of these studies makes this finding difficult to evaluate). No LHON mutations were identified from 1,436 matched controls, although this was not statistically different from the mutation rate in the patients with MS (p = 0.066). The mutation rate in the patients with MS from this meta-analysis was not statistically different compared with the prevalence rates for the primary LHON mutations (homoplasmic or heteroplasmic) previously reported in the population.14
Table 3.
Summary of all studies screening patients with MS for primary LHON mtDNA mutations
DISCUSSION
Here, we describe 12 LHON mtDNA mutation carriers with clinically definite MS prospectively ascertained from the BNSU survey, and 44 additional cases from the published literature. Within this group, there was a broad range of features, from typical RRMS without visual involvement, to patients with severe ocular involvement and varying degrees of neurologic disability attributable to MS. On the whole, most patients had severe visual impairment (so severe as to be registered legally blind in 50%) and the phenotype of RRMS predominated, being present in 70.2%. Despite the limitations of our study, which include the selection bias of cases referred to neurologists engaged with the BNSU, the inherent publication bias of case reports, and the language limitations of our literature review, we observed a remarkably consistent phenotype between our study group and the previously reported cases.
How do the reported patients with LHON-MS differ from classic LHON, and typical MS? Compared with classic LHON, the age at onset and bilateral sequential visual involvement were typical, as was the lack of visual recovery. The distribution of 3 primary mtDNA LHON mutations also fell within the range previously reported in population-based epidemiologic studies.5−7 However, these patients with LHON-MS differ significantly from the standard population in LHON regarding the predominance of affected women (2.1:1), the large proportion of patients with more than 2 visual events (25%) or persisting unilateral visual loss (8.5%), and the long time interval before both eyes became affected (average 1.66 years, and up to 17 years). These would be extremely unusual for LHON in which typically the sex ratio favors men by a ratio of 4:1, patients have only 2 visual events, and both eyes are almost invariably affected within 6 months (at most 1 year).15 This case series demonstrates that slightly more than half of all LHON-MS cases present with an atypical phenotype for LHON, based on the above uncharacteristic features for classic LHON (table 2).
When comparing LHON-MS with MS, shared features are age at onset, predilection for women, and predominance of the RRMS phenotype (71.1%). However, important differences from MS in our study are the high proportion of patients with visual involvement (96%), lack of ocular pain in most patients (only 10 patients reported ocular pain), absence of visual recovery in most patients (72.1%), and visual outcome resulting in registrable blindness in 50%. This is significantly more prevalent and severe than in typical MS, whereby 50% of patients with MS are expected to have visual involvement through the entire course of their illness and, of these, 85% to 95% recover to visual acuity better than 6/9.16 Furthermore, optic neuritis is ordinarily accompanied by ocular pain in more than 90% of patients.17 LHON-MS does, therefore, appear to have a clinical phenotype distinct from both LHON and MS, implying a mechanistic interaction between the 2 disorders.
In the United Kingdom, LHON mtDNA mutations are found in 1/300 of the population (1/1,000 of the population in homoplasmic state),14 and MS affects 2.6/1,000.18 This suggests that approximately 1 in 115,000 of the UK population will harbor both a LHON mutation and develop MS by chance. However, if we assume that only homoplasmic LHON mutations are associated with disease, then homoplasmic LHON mutations overlap with MS in approximately 1 in 400,000 of the UK population. Based on the current population of approximately 63 million from the 2011 census, this equates to 150 individuals, which is far greater than the number ascertained through our national survey. The cases we have identified could therefore represent the chance finding of relatively common mtDNA mutations and a relatively common neuroinflammatory disorder. Based on these observations, it would be unwise to conclude that LHON mtDNA mutations predispose to MS. This is supported by several negative studies screening MS cohorts for LHON mtDNA mutations (table 3).
How can we reconcile this conclusion with the distinct phenotype of LHON-MS? There are several possibilities: 1) that the mtDNA mutations modify the phenotype of MS, causing “atypical optic neuritis,” which is painless, severe, and irreversible; 2) that the genetic and environmental factors that predispose to MS, enriched in women, precipitate acute LHON in mtDNA mutation carriers who would otherwise have remained asymptomatic; or 3) a combination of the above. In other words, that having the LHON mtDNA mutation would direct a coincidental inflammatory response to affect the vulnerable anterior visual pathway in individuals already susceptible to develop MS. In this case, there would be interdependence, and an interplay causing this unusual phenotype. A similar scenario may also exist in patients with the mitochondrial optic neuropathy due to OPA1 mutations, whereby patients have been described with coincident MS.19 Understanding the mechanism of this interaction is likely to provide novel insights into the pathophysiology of both disorders.
LHON mtDNA mutations cause a defect of respiratory chain complex I,20 and given the recent observation of reduced respiratory chain activity in both acute and chronic MS lesions,21,22 it is conceivable that the 2 disorders interact at the biochemical level, through a defect of ATP (adenosine triphosphate) synthesis. However, LHON mtDNA mutations also sensitize cells to apoptotic cell death, perhaps exposing neurons to the downstream consequences of an inflammatory insult. There is also evidence that mtDNA codes for a maternally inherited human leukocyte antigen,23 and that mtDNA itself can be proinflammatory if released into the circulation. It is therefore possible that the underlying sequence variations themselves modify the inflammatory response. Intriguingly, 2 of the new cases we report are related (mother and daughter), and 5 of the published cases described additional affected family members with MS. This raises the possibility of an inherited predisposition to LHON-MS, but it is difficult at this stage to know whether this simply reflects the coinheritance of the LHON mtDNA mutation on a high-risk nuclear genetic background, or whether additional factors come in to play, such as the background mtDNA haplogroup.
With this in mind, how should we treat patients with LHON-MS? There is evidence suggesting that idebenone may limit progression in patients with LHON who have discordant visual acuity and are presumably early in their disease course.24 There is also lengthy experience with disease-modifying agents in MS, although these have not been shown to have any effect on long-term disability progression.25 Based on our own experience, this is a difficult and emotive issue, and the data are insufficient to offer clear guidance at present. Having said that, idebenone is safe in LHON, could prevent visual disability, and it led to visual improvement in one26 of two reported case reports with LHON-MS.26,27 Others reported the use of mitoxantrone,27,28 with improvement in one patient.28 Mitoxantrone is considered to be a therapeutic option in severe MS for selected patients, but its use is difficult to justify because of its severe short- and long-term adverse reactions in the context of safer alternatives.29 In any event, the presence of an LHON mutation in a patient with MS indicates a high likelihood of severe visual disability, and therefore early disease-modifying therapy to prevent relapses may be preferable despite the uncertainty relating to long-term benefits. Furthermore, a recent study indicates that a portion of patients with MS have improvements in visual evoked potentials on treatment with 4-aminopyridine,30 and this LHON-MS patient group with severe visual prognosis may be a suitable subpopulation of MS for study with this agent.
In the absence of any evidence in this situation, treatment should be determined on a case-by-case basis in a center with expertise in the management of MS. There is a clear need to evaluate these different treatment approaches in a prospective, objective way. This will be extremely challenging, given the difficulty of studying treatment effects in rare diseases, and the size of the studies needed to demonstrate a treatment effect in MS. Identifying a reliable, clinically relevant biomarker would provide a valuable tool in developing new treatments for LHON-MS, enabling open-labeled studies as a starting point for this devastating disease.
Supplementary Material
ACKNOWLEDGMENT
G.P. is the recipient of a Bisby Fellowship from the Canadian Institutes of Health Research. A.B. is National Institute for Health Research (NIHR) Academic Clinical Fellow. P.Y.-W.-M. is a Medical Research Council (MRC, UK) Clinician Scientist. P.F.C. is an Honorary Consultant Neurologist at Newcastle upon Tyne Foundation Hospitals NHS Trust, is a Wellcome Trust Senior Fellow in Clinical Science (084980/Z/08/Z), and a UK NIHR Senior Investigator. P.F.C. receives additional support from the Wellcome Trust Centre for Mitochondrial Research (096919Z/11/Z), the Medical Research Council (UK) Centre for Translational Muscle Disease research, the Association Française contre les Myopathies, and EU FP7 TIRCON, and the NIHR Newcastle Biomedical Research Centre based at Newcastle upon Tyne Hospitals NHS Foundation Trust and Newcastle University. D.A.S.C. is supported by the Cambridge NIHR Biomedical Research Centre. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health. The authors thank the British Neurological Surveillance Unit and Association of British Neurologists for their assistance with this study. With thanks to Dr. Roger Barker, Dr. Simon Hammans, Dr. George Bhima, Dr. Owen Pearson, Dr. Neil Robertson, Dr. Cris Constantinescu, and Dr. James Overell who referred patients for this study. With thanks to Dr. Maria A Rocca, Prof. M. Filippi, and Prof. Jacqueline Palace who provided additional information regarding previously published cases.
GLOSSARY
- BNSU
British Neurological Surveillance Unit
- LHON
Leber hereditary optic neuropathy
- MS
multiple sclerosis
- mtDNA
mitochondrial DNA
- PPMS
primary progressive multiple sclerosis
- RRMS
relapsing-remitting multiple sclerosis
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Gerald Pfeffer: data acquisition and analysis, manuscript authorship/revision. Ailbhe Burke: data acquisition and analysis, manuscript revision. Patrick Yu-Wai-Man: data acquisition and analysis, manuscript revision. D. Alastair S. Compston: study concept and design, data acquisition and analysis, manuscript revision. Patrick F. Chinnery: study concept and design, data acquisition and analysis, manuscript authorship/revision.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Kirkman MA, Korsten A, Leonhardt M, et al. Quality of life in patients with Leber hereditary optic neuropathy. Invest Ophthalmol Vis Sci 2009;50:3112–3115 [DOI] [PubMed] [Google Scholar]
- 2.Harding AE, Sweeney MG, Miller DH, et al. Occurrence of a multiple sclerosis-like illness in women who have a Leber's hereditary optic neuropathy mitochondrial DNA mutation. Brain 1992;115(pt 4):979–989 [DOI] [PubMed] [Google Scholar]
- 3.Kellar-Wood H, Robertson N, Govan GG, Compston DA, Harding AE. Leber's hereditary optic neuropathy mitochondrial DNA mutations in multiple sclerosis. Ann Neurol 1994;36:109–112 [DOI] [PubMed] [Google Scholar]
- 4.Bhatti MT, Newman NJ. A multiple sclerosis-like illness in a man harboring the mtDNA 14484 mutation. J Neuroophthalmol 1999;19:28–33 [PubMed] [Google Scholar]
- 5.Man PY, Griffiths PG, Brown DT, Howell N, Turnbull DM, Chinnery PF. The epidemiology of Leber hereditary optic neuropathy in the North East of England. Am J Hum Genet 2003;72:333–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Puomila A, Hamalainen P, Kivioja S, et al. Epidemiology and penetrance of Leber hereditary optic neuropathy in Finland. Eur J Hum Genet 2007;15:1079–1089 [DOI] [PubMed] [Google Scholar]
- 7.Spruijt L, Kolbach DN, de Coo RF, et al. Influence of mutation type on clinical expression of Leber hereditary optic neuropathy. Am J Ophthalmol 2006;141:676–682 [DOI] [PubMed] [Google Scholar]
- 8.Vanopdenbosch L, Dubois B, D'Hooghe MB, Meire F, Carton H. Mitochondrial mutations of Leber's hereditary optic neuropathy: a risk factor for multiple sclerosis. J Neurol 2000;247:535–543 [DOI] [PubMed] [Google Scholar]
- 9.Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2011;69:292–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bunce C, Wormald R. Causes of blind certifications in England and Wales: April 1999–March 2000. Eye 2008;22:905–911 [DOI] [PubMed] [Google Scholar]
- 11.Carrara F, Eoli M, La Mantia L, Zeviani M. The contribution of LHON mitochondrial DNA mutations to multiple sclerosis. Am J Hum Genet 1995;57:A336 [Google Scholar]
- 12.Kalman B, Rodriguez-Valdez JL, Bosch U, Lublin FD. Screening for Leber's hereditary optic neuropathy associated mitochondrial DNA mutations in patients with prominent optic neuritis. Mult Scler 1997;2:279–282 [DOI] [PubMed] [Google Scholar]
- 13.Leuzzi V, Carducci C, Lenza M, et al. LHON mutations in Italian patients affected by multiple sclerosis. Acta Neurol Scand 1997;96:145–148 [DOI] [PubMed] [Google Scholar]
- 14.Elliott HR, Samuels DC, Eden JA, Relton CL, Chinnery PF. Pathogenic mitochondrial DNA mutations are common in the general population. Am J Hum Genet 2008;83:254–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Man PY, Turnbull DM, Chinnery PF. Leber hereditary optic neuropathy. J Med Genet 2002;39:162–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McDonald WI, Barnes D. The ocular manifestations of multiple sclerosis: 1: abnormalities of the afferent visual system. J Neurol Neurosurg Psychiatry 1992;55:747–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Optic Neuritis Study Group The clinical profile of optic neuritis: experience of the Optic Neuritis Treatment Trial. Arch Ophthalmol 1991;109:1673–1678 [DOI] [PubMed] [Google Scholar]
- 18.Hirst C, Ingram G, Pickersgill T, Swingler R, Compston DA, Robertson NP. Increasing prevalence and incidence of multiple sclerosis in South East Wales. J Neurol Neurosurg Psychiatry 2009;80:386–391 [DOI] [PubMed] [Google Scholar]
- 19.Yu-Wai-Man P, Griffiths PG, Gorman GS, et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain 2010;133(pt 3):771–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carelli V, Ghelli A, Ratta M, et al. Leber's hereditary optic neuropathy: biochemical effect of 11778/ND4 and 3460/ND1 mutations and correlation with the mitochondrial genotype. Neurology 1997;48:1623–1632 [DOI] [PubMed] [Google Scholar]
- 21.Mahad D, Ziabreva I, Lassmann H, Turnbull D. Mitochondrial defects in acute multiple sclerosis lesions. Brain 2008;131(pt 7):1722–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahad DJ, Ziabreva I, Campbell G, et al. Mitochondrial changes within axons in multiple sclerosis. Brain 2009;132(pt 5):1161–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dabhi VM, Lindahl KF. MtDNA-encoded histocompatibility antigens. Methods Enzymol 1995;260:466–485 [DOI] [PubMed] [Google Scholar]
- 24.Klopstock T, Metz G, Yu-Wai-Man P, et al. Persistence of the treatment effect of idebenone in Leber's hereditary optic neuropathy. Brain 2013;136(pt 2):e230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katrych O, Simone TM, Azad S, Mousa SA. Disease-modifying agents in the treatment of multiple sclerosis: a review of long-term outcomes. CNS Neurol Disord Drug Targets 2009;8:512–519 [DOI] [PubMed] [Google Scholar]
- 26.Cortelli P, Montagna P, Pierangeli G, et al. Clinical and brain bioenergetics improvement with idebenone in a patient with Leber's hereditary optic neuropathy: a clinical and 31P-MRS study. J Neurol Sci 1997;148:25–31 [DOI] [PubMed] [Google Scholar]
- 27.Perez F, Anne O, Debruxelles S, et al. Leber's optic neuropathy associated with disseminated white matter disease: a case report and review. Clin Neurol Neurosurg 2009;111:83–86 [DOI] [PubMed] [Google Scholar]
- 28.Buhmann C, Gbadamosi J, Heesen C. Visual recovery in a man with the rare combination of mtDNA 11778 LHON mutation and a MS-like disease after mitoxantrone therapy. Acta Neurol Scand 2002;106:236–239 [DOI] [PubMed] [Google Scholar]
- 29.Marriott JJ, Miyasaki JM, Gronseth G, O'Connor PW; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology Evidence Report: the efficacy and safety of mitoxantrone (Novantrone) in the treatment of multiple sclerosis: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2010;74:1463–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horton L, Conger A, Conger D, et al. Effect of 4-aminopyridine on vision in multiple sclerosis patients with optic neuropathy. Neurology 2013;80:1862–1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanefeld FA, Ernst BP, Wilichowski E, Christen HJ. Leber's hereditary optic neuropathy mitochondrial DNA mutations in childhood multiple sclerosis. Neuropediatrics 1994;25:331. [DOI] [PubMed] [Google Scholar]
- 32.Nishimura M, Obayashi H, Ohta M, Uchiyama T, Hao Q, Saida T. No association of the 11778 mitochondrial DNA mutation and multiple sclerosis in Japan. Neurology 1995;45:1333–1334 [DOI] [PubMed] [Google Scholar]
- 33.Kalman B, Lublin FD, Alder H. Mitochondrial DNA mutations in multiple sclerosis. Mult Scler 1995;1:32–36 [DOI] [PubMed] [Google Scholar]
- 34.Sartore M, Grasso M, Piccolo G, et al. Leber's hereditary optic neuropathy (LHON)-related mitochondrial DNA sequence changes in Italian patients presenting with sporadic bilateral optic neuritis. Biochem Mol Med 1995;56:45–51 [DOI] [PubMed] [Google Scholar]
- 35.Mayr-Wohlfart U, Paulus C, Henneberg A, Rodel G. Mitochondrial DNA mutations in multiple sclerosis patients with severe optic involvement. Acta Neurol Scand 1996;94:167–171 [DOI] [PubMed] [Google Scholar]
- 36.Chalmers RM, Robertson N, Compston DAS, Harding AE. Sequence of mitochondrial DNA in patients with multiple sclerosis. Ann Neurol 1996;40:239–243 [DOI] [PubMed] [Google Scholar]
- 37.Kalman B, Lublin FD, Alder H. Characterization of the mitochondrial DNA in patients with multiple sclerosis. J Neurol Sci 1996;140(1–2):75–84 [DOI] [PubMed] [Google Scholar]
- 38.Wilichowski E, Ohlenbusch A, Hanefeld F. Characterization of the mitochondrial genome in childhood multiple sclerosis: II: multiple sclerosis without optic neuritis and LHON-associated genes. Neuropediatrics 1998;29:307–312 [DOI] [PubMed] [Google Scholar]
- 39.Ohlenbusch A, Wilichowski E, Hanefeld F. Characterization of the mitochondrial genome in childhood multiple sclerosis: I: optic neuritis and LHON mutations. Neuropediatrics 1998;29:175–179 [DOI] [PubMed] [Google Scholar]
- 40.Kalman B, Li S, Chatterjee D, et al. Large scale screening of the mitochondrial DNA reveals no pathogenic mutations but a haplotype associated with multiple sclerosis in Caucasians. Acta Neurol Scand 1999;99:16–25 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.