

Abstract
B cell ADAM10 is required for the development and maintenance of proper secondary lymphoid tissue architecture; however, the underlying mechanism remains unclear. In this study, we show disturbances in naïve lymph node architecture from B cell specific ADAM10 deficient mice (ADAM10B−/−) including loss of B/T compartmentalization, attenuation of FDC reticula, excessive collagen deposition, and increased HEV formation. Because TNFα signaling is critical for secondary lymphoid tissue architecture, we examined compensatory changes in ADAM17 and TNFα in ADAM10B−/− B cells. Surprisingly, defective follicular development in these mice was associated with increased rather than decreased TNFα expression. Here, we describe an increase in TNFα message, mRNA stability, soluble protein release, and membrane expression in ADAM10B−/− B cells compared to WT, which coincides with increased ADAM17 message and protein. To assess the mechanistic contribution of excessive TNFα to abnormal lymphoid architecture in ADAM10B−/− mice, we performed a bone marrow reconstitution study. Rectification of WT architecture was noted only in irradiated WT mice reconstituted with ADAM10B−/− + TNFKO bone marrow due to normalization of TNFα levels not seen in ADAM10B−/− alone. We conclude that ADAM17 overcompensation causes excessive TNFα shedding and further upregulation of TNFα expression, creating an aberrant signaling environment within B cell cortical regions of ADAM10B−/− lymph nodes, highlighting a key interplay between B cell ADAM10 and ADAM17 with respect to TNFα homeostasis.
Introduction
A disintegrin and metalloproteinases (ADAMs) are a family of zinc dependent proteinases known to be involved in ectodomain cleavage and regulated intramembrane proteolysis of transmembrane proteins. Of all of the ADAMs, ADAM10 and ADAM17, commonly referred to as tumor necrosis factor alpha (TNFα) converting enzyme (TACE), are known to be most closely related with regards to structure and share many overlapping substrate specificities (1,2). Classically, ADAM17 is thought to orchestrate inflammatory responses as the principle, physiological sheddase of pro-TNFα; however, ADAM10 can also cleave membrane TNFα when ADAM17 is not present (3). Additionally, ADAM10 is crucial for functional and phenotypic maturation of the immune system. We have shown it is critical in Notch2-mediated marginal zone B cell development and CD23-mediated regulation of allergic diseases (4,5). Lastly, while we have previously reported that B cell ADAM10 is required for maintenance of proper secondary lymphoid tissue architecture, formation of germinal centers, as well as optimal class-switched antibody (Ig) production, the underlying mechanism was unclear (6).
TNFα is a key proinflammatory cytokine, which exists as a 26kDa transmembrane protein (mTNFα) before it is shed from the surface as a 17kDa soluble molecule (sTNFα) (7). Tristetraprolin (TTP), also known as ZFP36, is a low-abundance cytosolic zinc finger protein induced by lipopolysaccharide (LPS) and is critical for mRNA degradation of multiple mRNA targets including TNFα (8,9). TTP deficient mouse models portray the downstream consequences of increased TNFα mRNA stability including inflammatory arthritis, autoimmunity, and cachexia (10,11). In addition, B cell-TNFα has been implicated in the functional decline of aging B cells where increased TNFα production is inversely correlated with response to stimulation in vitro by LPS. Interestingly, aging B cells additionally exhibit increased TTP, which causes reduced optimal class switched antibody production by downregulating E47 and activation induced cytidine deaminase (AID). The paradoxical increase of both TTP and TNFα in unstimulated B cells from old mice may reflect increased TNFα transcription by these B cells to overcome elevated TTP, thus placing them in a preactivated state that is less susceptible to subsequent stimulation (12).
The role of TNFα in maintaining proper secondary lymphoid tissue architecture is indisputable, and ADAM10 also seems to be involved in this maintenance. Both B cell specific ADAM10 deficient (ADAM10B−/−) and global TNFα deficient mice exhibit disorganized follicular dendritic cell (FDC) networks, aberrant germinal centers, and lack of splenic B cell follicles (13). Furthermore, using B cells that express a non-cleavable form of mTNFα showed that adequate levels of B cell produced sTNFα was critical for maintaining secondary architecture in the lymph node, spleen and Peyer’s patches and for IgG production against T dependent antigens (14). While it is clear that regulation of B cell TNFα is required for proper follicular architecture and B cell function, the role of TNFα cleaving enzymes (ADAM10 and ADAM17) has yet to be explored.
Here, we investigate the hypothesis that compensatory over-expression of ADAM17 following B cell specific ADAM10 deletion mediates excessive TNFα levels in ADAM10B−/− mice, ultimately providing the mechanism underpinning the aberrant secondary lymphoid tissue architecture in these mice.
Materials and methods
Mice
All mice were housed in the Virginia Commonwealth University Molecular Medicine Research Building Barrier Facility in accordance with institutional and National Institutes of Health guidelines. All animal care and experimental protocols were approved by the Virginia Commonwealth University Institutional Animal Care and Use Committee. C57Bl/6 ADAM10B−/− (CD19-cre+) mice were generated as previously described and compared to littermate WT controls (CD19-cre−) (4). TNFα deficient (TNFαKO) mice were purchased from Jackson Laboratory (no. 005540, B6.129S-Tnf) for use in bone marrow reconstitution. Healthy male and female mice age 6–12 weeks were used in all experimentation except bone marrow reconstitution where only WT female mice age 6–8 weeks were reconstituted with sex matched bone marrow cells.
B cell isolation, reagents, in vitro activation, and ELISA
Spleen was crushed, filtered through 40μm Nylon Mesh (Fisherbrand), and RBC lysed with ACK Lysing Buffer (Quality Biological Inc.). B cells were isolated by positive selection (B220+) using magnetic beads and following manufacturer’s protocol (Miltenyi). B cells were grown in complete RPMI-1640 medium containing 10% heat inactivated (56 °C, 30 min) fetal bovine serum (Gemini Bio-Products, West Sacramento, CA), 2 mM L-glutamine, 50 μg/mL penicillin, 50 μg/mL streptomycin, 1 mM sodium pyruvate, 50 μM 2-mercaptoethanol, 1x non-essential amino acids, and 20 mM HEPES buffer (all from Invitrogen Carlsbad, CA) and stimulated in vitro for 1, 3, or 5 days with 1000 units IL-4 and either 50 μg/mL LPS (Lipopolysaccharides from Escherichia coli 0111:B4, Sigma) or 1.25μg/mL purified anti-mouse CD40 (no. 102902, Biolegend).
Proliferation was assessed after 72 hours of growth and a 24 hour pulse of [H3]-thymidine, 1μCi/well (Perkin Elmer) was used. Plates were then harvested using a Filtermate cell harvester onto GFC plates and analyzed using Topcount Plate Counter (Perkin Elmer, Waltham, MA). Soluble TNFα from B cell supernatants was determined by mouse quantitative ELISA kit (eBioscience 88-7324-88) according to manufacturers’ instructions.
RT-PCR, qPCR, and western blotting
Total RNA was extracted from naive and stimulated total B cells using TRIzol reagent (Invitrogen) according to manufacturer’s protocol and RNA concentration quantified by a ND-100 NanoDrop spectrophotometer. RNA (400 ng/μL) was reverse transcribed using an iScript cDNA Synthesis Kit (Bio-Rad). Real-time quantitative PCR was performed with a real-time PCR machine (iQ5; Bio-Rad Laboratories). Primers and probes used for TaqMan quantitative PCR assay (all from Applied Biosystems) were the following: TNFα (Mm00443258), ADAM10 (Mm00545742), ADAM17 (Mm00456428) TTP/Zfp36 (Mm00457144), MMP13 (Mm00439491) and 18s (Mm03928990). Fold variation was determined using the delta delta Ct (ΔΔCt) method of analysis (15). Protein lysates were made according to the manufacturer’s protocol using Cell Lysis Buffer (no. 9803, Cell Signaling). Equal amounts of protein were loaded onto Novex NuPage 10% Bis-Tris gels (Invitrogen), run for 35 minutes at 200 volts, transferred to nitrocellulose membrane and equal transfer verified by Ponceau S (Sigma) staining. Blots were probed with primary anti-mouse antibodies: anti-β-actin peroxidase (no. A3854, Sigma) or anti-ADAM17 (no. 2051, Abcam). Goat anti-rabbit IgG, HRP conjugate secondary antibody was used for ADAM17 blots and signal detected with SuperSignal West Pico Chemiluminescent Substrate (no. 34080, Thermo Scientific).
Flow cytometry with tyramide signal amplification
Single cell suspensions of naïve or stimulated B cells were stained using Tyramide Signal Amplification (TSA) Kit #26 with HRP streptavidin (no. T20936, Molecular Probes) and one of the following Ig: anti-mouse FITC- conjugated B220 or PE-conjugated B220 (Biolegend). Kit reagents were prepared according to manufacturer’s protocol and tyramide amplification using the “Peroxidase Labeling assay” was performed with the following modifications: Cells were incubated with blocking reagent (10 μg anti-mouse unlabeled CD16/32 (2.4G2)) for 15 minutes; stained with biotin anti-mouse TNFα primary antibody (Biolegend), and following tyramide labeling, cells were washed twice and stained with anti-mouse B220 (see above) for 30 minutes and examined on a BD Canto Flow analyzer, data analysis was with FCS Express, v. 4.
Bone marrow reconstitution
Bone marrow cells were isolated as previously described with the following modifications (16). Briefly, 2 femurs and 2 tibias from each mouse (WT (CD45.2), ADAM10B−/− (CD45.2), or TNFαKO) were centrifuged, RBC lysed with ACK Lysing Buffer (Quality Biological Inc.), bone marrow cells counted, and 5 million cells were i.v. injected. For 50/50 mixtures, such as ADAM10B−/− + TNFKO, 100μL/2.5 million cells from each were used to prepare the final injection mixture.
B6-Ly5.2/Cr (CD45.1) congenic mice from NCI/NIH were pretreated 5 days before irradiation with 100mg/L (concentration 0.01%) with Enrofloxacin (Baytril) in sterile water. CD45.1 mice were anesthetized using a 100μl i.p. injection of ketamine/xylazine in PBS at a dose of 80 mg/kg and 8 mg/kg, respectively. This was followed by two doses of 550 cGy irradiation, separated by a 2 hour rest period, using a MDS Nordion Gammacell 40 research irradiator with a Cs-137 source. Following irradiation, mice were reconstituted by i.v. injection of the indicated bone marrow cells as described above. After 6 weeks of reconstitution, mice were footpad immunized in 2 ipsilateral paws with 10μg 4-hydroxy-3-nitrophenylacetyl coupled to keyhole limpet hemocyanin (NP-KLH) at a ratio of 27:1 (Biosearch Technologies) in 4mg alum. Draining and non-draining popliteal and axillary lymph nodes were dissected at 14 days post immunization and analyzed by immunohistochemistry.
Immunohistochemistry, tyramide amplification, and confocal microscopy
Ten μm thickness frozen sections were cut from the excised mouse LNs, fixed in absolute acetone, air-dried and blocked with serum-free protein block (Dako, X0909). The sections were dual and triple-labeled for FDCs (PE anti-mouse CD21/CD35, Biolegend, 123410), B cells (Alexa Fluor 647 anti-mouse/human CD45R/B220, Biolegend, 103226), High Endothelial Venules (HEVs) (anti-mouse/human PNAd, Biolegend, 120804), T cells (Rat Anti-Mouse CD90.2/Thy-1.2-PE, Southern Biotech, 1750-09L), collagen type 1 (Abcam, ab21286), and TNFα (Abcam, ab34674). The Ig concentrations ranged between 5–10 μg/ml. Sections were mounted with anti-fade mounting medium, Vectashield (Vector Laboratories), cover-slipped, and examined with a Leica TCS-SP2 AOBS confocal laser scanning microscope. Three lasers were used: Argon (488nm); HeNe (543nm); HeNe (633nm) [far red emission is shown as pseudo-blue]. Parameters were adjusted to scan at 1024 × 1024 pixel density and 8-bit pixel depth. Emissions were recorded in two or three separate channels, and digital images were captured and processed with Leica Confocal, LCS Lite software; and Image-J for color separation and quantitative assessment of immunohistochemistry.
TNFα labeling was enhanced using Fluorescein-Tyramide signal amplification (TSA Plus Fluorescein System, Perkin elmer, NEL741001KT). Briefly, after quenching endogenous peroxidase using 1% H2O2, anti-TNFα Ig was applied for 2 hrs, washed, then HRP-conjugated secondary Ig was added for 1 hr. After washing, HRP was allowed to catalyze the deposition of Fluorescein-labeled tyramide for 10 minutes, then washed, mounted, and examined.
Statistical analysis
The p values were calculated using unpaired two-tailed Student t tests in GraphPad Prism. Error bars represent the SEM between samples. A p value <0.05 is considered significant.
Results
C57Bl/6 ADAM10B−/− mice exhibit abnormal lymph node architecture and excessive TNFα in B cell regions
Our lab previously described secondary lymphoid tissue architectural defects in immunized and naïve lymph nodes isolated from C57Bl/6 ADAM10B−/− mice. These defects included improper localization of B and T cells, reduced germinal center formation, and a decrease in follicular dendritic cell (FDC) networks (6). These architecture aberrancies bore some similarity with secondary lymphoid tissue defects noted in early studies of global TNFα deficient mice. Specifically, lack of splenic B cell follicles, disorganized FDC networks, and aberrant germinal centers were noted (13). Recent studies using B cells only capable of expressing mTNFα showed that sTNFα produced by B cells is required for maintaining secondary architecture in lymph node, spleen, Peyers patches and for IgG production against T dependent antigens (14). Taken together, ADAM10 and TNFα seem to reciprocally interact to maintain proper secondary lymphoid architecture and permit class switched antibody production. Fig 1 shows immunohistochemistry analysis of lymph nodes from naïve (non-immunized) WT and ADAM10B−/− mice. While our initial studies had indicated a relatively normal architecture in ADAM10B−/− nodes the absence of immunization (6), additional analysis revealed that not only are lymph node FDC networks largely absent and B/T boundaries intermingled (Fig 1A, 1B), but other abnormalities can also be seen. These changes include excessive collagen deposition as well as an increase in high endothelial venules (HEVs), especially within B cell cortical regions (Fig 1B, 1C). Most striking, however, was the dramatic increase in TNFα within the B cell regions of ADAM10B−/− nodes (Fig 1D). Given the data in Fig 1 and previous reports of secondary lymphoid tissue architecture abnormalities in TNFα knockouts and those only capable of expressing mTNFα, it appears that not only subnormal but also excessive TNFα levels in the lymph node cortices may lead to disruption of normal follicular architecture.
Fig 1. Naïve ADAM10B−/− lymph nodes (LNs) display abnormal follicular architecture.
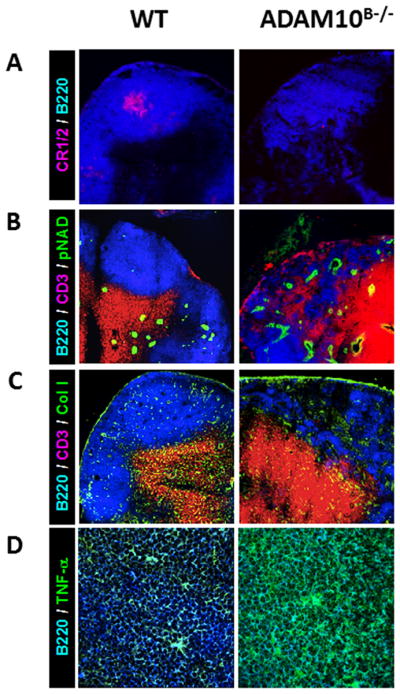
Compared to WT, ADAM10B−/− mice (A) lack well developed FDC reticula (red, CR1/2) in the B cell follicle (blue, B220), (B) lack B cell/T cell (red, CD3) segregation with more HEVs (green, pNAD) in the LN cortex, (C) display more collagen (green) deposition in the B cell follicle, and (D) express higher levels of TNFα than WT mice. Scale bar = 50 mm, and micrographs are representatives of at least 3 LNs.
B cells from C57Bl/6 ADAM10B−/− mice exhibit increased expression, stability and production of TNFα
Given the excessive TNFα staining in B cell regions of C57Bl/6 ADAM10B−/− lymph nodes (Fig 1), we further analyzed differences in TNFα expression and production in B cells purified from both ADAM10B−/− and WT mice. As can be seen in Fig 2A and 2B, flow analysis for mTNFα revealed that both naïve and stimulated ADAM10B−/− B cells exhibit increased mTNFα. Furthermore, ELISA analysis of supernatants from ADAM10B−/− B cells cultured with LPS/IL4 (Fig 2C) or anti-CD40/IL-4 (Fig 2D) for 1, 3, or 5 days all showed significantly higher sTNFα production compared to WT.
Fig 2. Increased TNFα surface expression and production in ADAM10B−/− B cells.

Naïve (A) and 5 day stimulated (B) (LPS + IL-4) WT (black thin line) and ADAM10B−/− (black bold line, A10KO) live B cells were analyzed for co-expression of TNFα using tyramide signal amplification. Black bars (overlay plots) indicate B cells staining high in mTNFα, represented in bar graphs (A,B). N=9 per group, 3 independent studies. (C,D) Supernatants were harvested on day 1, 3, or 5 from WT (white) or ADAM10B−/− (A10KO, black) B cell cultures stimulated with LPS + IL-4 (C) or anti-CD40 + IL-4 (D) and sTNFα determined by ELISA. N=9 per group, 3 independent studies. *indicates p<0.05, ** indicates p<0.005.
Relative (Fig 3A) and absolute (Fig 3B) message analysis by qPCR indicated a strong increase in TNFα message in ADAM10B−/− compared to WT B cells post stimulation in vitro. Furthermore, TTP is known to promote TNFα mRNA degradation (8,9). We, therefore, analyzed TTP message expression in both WT and ADAM10B−/− B cells and found it to be 2 fold higher in WT B cells, which also produce less sTNFα and mTNFα (Fig 2, Fig 3C). The combined results of increased TNFα message as well as soluble protein, suggests a possible feedback mechanism in which increased TNFα shedding upregulates further TNFα production. In any case, it is clear that TNFα is increased at both the message and protein level in ADAM10B−/− B cells.
Fig 3. Increased TNFα gene expression and message stability in ADAM10B−/− B cells.

Naïve (3A [checkered], 3B [left], 3C [checkered]) or 5 day stimulated (LPS + IL-4) (3A [stripe], 3B [right], 3C [stripe]) B cells were analyzed by qPCR for TNFα (A, B) and TTP (C) message normalized to 18s. Data presented as fold change of ADAM10B−/− over WT (A) or fold change of WT over ADAM10B−/− (C) using the (ΔΔ)Ct method of analysis. Absolute RNA quantification for TNFα normalized to 18s in WT (white) and ADAM10B−/− (black) B cells appears in (B). N=9 per group, 3 independent studies. *indicates p<0.05, ***indicates p<0.0005.
ADAM10B−/− B cells exhibit higher ADAM17 gene and protein expression
ADAM17 is known to be the principle sheddase of the membrane expressed pro-TNFα. Since, ADAM10B−/− B cells exhibit higher expression and production of TNFα, we next examined ADAM17 expression and function by analyzing ADAM17 message (Fig 4A, B) and protein levels (Fig 4C, D) in WT compared to ADAM10B−/− B cells. While both naïve and stimulated ADAM10B−/− B cells express significantly more ADAM17 message (Fig 4A, B), relative gene expression analysis showed that in the naïve state, ADAM10B−/− B cells express 2 times higher ADAM17 compared to WT which increased to 4 fold higher expression upon stimulation (Fig 4A). Similarly, absolute RNA quantification revealed that naïve ADAM10B−/− B cells exhibit significantly increased ADAM17 RNA expression, which increases further upon stimulation (Fig 4B). Western blot analysis, furthermore, showed a 2.3 fold increase in ADAM17 protein levels in naïve ADAM10B−/− B cells over WT, which too increased upon stimulation to 5 fold (Fig 4C, D). Upon overexposure of the blots, both the precursor and glycosylated forms of ADAM17 were seen (data not shown).
Fig 4. Increased ADAM17 message and protein levels in ADAM10B−/− B cells.

Naïve (4A [checkered], 4B [left], 4C) or 5 day stimulated (4A [stripe], 4B [right], 4D) (LPS + IL-4) B cells were analyzed by qPCR and western blotting. (A) Relative ADAM17 expression normalized to 18s presented as fold change of ADAM10B−/− (A10KO) over WT using the (ΔΔ)Ct method of analysis. (B) Absolute quantification of ADAM17 RNA normalized to 18s. N=9 mice per group, 3 independent studies. Band densitometry of naïve (C) and stimulated (D) B cells represents ADAM17 (~93kDa, mature form) normalized to actin (~42kDa). N=6 per group total, 4 independent studies. *indicates p<0.05.
In addition, Vandenbroucke et al. recently established that matrix metalloproteinase MMP13 also cleaves TNFα at least in intestinal epithelium (17). qPCR analysis of MMP13 in naïve B cells, however, showed there was no difference between WT and ADAM10B−/− B cells (ΔΔCt = 1.04; fold change ADAM10B−/− over WT). Western blot, furthermore, failed to show MMP13 protein in naïve WT and ADAM10B−/− B cells compared to positive control, RAW 264.7 macrophages (data not shown). This finding is in agreement with the report that significant levels of MMP13 are not found in B cells (18). Taken together, Figures 1–4 demonstrate that ADAM10 deletion from B cells results in a compensatory increase in ADAM17 expression and activity leading to excessive TNFα cleavage. The aberrant signaling environment created by this compensatory effect is an excellent candidate to explain the abnormal lymphoid tissue architecture in our ADAM10B−/− model and was thus further explored.
Reconstitution of irradiated C57Bl/6 WT with combination ADAM10B−/− + TNFKO bone marrow rectifies lymph node follicular abnormalities
In order to assess whether TNFα is involved in the mechanism underlying lymph node tissue abnormalities in ADAM10B−/− mice, we performed a bone marrow chimera experiment in which irradiated CD45.1 WT C57Bl/6 mice were reconstituted with one of the following bone marrow combinations: (1) WT (CD45.2) alone; (2) ADAM10B−/− (CD45.2) alone; (3) TNFα deficient (TNFKO) alone; (4) 50/50 mix ADAM10B−/− + WT (CD45.1); or (5) 50/50 mix ADAM10B−/− + TNFKO. Following 6 weeks of reconstitution, mice were bled and analyzed for successful reconstitution and CD45.2 cells predominated (Supplemental Fig 1). The mice were then footpad immunized with a T dependent antigen, NP-KLH, and draining lymph nodes were assessed by immunohistochemistry 14 days post-immunization (Fig 5A–H). As expected, lymph nodes from WT mice reconstituted with WT (CD45.2) bone marrow exhibited normal lymph node architecture (Fig 5D) and TNFα levels (Fig 5G), which was comparable to non-irradiated WT nodes (Fig 5A, 5G). Those reconstituted with ADAM10B−/− bone marrow (Fig 5B, 5G), however, had a similar phenotype to those of our ADAM10B−/− mice (Fig 1): loss of B cell/T cell segregation, decreased FDC networks, and increased TNFα. Those reconstituted with TNFKO bone marrow alone (Fig 5C) exhibit no TNFα staining but do still demonstrate FDC networks as these are resistant to irradiation. Furthermore, reconstitution with 50/50 mix ADAM10B−/− + WT CD45.1 still yielded abnormal lymph node architecture (Fig 5E) and high TNFα staining (Fig 5G) similar to ADAM10B−/−. Thus, the amount of TNFα made by B cells from this combination is still too high. Interestingly, when WT mice were reconstituted with combination ADAM10B−/− + TNFKO bone marrow (Fig 5F, 5G), lymph node tissue architecture and TNFα staining returned to WT levels. TNFα staining is further compared and quantified by mean gray values in multiple equal areas (at least 6) in Fig 5H. Here, mice reconstituted with ADAM10B−/− alone as well as ADAM10B−/− + WT CD45.1 exhibit significantly more TNFα staining compared to WT and ADAM10B−/− + TNFKO (Fig 5H). We reason that while B cells from ADAM10B−/− mice produce excessive TNFα, those from the TNFKO mice produce none, thus averaging to a normal, WT range allowing proper TNFα signaling to occur.
Fig 5. Reconstitution of irradiated WT naïve LNs with ADAM10B−/− + TNFKO bone marrow restores normal follicular architecture and rectifies structural abnormalities induced by ADAM10B−/− reconstitution alone.

Draining lymph node sections from non-irradiated WT (A, G (CD45.1 WT)) and irradiated CD45.1 WT reconstituted with ADAM10B−/− (B, G (Reconst with ADAM10KO)), TNFKO (C), WT CD45.2 (D, G (Reconst with CD45.2 WT)), ADAM10B−/− + WT CD45.1 (E, G (Reconst with ADAM10KO and CD45.1 WT)), ADAM10B−/− + TNFKO (F, G (Reconst with ADAM10KO and TNFKO)) were compared in (A–F) with regards to B/T compartmentalization (left column), FDC reticular development (middle column), and TNFα staining (right column). In (A–G) the following stains were used: B cells (blue, B220), T cells (red, Thy1.2), Collagen 1 (green), FDCs (red, CR1/2), and TNFα (green). White boxes in (A–F, right column) are magnified in (G, left column), with separation of the TNFα labelling (middle column) and measurements (histograms, right column). (H) TNFα expression (mean gray values) in multiple equal areas (at least 6) have been calculated and the results expressed as mean ± SD. Using unpaired T test, the p value between CD45.1 WT and reconstitution with CD45.2 WT is 0.33; CD45.1 WT and reconstitution with ADAM10KO and TNFKO is 0.36; and reconstitution with ADAM10KO and ADAM10KO and CD45.1 WT is 0.2. Micrographs are representative of at least 3 LNs from 3 independent experiments. Scale bar in A–F = 50 mm, in G = 20 mm.
Discussion
This study provides evidence that B cell TNFα and lymphoid tissue architecture are regulated by the orchestrated interplay between ADAM10 and ADAM17. Meyzk-Kopec et al. previously described, in mouse embryonic fibroblasts (MEFs), a compensatory relationship between ADAM10 and ADAM17 with regards to TNFα cleavage. In that study, ADAM17 deficient MEFs still exhibited TNFα cleavage due to a compensatory increase in ADAM10 (3). Here we show, in a B cell targeted model, that in the absence of B cell ADAM10, ADAM17 is overexpressed above WT levels (Fig 4). This compensatory change in B cell ADAM17 results in increased TNFα shedding, gene expression, mRNA stability, and surface expression, and thus skews the cytokine environment within B cell follicles (Fig 2, 3). While the absence of B cell sTNFα is known to cause lymph node architecture defects, here we demonstrate that excessive sTNFα also dramatically disrupts lymph node follicular architecture (14).
In addition to B cells, T cells, FDCs, and tingible body macrophages contribute to TNFα expression in secondary lymphoid tissues (19–21). Cooperation between these sources is needed to maintain TNFα homeostasis, which is essential for lymphoid tissue architectural maintenance and ultimately B cell humoral responses. Tumanov et al. clearly demonstrated this concept, showing impaired lymph node organization and B cell antibody production post immunization when either B or T cell TNFα was inhibited (14). Furthermore, therapeutic inhibition of TNFα with etanercept inhibited the maintenance of FDC networks, resulting in decreased germinal center responses and antibody production (22). While insufficient TNFα levels lend to disorganization and stunted B cell function, an example of pathologic overexpression of B cell TNFα is seen in B cell chronic lymphocytic leukemia (B-CLL). Leukemic B cells overexpress TNFα, which then acts in an autocrine fashion to upregulate its own mRNA and protein expression (23). Enhanced leukemic cell TNFα signaling leads to upregulation of MMP-9, a key protease involved in pro-angiogenic pathways needed for cancer metastasis (24).
Possible candidates responsible for defects characterizing ADAM10B−/− lymph nodes are TNFα and lymphotoxin (LTα, LTβ). These structurally homologous and genetically linked cytokines have been studied individually and as double deficient mouse models, in an attempt to tease out the contributions of each to secondary lymphoid tissue microarchitecture development and maintenance (25). LTα deficient mice lack lymph nodes and Peyer’s patches and exhibit abnormal splenic architecture including loss of B/T segregation and a complete absence of FDC networks, germinal center formation, and marginal zone B cells (26,27). LTα as a soluble homotrimer (LTα3) is also known to play an integral role in lymphoid organization including B/T segregation by binding to the TNFα receptor, p55TNFR-1, suggesting that blockade of this interaction would result in a similar phenotype to ADAM10B−/− lymph nodes (28). LTβ deficient mice, however, experience more mild disruption as they retain mesenteric and cervical lymph node development and maintain B/T segregation, FDC networks, and germinal center formation in spleen (29). More similar to LTα deficient mice, TNFα knockouts lack FDC networks in B cell follicles and fail to form germinal centers post immunization (13,30,31). Furthermore, it is known that without TNFα or its receptor p55TNFR-1, B cell follicles and FDC networks fail to form in peripheral lymph nodes, Peyer’s patches, and spleen; however, the effects of B cell TNFα overexpression are unreported (13,32).
Because p55TNFR-1 binds both TNFα and LTα3, it is reasonable to conclude that excessive TNFα could outcompete LTα3 for binding to this receptor, resulting in noted defects in B/T segregation. Furthermore, it has been shown that local cytokine factors such as TNFα contribute to HEV neogenesis (33). Excessive B cell TNFα could, therefore, explain the induction of increased cortical HEV neogenesis resulting in increased T cell recruitment via CCL21/CCR7 interactions and ultimate aberrancies in B/T segregation (Fig 1). Alternatively, Blobel et al. recently demonstrated that a conditional knockout model where ADAM17 was selectively deleted from endothelial cells and pericytes had significantly inhibited pathological neovascularization (34). Therefore, ADAM17 overexpression by ADAM10B−/− B cells may be directly involved in increased follicular HEVs.
In addition, we reason that defective follicular remodeling resulted in excessive collagen deposition associated with the lack of FDC reticula. Bajénoff and Germain et al. elegantly showed that FDC development replaces fibroblastic reticular cells during B cell follicle maturation (35). Therefore, lack of FDC development in ADAM10B−/− follicles could have resulted in the persistence of collagen-producing fibroblasts. Furthermore, in a Notch-dependent pathway, ADAM17 overactivation has been implicated in fibroblast activation, excessive collagen formation, and fibrosis (36). Increased ADAM17 in ADAM10B−/− B cells could lead to aberrant Notch signaling and increased fibrosis within the lymph node (Fig 1). Given the pro-fibrotic properties of ADAM17, our result that ADAM17 overcompensates for ADAM10 deficiency must be well-considered prior to attempting ADAM10 therapeutic neutralization.
The most conclusive data furthering the mechanistic contribution of TNFα over other ligands, however, is the rectification of WT architecture in irradiated WT mice reconstituted with ADAM10B−/− + TNFKO bone marrow (Fig 5F, G). Compared to those reconstituted with ADAM10B−/− bone marrow alone (Fig 5B, G) or a 50/50 mix of ADAM10B−/− + WT CD45.1 (Fig 5E, G), these produce an appropriate level of TNFα considering ADAM10B−/− B cells make too much and TNFKO B cells make none; thus, further supporting the mechanistic contribution of TNFα. With the combination ADAM10B−/− + TNFKO model, it is important to note that all cells in the TNFKO do not make TNFα, which may contribute to the effect seen.
To conclude, this study first reveals compensatory changes in B cell ADAM17 in the absence of ADAM10. This finding has substantial implications in therapeutic design where specific targeting of one ADAM may lead to changes in other closely related ADAMs. In this scenario, compensation by ADAM17 would lead to excessive cleavage of TNFα or other ligands resulting in foreseeable albeit undesirable side effects. It is important to study the interaction between ADAM10 and ADAM17 on other cell types to further elucidate other potential complications. Secondly, in pathologic conditions where TNFα is directly implicated in pathology such as insulin resistance and rheumatoid arthritis, this study suggests that ADAM10 and ADAM17 levels should be monitored as a direct indication of disease susceptibility and severity. Lastly, here we demonstrate that compensatory increases in ADAM17 occur in ADAM10B−/− B cells leading to increased production of TNFα, which ultimately underlies architectural aberrancies noted in ADAM10B−/− lymph nodes. This finding lends new insight to the discussion regarding how B cell TNFα helps regulate secondary lymphoid tissue organization and how a proper ADAM10/ADAM17 ratio is needed to ultimately control TNFα signaling.
Supplementary Material
Acknowledgments
Authors wish to thank Lee Dean, Rebecca K. Martin, and Julie Farnsworth for technical assistance and Dr. Suzanne Barbour for helpful comments on the manuscript.
Abbreviations used in this article
- ADAM
A disintegrin and metalloproteinase
- ADAM10B−/− or A10KO
B cell specific ADAM10 knockout mouse model
- B/T
B lymphocyte/T lymphocyte
- FDC
follicular dendritic cell
- WT
wild type
- KO
knock out
- sTNFα
soluble molecule TNFα
- mTNFα
membrane TNFα
- TTP
Tristetraprolin
- NP-KLH
4-hydroxy-3-nitropehnylacetyl coupled to keyhole limpet hemocyanin
- LN
lymph node
- LT
lymphotoxin
- HEV
high endothelial venule.
Footnotes
The work supported by grant RO1AI18697 from NIAID/NIH and a bridge grant from the VCU School of Medicine. Flow cytometry support from the Massey Cancer Center Core P30 CA16059 is also acknowledged.
Reference List
- 1.Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Mol Aspects Med. 2008;29:258–289. doi: 10.1016/j.mam.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Le Gall SM, Bobe P, Reiss K, Horiuchi K, Niu XD, Lundell D, Gibb DR, Conrad D, Saftig P, Blobel CP. ADAMs 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as transforming growth factor alpha, L-selectin, and tumor necrosis factor alpha. Mol Biol Cell. 2009;20:1785–1794. doi: 10.1091/mbc.E08-11-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mezyk-Kopec R, Bzowska M, Stalinska K, Chelmicki T, Podkalicki M, Jucha J, Kowalczyk K, Mak P, Bereta J. Identification of ADAM10 as a major TNF sheddase in ADAM17-deficient fibroblasts. Cytokine. 2009;46:309–315. doi: 10.1016/j.cyto.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Gibb DR, El SM, Kang DJ, Rowe WJ, El SR, Cichy J, Yagita H, Tew JG, Dempsey PJ, Crawford HC, Conrad DH. ADAM10 is essential for Notch2-dependent marginal zone B cell development and CD23 cleavage in vivo. J Exp Med. 2010;207:623–635. doi: 10.1084/jem.20091990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weskamp G, Ford JW, Sturgill J, Martin S, Docherty AJ, Swendeman S, Broadway N, Hartmann D, Saftig P, Umland S, Sehara-Fujisawa A, Black RA, Ludwig A, Becherer JD, Conrad DH, Blobel CP. ADAM10 is a principal ‘sheddase’ of the low-affinity immunoglobulin E receptor CD23. Nat Immunol. 2006;7:1293–1298. doi: 10.1038/ni1399. [DOI] [PubMed] [Google Scholar]
- 6.Chaimowitz NS, Martin RK, Cichy J, Gibb DR, Patil P, Kang DJ, Farnsworth J, Butcher EC, McCright B, Conrad DH. A disintegrin and metalloproteinase 10 regulates antibody production and maintenance of lymphoid architecture. J Immunol. 2011;187:5114–5122. doi: 10.4049/jimmunol.1102172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacEwan DJ. TNF ligands and receptors - a matter of life and death. 135. 2002. pp. 855–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frasca D, Landin AM, Alvarez JP, Blackshear PJ, Riley RL, Blomberg BB. Tristetraprolin, a negative regulator of mRNA stability, is increased in old B cells and is involved in the degradation of E47 mRNA. J Immunol. 2007;179:918–927. doi: 10.4049/jimmunol.179.2.918. [DOI] [PubMed] [Google Scholar]
- 9.Lai WS, Parker JS, Grissom SF, Stumpo DJ, Blackshear PJ. Novel mRNA targets for tristetraprolin (TTP) identified by global analysis of stabilized transcripts in TTP-deficient fibroblasts. Mol Cell Biol. 2006;26:9196–9208. doi: 10.1128/MCB.00945-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Phillips K, Kedersha N, Shen L, Blackshear PJ, Anderson P. Arthritis suppressor genes TIA-1 and TTP dampen the expression of tumor necrosis factor alpha, cyclooxygenase 2, and inflammatory arthritis. Proc Natl Acad Sci U S A. 2004;101:2011–2016. doi: 10.1073/pnas.0400148101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, Schenkman DI, Gilkeson GS, Broxmeyer HE, Haynes BF, Blackshear PJ. A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity. 1996;4:445–454. doi: 10.1016/s1074-7613(00)80411-2. [DOI] [PubMed] [Google Scholar]
- 12.Frasca D, Romero M, Diaz A, Alter-Wolf S, Ratliff M, Landin AM, Riley RL, Blomberg BB. A molecular mechanism for TNF-alpha-mediated downregulation of B cell responses. J Immunol. 2012;188:279–286. doi: 10.4049/jimmunol.1003964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pasparakis M, Alexopoulou L, Episkopou V, Kollias G. Immune and inflammatory responses in TNF alpha-deficient mice: a critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J Exp Med. 1996;184:1397–1411. doi: 10.1084/jem.184.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tumanov AV, Grivennikov SI, Kruglov AA, Shebzukhov YV, Koroleva EP, Piao Y, Cui CY, Kuprash DV, Nedospasov SA. Cellular source and molecular form of TNF specify its distinct functions in organization of secondary lymphoid organs. Blood. 2010;116:3456–3464. doi: 10.1182/blood-2009-10-249177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dussault AA, Pouliot M. Rapid and simple comparison of messenger RNA levels using real-time PCR. Biol Proced Online. 2006;8:1–10. doi: 10.1251/bpo114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gommerman J, Rojas O. Creation of mixed bone marrow chimeras with appropriate controls. 2011. [Google Scholar]
- 17.Vandenbroucke RE, Dejonckheere E, Van HF, Lodens S, De RR, Van WE, Staes A, Gevaert K, Lopez-Otin C, Libert C. Matrix metalloproteinase 13 modulates intestinal epithelial barrier integrity in inflammatory diseases by activating TNF. EMBO Mol Med. 2013;5:932–948. doi: 10.1002/emmm.201202100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bar-Or A, Nuttall RK, Duddy M, Alter A, Kim HJ, Ifergan I, Pennington CJ, Bourgoin P, Edwards DR, Yong VW. Analyses of all matrix metalloproteinase members in leukocytes emphasize monocytes as major inflammatory mediators in multiple sclerosis. Brain. 2003;126:2738–2749. doi: 10.1093/brain/awg285. [DOI] [PubMed] [Google Scholar]
- 19.Aggarwal BB, Gupta SC, Kim JH. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood. 2012;119:651–665. doi: 10.1182/blood-2011-04-325225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thacker TC, Zhou X, Estes JD, Jiang Y, Keele BF, Elton TS, Burton GF. Follicular dendritic cells and human immunodeficiency virus type 1 transcription in CD4+ T cells. J Virol. 2009;83:150–158. doi: 10.1128/JVI.01652-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCall JL, Yun K, Funamoto S, Parry BR. In vivo immunohistochemical identification of tumor necrosis factor/cachectin in human lymphoid tissue. Am J Pathol. 1989;135:421–425. [PMC free article] [PubMed] [Google Scholar]
- 22.Anolik JH, Ravikumar R, Barnard J, Owen T, Almudevar A, Milner EC, Miller CH, Dutcher PO, Hadley JA, Sanz I. Cutting edge: anti-tumor necrosis factor therapy in rheumatoid arthritis inhibits memory B lymphocytes via effects on lymphoid germinal centers and follicular dendritic cell networks. J Immunol. 2008;180:688–692. doi: 10.4049/jimmunol.180.2.688. [DOI] [PubMed] [Google Scholar]
- 23.Cordingley FT, Bianchi A, Hoffbrand AV, Reittie JE, Heslop HE, Vyakarnam A, Turner M, Meager A, Brenner MK. Tumour necrosis factor as an autocrine tumour growth factor for chronic B-cell malignancies. Lancet. 1988;1:969–971. doi: 10.1016/s0140-6736(88)91782-5. [DOI] [PubMed] [Google Scholar]
- 24.Bauvois B, Dumont J, Mathiot C, Kolb JP. Production of matrix metalloproteinase-9 in early stage B-CLL: suppression by interferons. Leukemia. 2002;16:791–798. doi: 10.1038/sj.leu.2402472. [DOI] [PubMed] [Google Scholar]
- 25.Allen CD, Cyster JG. Follicular dendritic cell networks of primary follicles and germinal centers: phenotype and function. Semin Immunol. 2008;20:14–25. doi: 10.1016/j.smim.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De TP, Goellner J, Ruddle NH, Streeter PR, Fick A, Mariathasan S, Smith SC, Carlson R, Shornick LP, Strauss-Schoenberger J. Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science. 1994;264:703–707. doi: 10.1126/science.8171322. [DOI] [PubMed] [Google Scholar]
- 27.Matsumoto M, Mariathasan S, Nahm MH, Baranyay F, Peschon JJ, Chaplin DD. Role of lymphotoxin and the type I TNF receptor in the formation of germinal centers. Science. 1996;271:1289–1291. doi: 10.1126/science.271.5253.1289. [DOI] [PubMed] [Google Scholar]
- 28.Kuprash DV, Qin Z, Ito D, Grivennikov SI, Abe K, Drutskaya LN, Blankenstein T, Nedospasov SA. Ablation of TNF or lymphotoxin signaling and the frequency of spontaneous tumors in p53-deficient mice. Cancer Lett. 2008;268:70–75. doi: 10.1016/j.canlet.2008.03.023. [DOI] [PubMed] [Google Scholar]
- 29.Koni PA, Sacca R, Lawton P, Browning JL, Ruddle NH, Flavell RA. Distinct roles in lymphoid organogenesis for lymphotoxins alpha and beta revealed in lymphotoxin beta-deficient mice. Immunity. 1997;6:491–500. doi: 10.1016/s1074-7613(00)80292-7. [DOI] [PubMed] [Google Scholar]
- 30.Fu YX, Chaplin DD. Development and maturation of secondary lymphoid tissues. Annu Rev Immunol. 1999;17:399–433. doi: 10.1146/annurev.immunol.17.1.399. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Wang J, Sun Y, Wu Q, Fu YX. Complementary effects of TNF and lymphotoxin on the formation of germinal center and follicular dendritic cells. J Immunol. 2001;166:330–337. doi: 10.4049/jimmunol.166.1.330. [DOI] [PubMed] [Google Scholar]
- 32.Pasparakis M, Alexopoulou L, Grell M, Pfizenmaier K, Bluethmann H, Kollias G. Peyer’s patch organogenesis is intact yet formation of B lymphocyte follicles is defective in peripheral lymphoid organs of mice deficient for tumor necrosis factor and its 55-kDa receptor. Proc Natl Acad Sci U S A. 1997;94:6319–6323. doi: 10.1073/pnas.94.12.6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Girard JP, Springer TA. High endothelial venules (HEVs): specialized endothelium for lymphocyte migration. Immunol Today. 1995;16:449–457. doi: 10.1016/0167-5699(95)80023-9. [DOI] [PubMed] [Google Scholar]
- 34.Weskamp G, Mendelson K, Swendeman S, Le GS, Ma Y, Lyman S, Hinoki A, Eguchi S, Guaiquil V, Horiuchi K, Blobel CP. Pathological neovascularization is reduced by inactivation of ADAM17 in endothelial cells but not in pericytes. Circ Res. 2010;106:932–940. doi: 10.1161/CIRCRESAHA.109.207415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bajenoff M, Germain RN. B-cell follicle development remodels the conduit system and allows soluble antigen delivery to follicular dendritic cells. Blood. 2009;114:4989–4997. doi: 10.1182/blood-2009-06-229567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kavian N, Servettaz A, Weill B, Batteux F. New insights into the mechanism of notch signalling in fibrosis. Open Rheumatol J. 2012;6:96–102. doi: 10.2174/1874312901206010096. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.