

Abstract
Quorum sensing (QS) is a cell-to-cell communication system and responsible for a variety of bacterial phenotypes including virulence and biofilm formation. QS is mediated by small molecules, autoinducers (AIs), including AI-2 that is secreted by both Gram (+/−) microbes. LsrR is a key transcriptional regulator that governs the varied downstream processes by perceiving AI-2 signal, but its activation via autoinducer-binding remains poorly understood. Here, we provide detailed regulatory mechanism of LsrR from the crystal structures in complexes with the native signal (phospho-AI-2, D5P) and two quorum quenching antagonists (ribose-5-phosphate, R5P; phospho-isobutyl-AI-2, D8P). Interestingly, the bound D5P and D8P molecules are not the diketone forms but rather hydrated, and the hydrated moiety forms important H-bonds with the carboxylate of D243. The D5P-binding flipped out F124 of the binding pocket, and resulted in the disruption of the dimeric interface-1 by unfolding the α7 segment. However, the same movement of F124 by the D8P′-binding did not cause the unfolding of the α7 segment. Although the LsrR-binding affinity of R5P (Kd, ~1 mM) is much lower than those of D5P and D8P (~2.0 and ~0.5 μM), the α-anomeric R5P molecule fits into the binding pocket without any structural perturbation, and thus stabilizes the LsrR tetramer. The binding of D5P, not D8P and R5P, disrupted the tetrameric structure and thus is able to activate LsrR. The detailed structural and mechanistic insights from this study could be useful for facilitating design of new anti-virulence and anti-biofilm agents based on LsrR.
Keywords: Quorum sensing, LsrR, Crystal structure, autoinducer-2 (AI-2), phospho-DPD, phospho-isobutyl-DPD, Biofilm
INTRODUCTION
Bacteria do not live alone, but in communities where they interact to adapt to changing environments. Quorum sensing (QS) is a bacterial cell-to-cell communication process, not confined to self-species reporting but extends to mediate inter-species behaviors. QS signaling is generally mediated by small molecules called autoinducers, which are synthesized and secreted by bacteria themselves1,2. The importance of QS has come to the forefront due to the potential of anti-QS agents acting as anti-virulence or anti-biofilm agents to curb recalcitrant bacterial infections3. The switch from a planktonic to sessile (biofilm) growth and the corresponding expression of many virulence factors has been attributed QS processes2,4. Biofilms protect bacteria for survival in otherwise hostile environments and enable their persistence in specific sites within a host. Of critical importance to both a bacterium’s survival and its host protection are the initial energy-consuming events that underpin these processes; understanding QS can therefore provide a basis for new antimicrobial therapies as well as new constructs of synthetic biology4.
Many autoinducer families have now been identified2,5,6: (i) AI-1 consists of an array of N-acyl homoserine lactones (AHLs)2; (ii) AI-2 is a term used to define (4S)-4,5-dihydroxy-2,3-pentanedione (DPD) and an equilibrium mixture of isomers, including a borate complex that was the first identified structure of AI-27; (iii) AI-3 has been postulated as a third general autoinducer among enteric bacteria, but its chemical structure and biosynthetic pathway have not been elucidated6; (iv) AIPs (autoinducer peptides) are encoded by agrD genes and are generated by post-translational peptidyl modifications5; (v) Indole, a metabolite of tryptophan pathway and which can be synthesized by many Gram (+/−) bacteria, has recently been proposed as a QS signaling molecule8. Although AI-1 and AI-3 systems are present in Gram (−) bacteria and the AIP system is found only among Gram (+) bacteria, the AI-2 system is present in both Gram (+/−) negative bacteria but only a handful of receptor proteins that perceive the AI-2 signal have been well characterized2,9,10. DPD (the diketone diol form of AI-2) is synthesized from S-ribosylhomocysteins (SRH) by LuxS. The synthesis of AI-2 is integrated into a general cellular metabolism pathway, since SRH is synthesized from S-adenosylmethionine (SAM), sequentially catalyzed by methyltransferases and Pfs11. The DPD molecule has several equilibrium isoforms in solution, in which two epimeric furanoses (R- and S-DHMF) result from the cyclization of DPD, and the further hydration of DHMF molecules produces R- and S-THMF12. The first visualized structure of AI-2 was as bound to LuxP in the S-THMF-borate form. In the presence of boric acid, S-THMF spontaneously forms the borate complex7.
LsrR is the repressor protein of lsr (LuxS-regulatory) operon and it seems to be a key regulator that senses the QS signaling mediated by AI-210,13,14. In enteric bacteria, AI-2 is first imported into the cytoplasm by the LsrABC transporter, where it is phosphorylated by LsrK. The resulting phospho-AI-2 (D5P) binds to the transcriptional regulator, LsrR, which triggers various downstream processes via the de-repression of LsrR. Although the mutation of luxS affects less than 50 genes, the deletion of lsrR and lsrK affects the expression of 146 and 149 genes, respectively. It has also been demonstrated that the deletion of lsrR or lsrK greatly decreased E. coli biofilm formation10,11. LsrR has also been proposed to be a target candidate for the development of anti-infectives13,15. There have been many efforts to develop AI-2 based QS inhibitors that target various AI-2 sensing proteins, such as LuxS, LuxPQ, LsrB, and LsrR16–20. Recently, various C1-alkyl analogs of DPD were synthesized that targeted the LsrR protein, and among them, isobutyl-DPD was identified to be the most potent in that it attenuated QS signaling in both E. coli and S. typhimurium after in vivo phosphorylation by LsrK21,22. The molecular details that contributed to an apparent differential interaction of AI-2 and its analogs to LsrR have been lacking. Moreover, and perhaps more importantly, it has been recently shown that the simultaneous treatment of isobutyl-DPD and the antibiotic gentamicin dramatically reduced biofilm formation and cleared extant biofilms of both E. coli and, non-AI-2 responding, P. aeruginosa14 and this supports the notion that the further development of small molecules that target LsrR as viable anti-biofilm agents for enteric bacteria would be fruitful endeavors.
Anti-QS drugs, unlike cytotoxic antibiotics, are expected to impose less evolutionary pressure for bacteria to develop drug-resistance, because anti-QS agents do not affect vitality4. Therefore, the development of a drug targeting QS componentry and its combined usage with traditional antibiotics could provide a promising tool for fighting bacterial infections. Due to the central role that LsrR plays in virulence factor production and biofilm formation among enteric bacteria, there has been growing interest to elucidate the structure of LsrR and to define mechanistically its mode of action. Here, we provide structures of E. coli LsrR, in complex with phospho-AI-2 as well as the phosphorylated isobutyl analog of AI-2. During the preparation of this manuscript, the crystal structure of apo-LsrR at 3.0 Å resolution was reported. The authors used computational methods to dock the original diketo form of phospho-DPD (D5P), which we now show not to be the correct ligand, into the active site of LsrR23. We present the crystal structures of the LsrR and N-terminal deleted LsrR (C-LsrR) proteins, in complex with ligands, which reveals the binding mode of phospho-DPD (D5P) as well as suggesting a mechanism that leads to the de-repression of LsrR upon D5P binding. The crystal structures of C-LsrR bound to D5P/phospho-isobutyl DPD (D8P) reveal that the bound form of the ligands is not the diketo form of D5P/D8P but rather the hydrated form of D5P/D8P, in which one of the keto groups in D5P/D8P is hydrated in the LsrR-D5P/D8P complex. The determination of the correct form of bound ligand in the LsrR complex is critical for efforts to develop new small molecules that target LsrR. The binding of D5P resulted in the disruption of one of two dimeric interfaces that is important for the transcriptional regulation of LsrR.
RESULTS
LsrR protein has two different inter-molecular interactions, of which the one mediated by the interface-1 is less stable but important for the tetrameric structure of LsrR by mediating the dimer-to-dimer contact
Overall structure determinations of various apo- and holo-proteins are described in the Experimental Procedures section and their statistics are summarized in Supporting Information (SI), Table S1. LsrR monomer contains two domains, the N-terminal DNA-binding domain (DBD, residues 14–60) and the C-terminal ligand-binding domain (LBD, residues 70–317). Although the asymmetric units of the apo-LsrR crystal (I222 symmetry) that was recently reported by Wu and co-workers contains 2 protein molecules23, those of our LsrR (C2) and C-LsrR (residues 53–317, P65) crystals contained 4 and 2 protein molecules, respectively (Figure 1). The overall structure consists of 14 α-helices and 10 β-strands, in which the α6 has a kinked shape and thus was divided into α6a and α6b (Figure 2). Interestingly, there are additional secondary structure elements annotated by β0 and α″1/2, which seems to be important for binding to a target DNA and a ligand, respectively (Figure 1B and C). The overall architecture of LsrR tetramer can be described as dimer of two dimers, very similar to the sorbitol operon regulator, SorC24. The crystal structure of LsrR shows the presence of two different inter-molecular interactions; (i) The first one (interface-1) stabilizes the dimer of subunit-A/B or subunit-C/D, and is the same dimeric interface of C-LsrR. The α7-β4-rigid loop-α8 segments form the interface-1, in which many hydrophobic residues stabilize the dimeric interaction (Figure 1D). (ii) The second one (interface-2) mainly consists of inter-molecular β0-sheet, but its geometry was somewhat different from ideal anti-parallel β-sheet, and provides a correct pair of N-terminal DNA-binding domains from subunit-A/D or from subunit-B/C (Figure 1A and C). The superposition of various subunit-A molecules from the different crystal structures of LsrR and C-LsrR showed that both interfaces-1 and -2 were relatively flexible to allow the inter-domain motions (Figure 1A, right inset).
Figure 1. Structural overviews of the LsrR tetramer and the C-LsrR dimer.

(A) The crystal asymmetric unit of LsrR and LsrR/R5P contains 4-subunits, and the dimer of two dimers (subunit-A/B and -C/D) results in the LsrR tetramer. The overlay of subunit-A from the dimeric structures of apo-LsrR, LsrR/R5P and apo-C-LsrR shows that the relative orientations of each domain (subunit-A vs. -B and -A vs. -D) are variable (right top). (B) The overlaid structures of C-LsrR and C-LsrR/R5P are almost identical, except for the flexible loop of residues Q215–S224 that formed short α-helical segments (α″1/2) in the presence of the bound ligand. The region of the residues G92–D97 is also very flexible in both apo- and holo-LsrR structures. (C) The magnified view of the subunit-A/B from the LsrR/R5P tetramer. The positions of tyrosine and tryptophan residues are indicated for better interpretation of the intrinsic fluorescence data (Figure 4). The dimeric interface-2 (i) between the subunit-A (or -C) and -D (or -B) mainly consists of the inter-subunit β-sheet (β0), but (D) the interface-1 between the subunit-A (or -C) and -B (or -D) consists of the α7-β4-rigid loop-α8 structure and seems to be stabilized by hydrophobic interactions.
Figure 2. Sequence alignment of various LsrR proteins.

Sequence alignment was performed for the various LsrR sequences from Gram (−) bacteria (E.coli, Escherichia coli; S.dyse, Shigella dysenteriae; S.typh, Salmonella typhimurium; A.acti, Aggregatibacter actinomycetemcomitans) and from Gram (+) bacteria (B.thur, Bacillus thuringiensis; S.dysg, Streptococcus dysgalactiae subsp). The mutated residues of the ligand-binding pocket for the fluorescence binding experiment are indicated using an arrow. The secondary structures of the subunit-A and -B from the structure of LsrR/R5P are shown in the top row.
The interface-1 appeared to be less stable than the interface-2, since the oligomeric state of LsrR in solution seemed to be mainly dimer and that of C-LsrR was monomer; the calculated MWs of C-LsrR monomer and dimer are 28 and 56 kDa, respectively. (i) The estimated MW of C-LsrR (40 μM) from the sedimentation equilibrium AUC experiment at 4°C was in the range of 36.0~32.4 kD for the ranged values of the partial specific volume (0.73–0.70), respectively, close to the value of the monomer (SI Figure S1). The presence of long unstructured region in C-LsrR (residues 53–66) likely decreases its partial specific volume25, which can further decrease the estimated MW. (ii) The AUC experiment of LsrR (20 μM) indicated that LsrR mostly exists as a dimer (SI Figure S1). We also identified that the elution of the LsrR protein in the SEC analysis became faster when the protein concentration was increased, consistent with the dimer-to-tetramer transition (SI Figure S2). (iii) The presence of the oligomeric equilibrium process and a possible higher inter-domain flexibility in both monomeric and dimeric states made it difficult to determine the exact solution model based on SAXS data and the X-ray crystal structure of LsrR. The molecular masses of LsrR were estimated to be 136 and 87 kDa, at 100 and 15 μM, respectively from their zero-angle scattering intensities, consistent with those of primarily tetrameric and dimeric forms. Low-resolution reconstructions fitted from the SAXS data of the 100 μM sample via DAMMIN are qualitatively consistent with the hollow tetrameric shape of the crystal structure (SI Figure S3). The fitting model exhibiting a somewhat larger central opening and tending to be flatter than the crystal structure suggested that the LsrR tetramer may be sampling more open geometries in solution, suggesting presence of conformational dynamics.
Analysis of the ligand-binding modes in the complex crystal structures of C-LsrR with ribose-5-phosphate (R5P), phospho-DPD (D5P), and phospho-isobutyl DPD (D8P)
Although D5P initiates QS signal transduction, D8P is a very potent inhibitor for LsrR and decreases biofilm formation by inhibiting the QS signal transduction14,21,22. On the other hand, R5P is a potential D5P analog in terms of chemical structure, and its complex crystal structure with LsrF has recently been reported26. LsrF has been suggested to be a class I aldolase mediating the decomposition of D5P. The electron density map of the α″1/2 segments was not well defined for both apo-C-LsrR and apo-LsrR (not shown), but their holo-forms clearly showed that two consecutive α-helical segments completely cover the ligand-binding pocket (Figure 1B). The OG1 atom of T220 in the α″2 segment forms a stable H-bond with the phosphate oxygen of all used ligands (R5P, D5P, and D8P) (Figure 2 and Figure 3). The innate flexibility of these segments makes it possible to gate the entrance of the ligands. Interestingly, the structure of R5P in the LsrF is a linear form but that of R5P in the LsrR was a cyclic form, in which the α-anomer was better fitted to the map compared to the β-anomer (SI Figure S4A). R5P could be fitted into the binding pocket of LsrR without any apparent structural perturbations. In contrast, D5P/D8P ligands could only be fitted into the binding pocket when the side chain F124 was flipped out, due to the presence of the steric crash originated from their linear conformation. We firstly located the native D5P/D8P molecules within the map of the ligand-binding pocket. However, detailed analyses of the interaction network between D5P/D8P and LsrR indicated that only the phosphate of these ligands mediated the ionic interaction with the NH4+ of K288 and the H-bonding with the HN of E126/A127and the OG of T220, and the total number of H-bonds mediated by D5P/D8P was less than that mediated by R5P. Using the diketo-form of D5P/D8P, we could not identify other H-bonds that were mediated by the other oxygen atoms of D5P/D8P. Although the carbonyl O3 of D5P/D8P was located in close proximity to the carboxyl group of D243, the charged state of the carboxylate could not form H-bond due to the absence of the mediating proton (Figure 3B and 3C). This suggested that perhaps D5P/D8P did not bind to LsrR as the diketo form, as proposed by Wu and co-workers23.
Figure 3. Ligand recognitions of LsrR for three different ligand molecules.

Detailed H-bond networks were analyzed for the structures of C-LsrR/R5Pα (A), C-LsrR/D5P′ (B), and C-LsrR/D8P′ (C). The chemical structures of all used ligands are also shown. The H-bonds meeting and not meeting exact criteria are indicated using cyan and orange colored lines, respectively. The bound D5P and D8P are not the diketone forms but rather the hydrated forms (D5P′ and D8P′). (D) The binding of D5P′ (left) and D8P′ (right) to LsrR induced the flipping out of F124, which generated the de novo hydrophobic pocket. The bulky isobutyl group of D8P′ is well fitted into this pocket.
Unexpectedly, we noticed the presence of an additional density (Fo-Fc) in the vicinity of C3 atom of both D5P and D8P that was likely resulting from the covalent attachment of an additional OH (SI Figure S4D). The presence of two OH groups on the same C3 atom of THMF - the circularized form of DPD has also been identified in the complex structures of (i) LsrB and R-THMF27 and (ii) LuxP and S-THMF-borate7. It has been suggested that the THMF-form of DPD has two OH group attached to the C3 atom and that the hydration of the keto moiety occurs after the cyclization of DPD (DHMF)27. Recently, it was shown that the hydration of the C3 keto moiety of DPD could also occur in the linear form28. D5P and D8P have a linear conformation due to the attachment of phosphate group into the OH5 of DPD, and the C3 atoms of the LsrR-bound D5P/D8P also seemed to carry two OH groups. Therefore, we generated modified molecules (D5P′ and D8P′) by adding one more OH group in the C3 atoms of D5P and D8P, respectively. The modified D5P′ and D8P′ can completely be fitted to their electron-density maps by relieving the planarity restraint imposed by the O3, C3, C2, and O2 atoms (SI Figure S4). This chemical modification can be critical for increasing the LsrR-binding affinity for D5P/D8P molecules via H-bond formation (Figure 3B and 3C). The geometry of two de novo generated OH groups of D5P′/D8P′ were optimal for the formation of two simultaneous H-bonds with the carboxylate of D243, in which the distances between two oxygens of the H-bonds were both ~2.7 Å. Moreover, several water-mediated H-bonds were also formed by the OH groups (Figure 3B and 3C). The binding of linear D5P′/D8P′ flipped out the aromatic ring of F124 and the newly appeared hydrophobic patch consisting of F124, L176, I221, Y226, and L245, well accommodated the bulky isobutyl group of D8P′ (Figure 3D). This could be the reason why D8P displayed higher binding affinity for LsrR than D5P did (vide infra).
Determination of the ligand-binding constants for the C-LsrR and LsrR proteins
We first assessed the binding affinities of LsrR for R5P, D5P, and D8P using the intrinsic fluorescence of protein. Time-course fluorescence experiments, following the Tyr fluorescence of C-LsrR showed that D5P or D8P binding to LsrR did follow simple binding kinetics. However, the intrinsic Trp fluoresces from W24 of LsrR (excitation wavelength, 295 nm) responded to the binding of D5P, but not D8P, in bi-phasic manner, making the interpretation of the fluorescence data difficult (Figure 4A). The Trp fluorescence of LsrR indicated that the fluorescence from the DNA-binding domain could be affected by both the direct D5P-binding and also by an additional indirect process (Figure 1C), which was slow and seemed to be related to the disruption of the interface-1 (vide infra). Therefore, we assessed all ligand-binding affinities using the C-LsrR constructs. The determined Kd values of C-LsrR for R5P, D5P, and D8P were 995±50, 2.1±0.3, and 0.88±0.14 μM, respectively (Figure 4B, left panel). The increasing Tyr fluorescence of C-LsrR by the addition of R5P suggests that upon R5P binding, the environments of the Tyr residues become less hydrophilic, probably due to the expulsion of water molecules.
Figure 4. The ligand-binding of the LsrR proteins was monitored by using the intrinsic Tyr/Trp fluorescence.

The quantum yield of Trp is generally 10-times larger than that of Tyr, and the LsrR protein has one Trp residue in the N-terminal DBD (refers to Figure 1C). It was possible to monitor the fluorescence mainly from Trp by using 295 nm excitation wavelength. The excitation wavelength at 280 nm was used for the experiment on the C-LsrR protein. (A) Time-courses of the fluorescence changes show that the binding of all three ligands (R5P, D5P, and D8P) impacted not only the fluorescence of the C-terminal LBD (right), but also that of the N-terminal DBD (left). The addition of the same amount of the buffer solution that was used for synthesizing D5P and D8P molecules did not change the fluorescences of LsrR and C-LsrR. Interestingly, the fluorescence intensity of Trp initially decreased and then slowly increased again during binding to D5P, which made it difficult to measure the D5P-binding affinity of LsrR. (B) All ligand binding affinities were assessed by using intrinsic Tyr fluorescence of C-LsrR (left), and the effects of several point-mutations in the ligand-binding pocket were also assessed for the D5P-binding (right).
For the fluorescence titration experiments, 2.5 μM of C-LsrR was used and since this value is closer to the obtained Kd values of C5P and C8P, there could have been some errors associated with the determination of Kd via the intrinsic fluorescence of C-LsrR. Therefore, we also confirmed the binding affinity of LsrR for D5P and D8P using ITC experiments, but this time using LsrR and not C-LsrR because C-LsrR protein slowly aggregated at room temperature. The determined Kd values for D5P and D8P were 1.78±0.13 and 0.43±0.15 μM, respectively, slighter lower than those from the fluorescence experiments (SI Figure S5). Interestingly, there was a significant difference in released heat upon the binding of D5P and D8P, even though similar concentrations of LsrR were used. The values of ΔH and ΔS obtained for the titration of D5P (ΔH, 0.98 kcal/mol; ΔS, 23.0 cal/mol/K) were about 5-times lower and 2-times higher than those obtained for that of D8P (ΔH, 5.3 kcal/mol; ΔS, 11.2 cal/mol/K), respectively. This difference might result from the induced unfolding of the α7 located in the dimeric interface-1 by the binding of D5P, not D8P, in which the exothermic ligand-binding was assumed to be compensated by the endothermic α7-melting (vide infra).
We mutated all the residues that can interact with these ligands, and then assessed their binding affinities for D5P using the intrinsic Tyr fluorescence of C-LsrR. The mutated residues (Q215A, T220A, D243A, and K288A) are well conserved in the LsrR family proteins (Figure 2). All the mutant C-LsrR proteins, except for the Q215A, had less affinity for D5P than the wild type. The effect of Q215 mutation was moderate, compared to the others (Figure 4B, right panel). Q215 is located in the front of the α″1 segment and forms a water-mediated H-bond with the D5P′. The significant decrease of the binding affinity in T220A mutant suggests that the flexible α″1/2 segments are important not only for gating the entrance of a ligand, but also for stabilizing its binding. The α″1/2 segments was not identified in the recent apo-LsrR structure by Wu and co-workers23. However, we could model these segments even in apo-LsrR structure although their electron-density map was very weak (not shown) and the resulting B-factor values were very high (SI Figure S6). It is likely that the α″1/2 segments are in equilibrium between open and closed states in solution and the presence of ligand molecules eventually stabilizes the closed state of the gate region.
The binding of D5P, not R5P and D8P, disturbed the half of the dimeric interface-1 including the α7 segment
The determination of the molecular mechanism by which LsrR initiates QS signaling mediated by D5P is important for developing a drug that reduces biofilm formation by inhibiting the QS process. The transcriptional regulator CbnR forms a tetramer as a dimer of two dimers, and its binding to a target DNA results in the DNA-bending29. The ligand-binding site of CbnR is located in between the DNA-binding and the regulatory domain, and the ligand-binding likely relaxes the bending of the target DNA through changing the quaternary structure. However, the D5P-binding site of LsrR is almost in the middle of the regulatory domain, and thus it is unlikely that LsrR uses a similar mechanism to CbnR. Based on the crystal structure of apo-LsrR and LsrR/R5P, we firstly assumed that the induced structure in the flexible loop (α″1/2) by the D5P-binding may provide de novo interaction surface that is able to recruit the N-terminal DNA-binding domain (DBD, residues 1–61). However, the HSQC-titration NMR experiment did not show any differential binding between 0.1 mM 15N-labled DBD and 0.2 mM C-LsrR in the presence or in the absence of 0.5 mM D5P (data not shown). Moreover, the structures of this gate region are almost identical for the complex structures of C-LsrR/R5P, C-LsrR/D5P, and C-LsrR/D8P.
The comparison of apo-C-LsrR structure and three ligand-bound structures with R5P, D5P′, and D8P′ showed that the D5P′-binding only caused a significant structural perturbation in the vicinity of the α7 segment that participates in the dimeric interface-1 (Figure 1D and Figure 5). The structure of C-LsrR/R5P was almost identical with that of apo-C-LsrR, except for the gate region (α″1/2) (Figure 5A). Interestingly, the binding of both D5P′ and D8P′ flipped the aromatic ring of F124 away from the binding pocket, but the D8P′-binding did not cause the unfolding of the α7 segment. The flipping out of F124 seemed to be an essential trigger for the structural change of LsrR to initiate QS signaling mediated by phospho-AI-2 (Figure 5C and 5D). The electron-density of C-LsrR/D8P′ in the region of the α7 segment was just attenuated and the overall structure did not change apparently. Although the overall electron density map of C-LsrR/D5P′ was not as clear compared with the C-LsrR/D8P′, the structural unfolding of the α7 segment was identified for the C-LsrR/D5P′, which abolished the hydrophobic surface consists of the residues M157, I160, L163, V169, and I171 (Figure 5B and 5C). Moreover, the noncrystallographic symmetry in two subunits of the dimeric C-LsrR/D5P′ structure was also broken for the region of the α7 segment (not shown). The B-factor values of this region were higher for the C-LsrR/D5P′ than for the C-LsrR/D8P′ and the difference between the two subunits was also larger for D5P′ (Figure 5E).
Figure 5. The dimeric interface-1 was disrupted by the binding of D5P′, but not by that of R5P and D8P′.

Molecular trajectories were generated for the structural changes (A) from apo-C-LsrR to C-LsrR/R5P, (C) from C-LsrR/R5P to C-LsrR/D5P′, and (D) from C-LsrR/R5P to C-LsrR/D8P′, respectively. The steric crash induced by the D5P′/D8P′-binding flipped out F124 from the binding pocket, but the cyclic conformation of R5P did not cause the same steric crash. The directions of structural movements are indicated using red and blue arrows. (A) The structure of C-LsrR was not changed by the R5P-binding, except for the α″1/2 segments. The surface of the dimeric interface-1 was decorated using yellow color, and its constituting secondary structures are shown as a ribbon presentation (inset). (B) The curved α7 segment of C-LsrR/R5P is supported by only several H-bonds, and G152 located in the front of the α7 segment has the alternative conformations (Psi-1 and Psi-2). The Psi-1 of G152 resulted in bad Phi angle of V153 in the Ramachandran plot. The structure of the α7 segment is important for maintaining the hydrophobic surface formed by M157, I160, L163, V169, and I171. (E) The B-factor (Cα) values indicate that the α7 segment became highly delocalized by the binding of D5P′. Error bar represents the difference of the B-factors between two subunits in the dimeric C-LsrR structures. (F) The population of the G152 Psi-1 orientation was predominant in the apo-C-LsrR and became close to a half in both C-LsrR/R5P and C-LsrR/D8P′. However, the only Psi-2 orientation (Psi-2*) was identified in the C-LsrR/D5P′.
The α7 segment does not have a stable α-helical geometry, being curved and supported by only several H-bonds (Figure 5A, inset and 5B) and overall B-factor values of this region are higher than the other structured regions (SI Figure S6). The C-LsrR protein displayed a broad and tailed elution during the SEC analysis (not shown), which may be related to the presence of structural heterogeneity resulting from the unfolded α7 segment. Interestingly, only one residue (V153) of the apo-C-LsrR structure (1.9 Å) had an unusual phi angle, which is exactly at the initiation of the α7 segment. The Fo-Fc maps of C-LsrR/R5P and C-LsrR/D8P′ indicated the presence of extra density in the difference position for the CO of G152 (Psi-2 in Figure 5B), for which the phi angle of V153 was in the allowed region of the Ramachandran plot. The Psi-1 angle was preferred in the apo-C-LsrR structure, and almost equal populations of two orientations were shown in the complex structures with R5P and D8P′ (Figure 5F). However, the CO of G152 was completely merged to the orientation-2 (Psi-2*) in the C-LsrR/D5P′ structure (Figure 5C and 5E).
The melting of the α7 segment by the D5P′-binding seemed to be a consequence of several sequential movements of structures (Figure 5C): (i) The flipping out of F124 initiated the structural transition by pushing the neighboring Y156 that caused overall outside movement of the α7 segment. (ii) The movement of F124 also generated the de novo space that made it possible for L176 to move closer to the binding pocket. (iii) The consequent inside movement of carbonyl oxygen of A174 that is located in the same rigid loop pushed G152 inside and thus the orientation of the CO is completely changed into the orientation-2. This shearing force could trigger the melting of the α7 segment that might have been originally strained and unstable. On the other hand, the flipping out of F124 by the D8P′-binding rather pushed out L176 due to the presence of the bulky isobutyl group, which is able to coincide overall motions of structural segments in the same direction (Figure 5D). All residues related to the movement of F124 are completely and relatively conserved in Gram (−) and Gram (+) LsrR family proteins, respectively (Figure 2).
The D5P-binding destabilized the tetrameric structure of LsrR in solution
We assessed the structural transition of LsrR induced by the bindings of these ligands using dynamic light scattering (DLS). The LsrR alone existed as mainly mono-disperse species, and the addition of R5P or D8P did not show any apparent changes in the size distribution compared to the LsrR alone. However, the presence of D5P in the solution clearly changed the DLS pattern of LsrR. The population and the molecular size of main species were reduced while larger ones appeared (Figure 6A). We additionally confirmed that the D5P-binding reduced the apparent molecular size of LsrR via SEC analysis (Figure 6B), and the elution time of the D5P-bound LsrR was comparable to that of the extrapolated 0 mM LsrR (SI Figure S2). This is well in agreement with the recent result by Wu and co-workers, that shows the tetramer-to-dimer transition of LsrR induced by the binding of D5P23. However, the binding of D8P and R5P did not reduce the elution times of LsrR, although the elution profile of the D8P-bound LsrR protein was somewhat broader and showed a larger tailing. Interestingly, the elution profile of the R5P-bound LsrR was narrower than that of the D5P-bound LsrR, and thus the R5P-binding seemed to increase the homogeneity of an oligomeric state. Therefore, LsrR seems to be in the concentration dependent dimer-to-tetramer equilibrium, and binding D5P likely shifts the equilibrium into towards the dimer by destabilizing the hydrophobic interface-1.
Figure 6. The molecular sizes of the LsrR protein in the absence and the presence of ligand molecules.<.
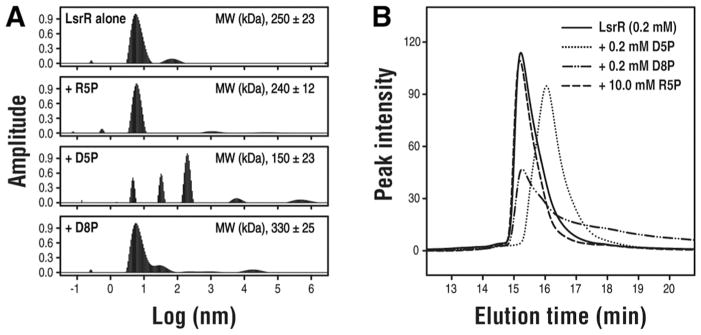
br>(A) The hydrodynamic radius (Rh) distributions and the intensity averaged MWs of LsrR were determined from the DLS experiments. (B) SEC analysis of the LsrR protein. The 0.2 mM LsrR was used for increasing the population of the tetramer. The ligands were added to the LsrR proteins (0.2 mM D5P, 0.2 mM D8P, and 10 mM R5P), and then the SEC analyses were performed after 30 min incubation. The molecular sizes of LsrR (15.22min, 158.6 kDa) were not changed by the binding of D8P (15.26 min, 155.4 kDa) and R5P (15.19 min, 160.6 kDa). However, the D5P-binding clearly reduced the molecular size (16.04 min, 106.5 kDa), and this value is very similar to the apparent MWGPC at 0 mM LsrR, was estimated to be a dimer (see the SI Figure S2).
DISCUSSION
The disruption of the α7 segment by the D5P-binding that destabilizes the tetrameric structure of LsrR due to significant decrease of the hydrophobic interface-1 is an interesting and unprecedented phenomenon in terms of transcriptional regulation and protein folding. Future work, involving molecular dynamics simulation, could help shed more light on the melting of the α7 segment upon D5P binding. The melting of the α7 segment was likely to be much slower process than the binding event of D5P as observed in the time-course of the fluorescence experiment of LsrR, which likely made it possible to capture the complex structure of C-LsrR/D5P′. The in vivo activities of the C1-alkyl analogs (DPD, ethyl-DPD, propyl-DPD, and higher alkyl analogs) are initiated by the phosphorylation by LsrK that has broad substrate specificity especially for the modification of C1 atom of DPD21,22. It is intriguing to note that D5P and phospho-ethyl-DPD (D6P) are agonists, but phospho-propyl-DPD (D7P) and D8P are antagonist22. It is possible that the binding circumstances of D6P and D7P may be in the middle of two extreme cases of D5P and D8P, in which the binding of D6P and D7P is more similar to the case of D5P and D8P, respectively.
It was unexpected to find out the presence of one more OH group in the C3 atom of the linear D5P′ and D8P′ molecules, however this modification is critical for the high specificity of QS molecule (D5P) during the LsrR-binding. The resulting two OH groups at C3 atom are able to concurrently form H-bonds with two oxygen atoms of the D243 carboxylate. A short H-bond has been shown to be important for the stability of protein and is generally observed between two carboxylate side-chain30,31. The two H-bond distances between D5P′/D8P′ and D243 of LsrR (both ~2.7 Å) are relatively short, likely corresponding to strong H-bonds. Moreover, each HO3 and HO3′ is also able to form H-bond with both O atoms in the carboxylate, which can share a similar characteristic to a strong bifurcated H-bond mediated by carboxylate group32. It is currently unknown if the pre-existing diketo D5P′/D8P′ in solution binds to LsrR or the binding of D5P/D8P into LsrR induces this chemical modification. Because phosphorylated AI-2 and analogs do not have long shelf-life, resolution of the above issue remains challenging.
Overall transcriptional regulation of the LsrR protein by the D5P-binding can be inferred from the previous studies on the SorC protein family including the CggR (central glycolytic genes regulator) and DeoR proteins24. As described in a recently published paper23, the architecture of the lsr operator region is similar to that of the CggR-regulated gapA operon33 and the two recognition sites of LsrR also do not have identical sequences33. Previous biophysical studies on the CggR/DNA complex using AUC and fluorescence anisotropy experiments showed that two CggR dimers cooperatively bind to both right and left half-operator with much higher affinity for the right one. The presence of the inducer molecule (fructose-1,6-bisphosphate) abolishes the cooperative DNA-binding of two CggR dimers by suppressing this protein-protein interaction34. The overall interaction of LsrR on the target DNA seemed to be very similar to that of CggR. Our crystal structure of C-LsrR/D5P′ shows that the interface-1 was disrupted and explains how the D5P-binding initiates the QS signaling. The presence of a weak LsrR dimer-to-dimer interaction that is mediated by the hydrophobic interface-1 could be a key feature of the D5P-mediated transcriptional regulation, which is able to provide the same cooperative binding of two dimeric LsrR molecules to the target DNA as that of the SorC family protein.
Although the binding affinity of R5P for LsrR (Kd, ~1 mM) is much lower than that of D5P/D8P, we speculate that some of phospho-sugar metabolites may also be involved in or interfere with QS regulation by using binding mode similar to that used by R5P. It has been reported that the intra-cellular concentrations of several phospho-sugar metabolites, such as hexose-phosphate, pentose-phosphate, and 3-Phosphoglycerate, are higher than 1 mM for the exponentially growing E. coli with consuming glucose35. Sugar metabolism may indirectly affect QS signaling by antagonizing the interaction of D5P to LsrR protein. The CggR protein displays specificity for a very diverse set of sugar phosphates (dihydroxyacetone phosphate, glucose-6-phosphate, glycerol-3-phosphate, etc.) in addition to fructose-1,6-bisphosphate, and has been suggested to sense the general metabolic state36. In terms of the design of AI-2 antagonist, the binding mode of R5P could also be utilized. The R5P-bidning does not flip out the F124 in the binding pocket that initiates the disruption of the interface-1, and the structure of LsrR/R5P is almost identical with that of apo-LsrR, except for the flexible gate including the α″1/2 segments. Although more detailed studies of the melting mechanism of the α7 segment are required, our studies show how the LsrR protein senses D5P and its analogs in a different manner. The relatively high resolution structures of the C-LsrR proteins for both apo- and holo (agonist and antagonist)-states, in addition to those of LsrR will provide a molecular basis for developing more potent antagonists of AI-2 that target to the LsrR protein.
CONCLUSIONS
The repression of the lsr operon seems to be maintained by the tetrameric LsrR proteins. The LsrR tetramer already exists in concentration-dependent equilibrium, and thus the oligmeric states of LsrR can be more practically regulated by the ligand-binding. Our crystal structures of the LsrR proteins in complex with the native signaling molecule (D5P′) and two antagonist molecules (R5P and D8P′) reveal a structural basis for the activation mechanism of the transcriptional regulator, LsrR. The original diketone forms of D5P and D8P are converted into their hydrated forms to form strong H-bonds with the carboxyl group of D243. The D5P′-binding resulted in the disruption of the hydrophobic dimeric interface-1, which was initiated by the flipping out of F124. On the other hand, the same movement of F124 induced by the D8P′-binding did not cause the same disruption due to the presence of the bulky isobutyl moiety at C1-position. It has been previously shown that C1-modified AI-2 molecules with short alkyl chains (methyl and ethyl) de-repress LsrR whereas propyl or higher C1 chains bind to LsrR to enhance its repression. Herein we provide a structural basis for this interesting phenomenon, whereby increasing carbon chain at C1-position switches an AI-2 analog from being an agonist to an antagonist. Although the binding affinity of R5P (Kd, ~1 mM) was lower than those of D5P and D8P (Kd, ~2.0 and ~0.5 μM), R5P is able to bind to LsrR without any structural perturbation. Therefore, the LsrR-binding mode of R5P could be utilized for the design of new anti-quorum sensing compounds based on LsrR.
MATERIALS AND METHODS
Protein crystallizations
Crystal screenings for C-LsrR (9~11 mg/ml) and LsrR (5~6 mg/ml) were performed with Hampton Research screen kits (Crystal screen I and II, Index, PEG/Ion, PEGRx, and SaltRx), and Emerald BioSystem screen kits (Wizard I and II). The Mosquito crystallization robot (TTP LabTech) was used to set the screens by the sitting-drop method at 20°C. The crystallization conditions were further optimized using the hanging-drop vapor diffusion method. Optimal crystals of C-LsrR and its SeMet-derivative were obtained by mixing 1 μl protein solution and 1 μl reservoir solution (pH 6.5, 5% PEG-3350, 0.1 M bis-tris, 0.1 M NaCl). The ligand-free crystals of LsrR were obtained using the crystallization buffer (pH 7.0, 0.1 M HEPES sodium, 15~17% isopropanol, 1~2% PEG-3350, 1~2% tacsimate).
Complex crystals of LsrR and C-LsrR with 5 mM D-ribose 5-phosphate (R5P) were grown with reservoir buffer (pH 6.5, 0.1 M bis-Tris, 2% isopropanol, 8% γ-butyrolactone, 10 mM R5P) and with buffer (pH 6.5, 0.1 M bis-tris, 9.1% PEG-3350, 10 mM barium chloride dehydrate, 10 mM R5P), respectively. Complex crystals of C-LsrR/D5P and C-LsrR/D8P were obtained by soaking the native crystals in the same crystallization buffer containing 0.15 mM D5P and 2.0 mM D8P, respectively and by sampling for various times. The crystal soaking solution was exchanged with freshly prepared D8P solutioneach hour. 35% ethylene glycol, in addition to the crystallization buffer, was used as a cryo-protectant and the crystals were directly frozen in the cold nitrogen stream of R-AXIS IV X-ray diffractometer (Rigaku).
Structure determinations of various apo/holo-LsrR and C-LsrR proteins
The crystal of apo-LsrR did not diffract well (3.2 Å, SI Table S1). Therefore, we prepared the truncated LsrR proteins in which the N-terminal DNA-binding domain was eliminated (C-LsrR). We crystalized C-LsrR and determined its 3-dimensional (3D) structure at 1.9 Å resolution after the SAD phasing (SI Table S1). We determined the complex structures of both LsrR and C-LsrR by crystalizing these proteins in the presence of 5 mM R5P, a potential D5P analog and additional soaking into the washing solution containing 30 mM R5P. Interestingly, the incorporation of R5P greatly increased the diffraction resolution of the LsrR/R5P crystal, probably due to a better crystal packing. We could complete the model building of LsrR/R5P (2.6 Å) and then finished that of apo-LsrR (3.2 Å). The resolution of the C-LsrR/R5P crystal (1.9 Å) was the same as that of the C-LsrR crystal (SI Table S1).
We also tried to determine the complex structure of LsrR/D5P and LsrR/D8P (phospho-isobutyl DPD) to obtain insight on the dissociation mechanism of LsrR from the target DNA after D5P-binding. Co-crystallization of LsrR and C-LsrR with D5P were not successful. Therefore, we tried to obtain the complex crystal by soaking the D5P into the apo-LsrR and C-LsrR crystals. The addition of only a small amount of D5P into the LsrR crystal immediately changed the crystal color to brown and yielded pronounced crystal cracks. Therefore, we focused on the more rigid C-LsrR crystal to obtain the D5P-bound complex crystals. Various time-course samplings of the crystals in addition to optimizing both the soaking time and the concentration of DPD in the crystal washing solution facilitated the capture of the structural transition of the C-LsrR protein that occurred upon D5P-binding. The C-LsrR crystals soaked in the washing solution containing 0.15 mM D5P were sampled at each 10 min increasing incubation time. The crystals color gradually changed into brown and their surface became rougher (not shown). The soaking of D5P for more than 0.5~1.0-h completely corroded the C-LsrR crystals leading to complete disappearance of their X-ray diffraction. A limited soaking of D5P clearly displayed the electron-density maps at two different positions of F124, which corresponded to the structures of apo- and holo-LsrR, respectively. Therefore, we tried to maximize the occupancy of D5P molecule in the C-LsrR crystal by increasing the soaking time until the diffraction ability of the crystal did not decrease appreciably. On the other hand, the same crystal could be soaked into 2.0 mM D8P solution for more than 8-h with slight diminution of the crystal’s X-ray diffraction. In the case of D8P, the color of the soaked crystal also changed into brown (not shown). D5P and D8P do not contain a strong chromophore and we currently do not have a hypothesis to account for the brown coloration. We were able to determine the complex structures of C-LsrR/D5P and C-LsrR/D8P in 2.3 and 2.1 Å resolutions, respectively (SI Table S1).
Measurements of the ligand-binding affinities for various LsrR proteins
Intrinsic fluorescence of Trp and Tyr residues was used to probe the ligand-binding process. LsrR has one Trp residue in the N-terminal DNA-binding domain; we focused on the Trp fluorescence for LsrR using an excitation wavelength of 295 nm. Conversely, C-LsrR does not have this Trp residue and thus the fluorescence of Tyr residues was measured to probe the ligand-binding process and excitation wavelength of 280 nm. LsrR (2.5 μM) and C-LsrR (2.5 μM) were prepared in buffer (pH 8.0, 50 mM Tris-HCl and 100 mM NaCl), and the fluorescence of LsrR proteins was measured using Cary Eclipse Fluorescence Spectrophotometer (Varian) while increasing the ligand concentrations.
Isothermal titration calorimetry (ITC) experiments were carried out using a MicroCal Auto-iTC200 calorimeter (GE Healthcare). LsrR was prepared in the sample cell and the D5P was loaded into the injectable syringe. All samples were prepared in the same buffer that was used for the fluorescence experiment. Titration measurements that consisted of 19 injections (2 μL) with 200 sec spacing were performed at 20°C while the syringe was stirred at 1000 rpm. Data analysis was done using the MicroCal Origin 7 program, and a linear heat contribution resulting from the dilution of additional supplements (ATP, AMP, phosphate, and etc.) in the D5P mixture was manually subtracted before the one-site binding model analysis.
Supplementary Material
Acknowledgments
This research was supported by bio-international cooperative research program from Chungcheongbuk-Do province and by high-field NMR research program from Korea Basic Science Institute (KBSI) to K.-S. Ryu, and was additionally supported by KBSI grant (T33410) to C.H.K. We thank Photon Factory in Japan for using the X-ray beam-lines and appreciate that Pohang Acceleratory Laboratory supported a travel stipend to Photon Factory. We thank Dr. Yun, Sijung (NIH/NIDDK) for valuable discussions about the structural transition and the molecular dynamic simulation. We also thank Camille Dreyfus foundation for a grant to H.O Sintim, NSF CHE0746446 and a GANN fellowship to J.A.I. Smith and the support of US Army DTRA (BO085PO008) and the National Science Foundation (BES-0124401). We gratefully acknowledge use of the shared scattering beamline resource allocated under the PUP-77 agreement between the National Cancer Institute and the Argonne National Laboratory and thank Drs. Soenke Seifert and Xiaobing Zuo (Argonne National Laboratory) for their support of the SAXS experiments. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under Contract W-31-109-ENG-38.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
The coordinates of all determined structures were deposited to the Protein Data Bank (apo-LsrR, 4L5I; LsrR/R5P, 4L5J; apo-C-LsrR, 4L4Y; C-LsrR/R5P, 4L51; C-LsrR/D5P, 4L4Z; C-LsrR/D8P, 4L50). Table S1, Figure S1–S6, and additional experimental methods are in the Supporting Information. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Fuqua WC, Winans SC, Greenberg EP. J Bacteriol. 1994;176:269–75. doi: 10.1128/jb.176.2.269-275.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waters CM, Bassler BL. Annu Rev Cell Dev Biol. 2005;21:319–46. doi: 10.1146/annurev.cellbio.21.012704.131001. [DOI] [PubMed] [Google Scholar]
- 3.Imperi F, Massai F, Facchini M, Frangipani E, Visaggio D, Leoni L, Bragonzi A, Visca P. Proc Natl Acad Sci USA. 2013;110:7458–63. doi: 10.1073/pnas.1222706110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Njoroge J, Sperandio V. EMBO Mol Med. 2009;1:201–10. doi: 10.1002/emmm.200900032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gospodarek E, Bogiel T, Zalas-Wiecek P. Pol J Microbiol. 2009;58:191–8. [PubMed] [Google Scholar]
- 6.Parker CT, Sperandio V. Cell Microbiol. 2009;11:363–9. doi: 10.1111/j.1462-5822.2008.01272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen X, Schauder S, Potier N, Van Dorsselaer A, Pelczer I, Bassler BL, Hughson FM. Nature. 2002;415:545–9. doi: 10.1038/415545a. [DOI] [PubMed] [Google Scholar]
- 8.Hu M, Zhang C, Mu Y, Shen Q, Feng Y. Indian J Microbiol. 2010;50:362–8. doi: 10.1007/s12088-011-0142-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xavier KB, Bassler BL. Nature. 2005;437:750–3. doi: 10.1038/nature03960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li J, Attila C, Wang L, Wood TK, Valdes JJ, Bentley WE. J Bacteriol. 2007;189:6011–20. doi: 10.1128/JB.00014-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang L, Li J, March JC, Valdes JJ, Bentley WE. J Bacteriol. 2005;187:8350–60. doi: 10.1128/JB.187.24.8350-8360.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meijler MM, Hom LG, Kaufmann GF, McKenzie KM, Sun C, Moss JA, Matsushita M, Janda KD. Angew Chem Int Ed Engl. 2004;43:2106–8. doi: 10.1002/anie.200353150. [DOI] [PubMed] [Google Scholar]
- 13.Roy V, Adams BL, Bentley WE. Enzyme Microb Technol. 2011;49:113–23. doi: 10.1016/j.enzmictec.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 14.Roy V, Meyer MT, Smith JA, Gamby S, Sintim HO, Ghodssi R, Bentley WE. Appl Microbiol Biotechnol. 2013;97:2627–38. doi: 10.1007/s00253-012-4404-6. [DOI] [PubMed] [Google Scholar]
- 15.Byrd CM, Bentley WE. Cell Res. 2009;19:1229–30. doi: 10.1038/cr.2009.122. [DOI] [PubMed] [Google Scholar]
- 16.Zhu P, Li M. Curr Med Chem. 2012;19:174–86. doi: 10.2174/092986712803414187. [DOI] [PubMed] [Google Scholar]
- 17.Ganin H, Tang X, Meijler MM. Bioorg Med Chem Lett. 2009;19:3941–4. doi: 10.1016/j.bmcl.2009.03.163. [DOI] [PubMed] [Google Scholar]
- 18.Smith JA, Wang J, Nguyen-Mau SM, Lee V, Sintim HO. Chem Commun (Camb) 2009:7033–5. doi: 10.1039/b909666c. [DOI] [PubMed] [Google Scholar]
- 19.Peng H, Cheng Y, Ni N, Li M, Choudhary G, Chou HT, Lu CD, Tai PC, Wang B. Chem Med Chem. 2009;4:1457–68. doi: 10.1002/cmdc.200900180. [DOI] [PubMed] [Google Scholar]
- 20.Lowery CA, Abe T, Park J, Eubanks LM, Sawada D, Kaufmann GF, Janda KD. J Am Chem Soc. 2009;131:15584–5. doi: 10.1021/ja9066783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gamby S, Roy V, Guo M, Smith JA, Wang J, Stewart JE, Wang X, Bentley WE, Sintim HO. ACS chem Biol. 2012;7:1023–30. doi: 10.1021/cb200524y. [DOI] [PubMed] [Google Scholar]
- 22.Roy V, Smith JA, Wang J, Stewart JE, Bentley WE, Sintim HO. J Am Chem Soc. 2010;132:11141–50. doi: 10.1021/ja102587w. [DOI] [PubMed] [Google Scholar]
- 23.Wu M, Tao Y, Liu X, Zang J. J Biol Chem. 2013;288:15878–87. doi: 10.1074/jbc.M112.417634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Sanctis D, McVey CE, Enguita FJ, Carrondo MA. J Mol Biol. 2009;387:759–70. doi: 10.1016/j.jmb.2009.02.017. [DOI] [PubMed] [Google Scholar]
- 25.Harpaz Y, Gerstein M, Chothia C. Structure. 1994;2:641–9. doi: 10.1016/s0969-2126(00)00065-4. [DOI] [PubMed] [Google Scholar]
- 26.Diaz Z, Xavier KB, Miller ST. PLoS One. 2009;4:e6820. doi: 10.1371/journal.pone.0006820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller ST, Xavier KB, Campagna SR, Taga ME, Semmelhack MF, Bassler BL, Hughson FM. Mol Cell. 2004;15:677–87. doi: 10.1016/j.molcel.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 28.Globisch D, Lowery CA, McCague KC, Janda KD. Angew Chem Int Ed Engl. 2012;51:4204–8. doi: 10.1002/anie.201109149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muraoka S, Okumura R, Ogawa N, Nonaka T, Miyashita K, Senda T. J Mol Biol. 2003;328:555–66. doi: 10.1016/s0022-2836(03)00312-7. [DOI] [PubMed] [Google Scholar]
- 30.Mortensen UH, Breddam K. Protein Sci. 1994;3:838–42. doi: 10.1002/pro.5560030513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langkilde A, Kristensen SM, Lo Leggio L, Molgaard A, Jensen JH, Houk AR, Navarro Poulsen JC, Kauppinen S, Larsen S. Acta Crystallogr D Biol Crystallogr. 2008;D64:851–63. doi: 10.1107/S0907444908017083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Torshin IY, Weber IT, Harrison RW. Protein Eng. 2002;15:359–63. doi: 10.1093/protein/15.5.359. [DOI] [PubMed] [Google Scholar]
- 33.Xue T, Zhao L, Sun H, Zhou X, Sun B. Cell Res. 2009;19:1258–68. doi: 10.1038/cr.2009.91. [DOI] [PubMed] [Google Scholar]
- 34.Zorrilla S, Doan T, Alfonso C, Margeat E, Ortega A, Rivas G, Aymerich S, Royer CA, Declerck N. Biophys J. 2007;92:3215–27. doi: 10.1529/biophysj.106.095109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, Rabinowitz JD. Nat Chem Biol. 2009;5:593–9. doi: 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rezacova P, Kozisek M, Moy SF, Sieglova I, Joachimiak A, Machius M, Otwinowski Z. Mol Microbiol. 2008;69:895–910. doi: 10.1111/j.1365-2958.2008.06318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.