

Abstract
We have previously shown that non-myeloablative total lymphoid irradiation/rabbit anti-thymocyte serum (TLI/ATS) conditioning facilitates potent donor-recipient immune tolerance following bone marrow transplantation (BMT) across major histocompatibility complex (MHC) barriers via recipient invariant natural killer T cell (iNKT cell)-derived IL-4-dependent expansion of donor Foxp3+ naturally occurring Treg (nTreg). Here we report a more specific mechanism. Wild-type (WT) BALB/c (H-2d) hosts were administered TLI/ATS and BMT from WT or STAT6−/− C57BL/6 (H-2b) donors. Donor nTreg following STAT6−/− BMT demonstrated no loss of proliferation in vivo, indicating that an IL-4 responsive population in the recipient rather than the donor drives donor nTreg proliferation. In GVHD target organs, three recipient CD11b+ cell subsets (Gr-1highCD11cneg; Gr-1intCD11cneg; and Gr-1lowCD11c+) were enriched early after TLI/ATS + BMT versus TBI/ATS + BMT. Gr-1lowCD11c+ cells induced potent H-2Kb+CD4+Foxp3+ nTreg proliferation in vitro in 72-hr MLR. Gr-1lowCD11c+ cells were significantly reduced in STAT6−/− and iNKT cell-deficient Jα18−/− BALB/c recipients after TLI/ATS + BMT. Depletion of CD11b+ cells resulted in severe acute GVHD, and adoptive transfer of WT Gr-1lowCD11c+ cells to Jα18−/− BALB/c recipients of TLI/ATS + BMT restored day 6 donor Foxp3+ nTreg proliferation and protection from CD8 effector T cell-mediated GVHD. Blockade of PD-L1 or PD-L2, but not CD40, TGF-β, Arginase 1, or iNOS inhibited nTreg proliferation in co-cultures of recipient-derived Gr-1lowCD11c+ cells with donor nTreg. Through iNKT-dependent Th2 polarization, myeloid-derived immunomodulatory DCs are expanded after non-myeloablative TLI/ATS conditioning and allogeneic BMT, induce PD-1 ligand dependent donor nTreg proliferation, and maintain potent graft-versus-host immune tolerance.
Keywords: Graft-versus-host disease, transplantation, tolerance, T cells, monocytes
Introduction
Prevention of graft-versus-host disease (GVHD) while maintaining graft-versus-tumor (GVT) activity remains the “holy grail” of allogeneic hematopoietic cell transplantation (HCT) (1, 2). To minimize transplant-associated toxicities including GVHD, regimens of reduced intensity conditioning (RIC) have been successfully applied to prepare the transplant recipient to immunologically accept an allogeneic bone marrow graft while allowing maintenance of GVT (3). We and others have shown that a recipient RIC regimen using total lymphoid irradiation and anti-thymocyte globulin (TLI/ATG) results in durable engraftment, profound GVHD protection in both children (4) and adults (5, 6), and maintenance of GVT in patients whose disease features rendered them at high risk for relapse (5, 6).
In the murine MHC mismatched [C57BL/6 (H-2b) → BALB/c (H-2d)] pre-clinical model of TLI and anti-thymocyte serum (TLI/ATS), we and others have shown that GVHD protection is driven by IL-4 secreting Th2-polarized recipient invariant natural killer T (iNKT) cells (7, 8, 9). Specifically, recipient iNKT cells which preferentially survive TLI conditioning due to their relative radioresistance (7,8,10) secrete IL-4, which in turn facilitates the potent in vivo expansion of donor-type naturally occurring regulatory CD4+CD25+Foxp3+ cells (nTreg) (11). nTreg expanded in vivo then regulate the donor effector CD8+ T-cell driven lethal acute GVHD seen when identical transplants are performed into conventional total body irradiation (TBI)-conditioned recipients. Our previous studies established that TLI/ATS results in post-BMT expansion of Foxp3+ nTreg and not merely peripheral expansion of induced Treg (iTreg), as CD25-depletion of the graft prior to BMT was confirmed at day 6 to result in loss of all expanding CD4+Foxp3+ cells at day 6 after BMT (11). Although earlier publications suggested that IL-4-driven STAT6 signaling could down-regulate FOXP3 gene expression in induced Treg (12,13), more recent publications support our findings by demonstrating that GATA3 may actually stabilize Foxp3 protein expression in nTreg (14,15). We sought to determine specific mechanisms by which recipient iNKT-derived IL-4 signaling could induce nTreg proliferation in vivo after TLI/ATS and allogeneic BMT. Defining the specific mechanism by which iNKT cells and Th2 polarizing conditioning in the recipient generate dono-type nTreg proliferation in this model would lay the foundation for future conditioning strategies designed to augment nTreg maintenance and expansion in vivo after allogeneic BMT. Here we demonstrate that the effect of recipient IL-4 on donor nTreg expansion in vivo early after TLI/ATS and BMT is not direct, but rather occurs via a critical recipient B220negCD11b+Gr-1lowCD11c+ regulatory dendritic cell (DC) subset fitting the immune phenotype of myeloid-derived immunomodulatory cells, maintenance and expansion of which after TLI/ATS + BMT is STAT6- and iNKT-dependent. Donor-type nTreg proliferation occurs independent of common regulatory pathways described in other CD11b+Gr-1low populations, including CD40/CD154 (CD40L), TGF-β STAT6 signaling, Arginase 1 (Arg1), or inducible nitric oxide synthase (iNOS), but requires contact-dependent signaling through PD-1 ligands. These recipient DCs induce potent proliferation of donor-type nTreg cells with stable expression of Foxp3, and blockade of the PD-1 ligand axis using monoclonal antibody treatment of recipients abrogates donor nTreg cell expansion after TLI/ATS and allogeneic BMT. Our studies link for the first time this regulatory TNF-α and iNOS-producing DC population with expansion of Foxp3+ nTreg both in vitro and in vivo, and identify a novel means by which non-myeloablative Th2-polarizing recipient conditioning may maintain durable donor-recipient immune tolerance after allogeneic BMT.
Materials and Methods
Mice
Wild-type (WT) (CD45.2+), CD45 congenic (CD45.1+), Arginase-1flox/flox (ARG1fl/fl), STAT6−/− BALB/c (H-2d), Foxp3-IRES-mRFP (FIR) and STAT6−/− C57BL/6 (H-2b) mice were purchased from Jackson Laboratories (Bar Harbor, Maine). iNKT-deficient Jα18−/− BALB/c mice were kind gifts from Dr. D. Umetsu (Harvard University) (16) and bred in our facility. Only male mice aged 8–12 weeks old were used for experiments (minimum starting weight 25 grams in the case of recipients of TLI). A BALB/c LysM-cre breeder mouse was a kind gift from Dr. P. Murray (St. Jude). LysM-cre × ARG1fl/fl BALB/c mice were bred in the St. Jude Animal Resource Facility. DNTGF-βRII × Foxp3-IRES-GFP (Foxp3GFP) C57BL/6 mice and Foxp3GFP mice (C57BL/6) were gifts from Dr. H. Chi (St. Jude) and Dr. A. Rudensky (Memorial Sloan-Kettering Cancer Center), respectively. All animals were housed, monitored, and euthanized on a pre-approved protocol reviewed annually by the St. Jude Institutional Animal Care and Use Committee (IACUC).
Irradiation
Total lymphoid irradiation (TLI) was delivered to the lymph nodes, thymus, and spleen with shielding of the skull, lungs, limbs, pelvis and tail as previously described (8–10, 11, 17). TLI was administered in 17 doses of 240 cGy each, beginning on day −24 prior to transplantation. Total body irradiation was delivered as a single dose (myeloablative as 800 cGy or non-myeloablative as 400 cGy) 24 hours before transplantation. Irradiation was performed with a Gulmay X-ray unit (Gulmay Medical, Suwanee, GA) (300kV, 10mA) at a rate of 100 mU/minute with a 0.75 mm Cu filter.
Rabbit anti-thymocyte serum (ATS)
Rabbit ATS was purchased from ACCURATE Laboratories (New York, NY) and complement adsorbed with BALB/c erythrocytes prior to use. Recipient mice were injected intraperitoneally with 0.05 ml ATS in 0.5 ml sterile normal saline on days −12, −10, and −8 before BMT (9,10).
Microscopic assessment of GVHD
Animals were sacrificed at day 6 after BMT for specific studies, or when moribund as per St. Jude Animal Welfare protocol guidelines. Tissue specimens were obtained at time of sacrifice from the skin, liver, spleen, mesenteric lymph nodes (MLN), and terminal 1 cm of descending colon measuring from the anal verge. Tissues were fixed in 10% formalin and embedded in paraffin blocks, and 4 to 5-µm sections cut and stained with hematoxylin and eosin. Microscopic images were obtained as described in detail previously (9,11). At time of histopathologic analysis of hematoxylin/eosin-stained sections, the skin, liver, and colon were assigned scores assessing severity of GVHD as per previously published criteria (9,11). The evaluating pathologist was blinded to the experimental groups. The cumulative and colonic GVHD scores represent the mean ± SEM in each group of animals.
Antibodies and flow cytometry
All cells were incubated with anti-CD16/CD32 (2.4G2, Becton Dickinson, San Diego, CA) prior to antibody staining to block FcR-γ II/III. The following conjugated mAbs were used: FITC anti-H-2Kb (clone AF6-88.5), PE-Cy7 anti-CD4 (clone GK 1.5), allophycocyanin-Cy7 (APC-Cy7) anti-CD8 (clone 53-6.7), APC-Cy7 anti-B220 (clone RA3-6B2), PerCP-Cy5.5 anti-CD11b (clone M1/70), eFluor™450 anti-Gr-1 (clone RB6-8C5), APC anti-CD11c (clone HL3), PE anti-CD103 (clone M290), PE anti-CD80 (clone 16-10A1), PE anti-CD86 (clone GL1), PE anti-CD124 (anti-IL4Rα) (clone mIL4R-MI), PE anti-CD1d (clone 1B1), PE anti-CD54 (ICAM-1) (clone 3E2), PE anti-CD252 (OX40L) (clone RM134L), PE anti-H-2Kd (clone SF1-1.1), FITC anti-Ly6C (clone AL21), and PE anti-CD8α (clone 53-6.7), FITC anti-TNF-α (clone MP6-XT22) (all from BD Biosciences); PerCP-Cy5.5 anti-TCRαβ, eFluor™450 anti-CD25 (clone eBio307), APC anti-Foxp3 (clone FJK-165), eFluor™450 anti-F4/80 (clone BM8), APC anti-CD115 (clone AFS98), biotin anti-PD-L1 (clone 1-111A), biotin anti-PD-L2 (clone 122), biotin anti-PD1 (clone J43), PE anti-CD40 (clone 1C10), APC anti-IAd (clone AMS-32.1) (all from eBioscience); Pacific Blue™ anti-Helios (clone 22F6, Biolegend); rabbit anti-iNOS (clone M19, Santa Cruz Biotechnology, Santa Cruz, CA), goat anti-rabbit PE (Southern Biotech, Birmingham, AL), and Live Dead Aqua® (LDA)(Invitrogen, Carlsbad, CA).
Donor T cell accumulation
At day 6 after BMT, single-cell suspensions from recipient spleen, mesenteric lymph nodes (MLN) and mononuclear cells from the liver and colon were prepared as described previously (9, 17). Cells were stained, samples analyzed with a BD LSR-II® instrument (BD Biosystems), and data analyzed using FlowJo® software (Treestar).
Donor CD4+CD25+Foxp3+ nTreg accumulation
Organs were harvested and single-cell suspensions prepared on day 6 as described previously (9,11). Cells were surface stained, fixed, permeabilized and stained with APC anti-mouse Foxp3 (clone FJK-16s) or APC rat IgG2a isotype control (both from BD Biosciences). To determine the proportion of proliferating CD4+Foxp3+ nTreg, cells were counter-stained with Pacific Blue™ anti-Helios (clone 22F6, Biolegend) or Pacific Blue™ hamster IgG isotype control (Biolegend) following fixation and permeabilization. Samples were analyzed on a BD LSR-II® instrument (BD Biosystems), and data analyzed using FlowJo® software (Treestar).
In vivo proliferation assays
WT BALB/c recipients were conditioned with TLI and ATS. Donor splenocytes in all experiments were labeled with Cell Proliferation dye eFluor® 450 (eBioscience) per manufacturer's instructions prior to infusion. On day 0, 60 × 106 labeled splenocytes and 50 × 106 bone marrow cells from donor mice (WT or STAT6−/− C57BL/6) were injected per conditioned recipient. Spleens of transplanted recipients were harvested on day 6 after BMT. Spleens from n = 2–3 recipient mice per analysis were pooled, stained, and analyzed using a 4-laser LSR-II® (BD Biosystems), setting the eFluor® 450 voltage threshold based on control C57BL/6 splenocytes labeled with Cell Proliferation dye eFluor® 450 (10uM) (eBioscience) and fixed on day 0.
Recipient CD11b+ cell accumulation after BMT
Day 6 single-cell suspensions from recipient spleen, MLN, and liver and intra-epithelial mononuclear cells from colon were isolated as described previously (9,11). Cells were stained and samples analyzed on a 4-laser BD LSR-II® instrument (BD Biosystems), and data analyzed using FlowJo® software (Treestar).
Cytokine profiling
CD11b+Gr-1highCD11cneg, CD11b+Gr-1intCD11cneg, and CD11b+Gr-1lowCD11c+ cells were sorted from gated H-2Kb-negB220neg at day 6 after BMT from pooled spleens of TLI/ATS conditioned WT recipients receiving WT C57BL/6 BMT. Cells were cultured in triplicate wells at a concentration of 4 × 104 cells/well in a 96 well plate in RPMI 1640® (Cellgro, Manassas, VA) supplemented with 10% fetal bovine serum (FBS, Atlanta Biological, Lawrenceville, GA); penicillin (100 U/ml), streptomycin (100 µg/ml), and L-glutamine (2 nM) (all from Hyclone, Logan, UT); and 2-mercaptoethanol (Sigma-Aldrich, St Louis, MO). Sorted populations were stimulated with 1 µg/ml E. coli lipopolysaccharide (LPS) (L26390, Sigma-Aldrich) for 72 hrs. Supernatant cytokine concentrations were analyzed using the mouse Milliplex™ MAP (Millipore). For assays of intracellular cytokine expression by FACS, the above sorted cell populations were stimulated for 12 hours with 1 ug/mL E. coli LPS with GolgiPlug™ (BD Biosciences) added after 7 h of culture. Cells were fixed, permeabilized (Fixation/Permeabilization kit, eBioscience) and stained with unlabeled rabbit iNOS (clone M-19, Santa Cruz Biotechnologies) and PE conjugated anti-rabbit IgG (Southern Biotech) and FITC conjugated TNF-α (clone MP6-XT22, BD Biosystems).
Light microscopy
Sorted CD11b+ population subsets were stained for morphological assessment using Protocol Hema 3 Giemsa Stain (Fisher Healthcare, Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s protocol. Photomicrographs were aquired with a 100 × Plan APO 1.4/NA lens and a Nikon DXM 1200 camera. Images were prepared using NIS Elements AR software (NIKON Instruments, Melville, NY).
In vivo Gr-1+ cell depletion
Recipient BALB/c mice were conditioned with TLI and ATS. Antibody clone RB6-8C5 (18) (BioXCell, West Lebanon, NH) or isotype negative control antibody (Rat IgG2b, BioXcell) was diluted in PBS to a final concentration of 200 µg/ml, and recipient mice injected intraperitoneally with 500 µl (100 µg/dose/mouse) on days −10, −8, −6, and −4 prior to BMT with WT C57BL/6 bone marrow cells (50 × 106) and spleen cells (60 × 106) injected via lateral tail vein on day 0. On day 6 after BMT, recipients were euthanized and tissue specimens harvested from the skin, liver, and terminal 1 cm of descending colon and hematoxylin/eosin stained sections scored for GVHD. The cumulative and colonic GVHD score represents the mean ± SEM in each experimental group.
In vitro proliferation assays
Responder splenocytes from C57BL/6 congenic (CD45.1+), Foxp3-IRES-mRFP (FIR), Foxp3GFP, or DNTGF-βRII × Foxp3-IRES-GFP C57BL/6 male mice were labeled with Cell Proliferaton dye eFluor® 450 (eBioscience) per manufacturer’s instructions. At day 6 after BMT, stimulator cells were sorted according to CD11b, Gr-1, and CD11c expression from pooled spleens of TLI/ATS conditioned WT BALB/c hosts receiving BMT from WT C57BL/6 donors. Single cell suspensions of splenocytes were enriched by CD11b positive selection (Cat #18770, StemCell™ Technologies, Vancouver, Canada), and the following populations sorted to > 97% purity: H-2Kb-negB220negCD11b+Gr-1high, H-2Kb-negB220negCD11b+Gr-1intCD11cneg and H-2Kb-negB220negCD11b+Gr-1lowCD11c+. Sorting was performed on a 4-laser FACSAria-II® (BD Biosystems). Responder cells (1 × 105) were cultured with each sorted population (1 ×105), in a U-bottom 96 well plate in 5% CO2 at 37° C. At 72 hours of co-culture, cells were pooled from 6 wells and stained for H-2Kb, CD4, and Foxp3 and analyzed by LSR-II® (BD Instruments). Voltage threshold was defined using eFluor®450-labeled responder splenocytes fixed at day 0 of incubation, and proliferation calculated using FlowJo® software (Treestar).
Inhibition of Arginase 1 (Arg1), inducible Nitric Oxide synthase (iNOS), and STAT3/5
In specific proliferation experiments, co-culture wells were treated with the Arg1 inhibitor, Nω-Hydroxy-nor-L-arginine (nor-NOHA, n-NOHA) (50 µM) or the iNOS inhibitor, NG-Monomethyl-L-arginine (L-NMMA) (5 µM) (both from EMD Biosciences, Darmstadt, Germany) or STAT3/5 blocking Jak2 inhibitor AG490 (25 µM) (Cayman Chemical, Ann Arbor, Michigan). All inhibitors were added at the beginning of cultures to both experimental co-cultures and control wells containing only responder cells.
In vitro antibody blocking assays
Responder cells were splenocyte single-cell suspensions from Foxp3-IRES-mRFP (FIR) C57BL/6 mice (1 × 105/well), and stimulator cells were sorted CD11b+Gr1lowCD11c+ cells (1 × 105/well) from WT BALB/c recipient spleens at day 6 after TLI/ATS and BMT from WT C57BL/6 donors. Anti-CD40 (clone FGK45.5, Miltenyi Biotec), or anti-PD-L1 (clone 10F.9G2, Biolegend) and anti-PD-L2 (clone TY25, Biolegend) blocking antibodes were added (5 µg/ml) to sorted CD11b+Gr1lowCD11c+ cells just prior to co-culture with responders. Rat IgG2a (5 µg/ml) (catalog #553926, BD Biosciences) was used as isotype control.
In vivo nTreg proliferation and accumulation following PD-L1 and PD-L2 blockade or CD11b+Gr-1lowCD11c+ cell adoptive transfer
WT BALB/c mice were treated with blocking antibodies against both PD-L1 (clone 10F.9G2, Biolegend) and PD-L2 (clone TY25, Biolegend) or isotype control antibodies (Rat IgG2a and Rat IgG2b, Biolegend) at 200 µg/dose/mouse in 400 uL PBS injected intraperitoneally on days −2, 0, +2 and +5. BMT consisted of WT C57BL/6 bone marrow cells (50 × 106) and CD45.1 congenic splenocytes (60 × 106) labeled with e450 proliferation dye (10uM) and infused IV via lateral tail vein on day 0. In separate experiments, iNKT-deficient Jα18−/− BALB/c recipients received TLI/ATS and adoptive transfer of 1 × 105 CD11b+Gr1lowCD11c+ cells sorted at day 6 from spleens of WT BALB/c recipients of TLI/ATS + BMT (details in Figure 3E). At day 6 after BMT, animals were euthanized and spleen, liver, colon, and MLN isolated and mononuclear cells prepared for FACS analysis. Voltage threshold for analysis was set using stained e450 dye-labeled donor splenocytes fixed at day 0, and proliferation was assessed on specific gated subsets using FlowJo version 9.4.10 (Treestar).
Figure 3. CD11b+Gr-1low recipient cells recovered at day 6 after TLI/ATS + BMT express CD11c and produce significant iNOS and TNF-α upon LPS stimulation.
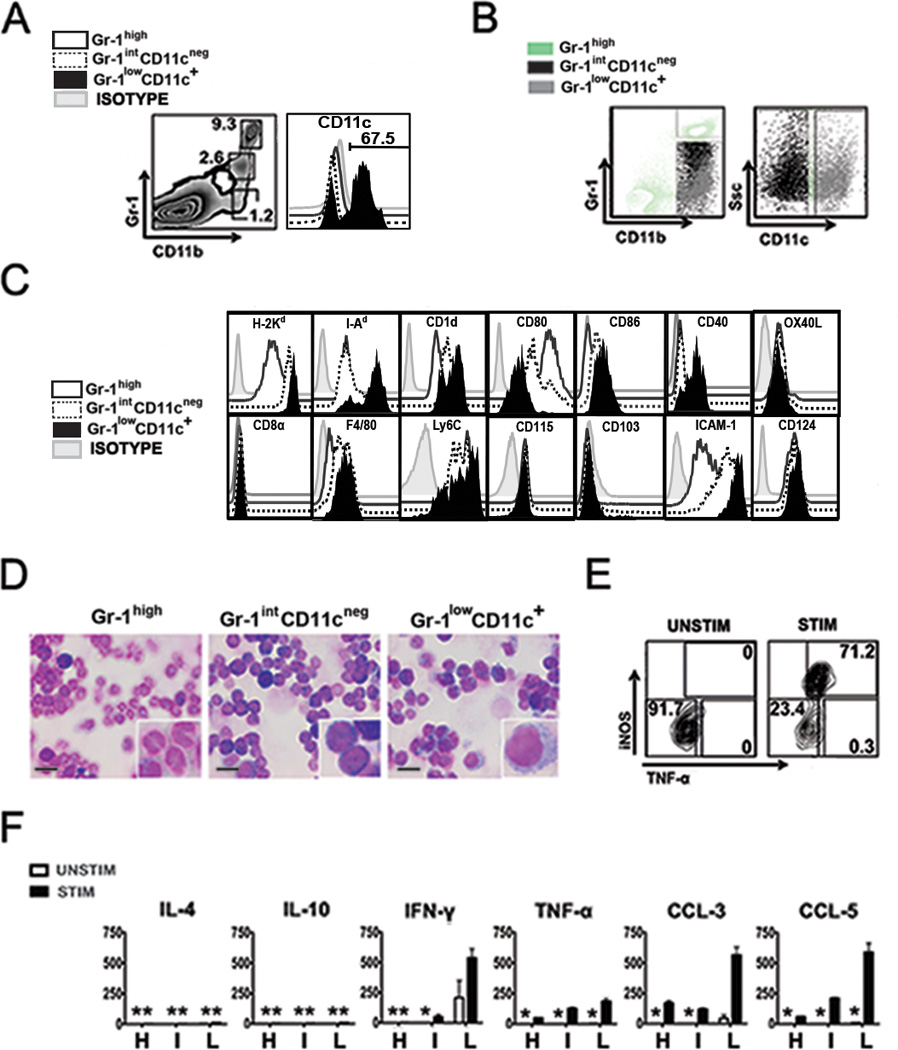
(A) Representative FACS plot (left panel) of CD11b and Gr-1 staining of gated H-2Kb-negB220neg cells from WT BALB/c recipient spleens at day 6 after TLI/ATS and infusion of BM + SPL from WT C57BL/6 donors. Right panel, CD11c expression on gated H-2Kb-negB220negCD11b+Gr-1high, H-2Kb-negB220negCD11b+Gr-1int, and H-2Kb-negB220negCD11b+Gr-1low populations. Percentage positive in the Gr-1lowCD11c+ population is shown above the gate. (B) Representative FACS plot (left panel) of CD11b and Gr-1 staining of gated H-2Kb-negB220neg cells following CD11b enrichment of splenocytes from WT BALB/c recipient spleens at day 6 after TLI/ATS and BM + SPL infusion from WT C57BL/6 donors, showing the gating strategy used to sort the Gr-1highCD11cneg population. Right panel, gating strategy used to sort Gr-1intCD11cneg and Gr-1lowCD11c+ subsets from the remaining CD11b+Gr-1int-low cells (C) FACS histograms of mean fluorescence intensity for each of the listed surface markers on recipient CD11b+ populations. Open histogram: isotype negative control; grey histogram: H-2Kb-negB220negCD11b+Gr-1highCD11cneg (Gr-1high); dashed histogram: H-2Kb-negB220negCD11b+Gr-1intCD11cneg (Gr-1intCD11cneg); black histogram: H-2Kb-negB220negCD11b+Gr-1lowCD11c+ (Gr-1lowCD11c+). (D) Representative photomicrographs (100×) with magnification (400×) showing morphology under light microscopy of FACS-sorted, Giemsa stained Gr-1high, Gr-1intCD11cneg, and Gr-1lowCD11c+ populations. Scalebar represents 10 um. (E) Representative FACS plot of intracellular iNOS and TNF-α staining of Gr-1lowCD11c+ cells sorted from spleens of recipient mice at 6 day after TLI/ATS + WT C57BL/6 BMT. Cells were cultured without (left panel) or with E. coli lipopolysaccharide (LPS) stimulation (right panel) for 12h, with the addition of Brefeldin A for the last 5 hours of culture. Representative of n = 3 experiments, n = 4 recipient mice per experiment. (F) Mean ± SEM concentration of cytokines (pg/mL) by 22-plex Luminex® assay of supernatants of day 6 sorted Gr-1high (H), Gr-1intCD11cneg (I), and Gr-1lowCD11c+ (L) cells. Means represent triplicate wells, n = 3 experiments, n = 3 mice per experiment. UNSTIM: without or STIM: with stimulation with LPS. *: mean < 20 pg/mL.
Statistical analysis
Statistical significance in survival between experimental groups was assessed using the log-rank test. Statistical significance in mean GVHD scores and mean absolute cell numbers between groups was assessed using the Mann-Whitney U test. For all tests, p < 0.05 was considered significant.
Results
TLI/ATS prevents donor CD8+ T cell-mediated acute GVHD after allogeneic BMT
WT BALB/c recipients of non-myeloablative TLI/ATS conditioning were protected from acute GVHD after transplantation of WT C57BL/6 bone marrow (50 × 106) and splenocytes (60 × 106) (BMT), while mice receiving 800 cGy (myeloablative) TBI + ATS (TBI 800/ATS) or control 400 cGy (non-myeloabative) TBI + ATS (TBI 400/ATS) developed lethal acute GVHD as demonstrated by survival curves (Supplementary Figure 1A), histopathologic GVHD scoring at day 6 after BMT (Supp. Fig. 1B), and mean weight curves (not shown). The doses of bone marrow and splenocytes were those utilized in prior murine studies (9,11), chosen to recapitulate the cumulative CD34+ hematopoietic stem cell dose and peripheral CD3+ T cell dose (2–3 × 108/kg) administered in clinical trials using TLI/ATG conditioning in adult patients with hematologic malignancies (5, 6). The difference in both cumulative and colonic GVHD scores was highly significant between either myeloablative or sub-myeloablative TBI/ATS and TLI/ATS groups (P < .001, TLI/ATS vs TBI 800/ATS; P < .001, TLI/ATS vs TBI 400/ATS). Representative photomicrographs of colon sections (200×) from each group and controls are shown in Supplementary Figure 1C.
Donor TCRαβ+CD8+ effector T cell accumulation is decreased and CD4+Foxp3+ Treg accumulation is increased in key GVHD target organs at day 6 after TLI/ATS + BMT as compared to TBI/ATS + BMT
TLI/ATS-conditioned recipients had dramatically reduced day 6 donor TCRαβ+CD8+ T cell accumulation in spleen, mesenteric lymph nodes (MLN), and colon when compared to either TBI 800/ATS or TBI 400/ATS groups (Figures 1A and 1B).
Figure 1. Non-myeloablative TLI/ATS but not TBI/ATS conditioning decreases donor TCRαβ+CD8+ effector T cell accumulation and enhances donor nTreg proliferation in a donor STAT6 independent fashion after MHC-mismatched allogeneic BMT.

(A) Representative FACS plots of CD4 and CD8 staining of gated H-2Kb+TCRαβ+ cells in spleen (top row), mesenteric lymph node (MLN) (middle row), and colon (bottom row) of recipients at day 6 after conditioning and transplantation. Percentage of cells in each gate is given above the gate. (B) Mean ± SEM absolute number (log 10) H-2Kb+TCRαβ+CD8+ cells in spleen (top panel), MLN (middle panel), and colon (bottom panel) of recipients at day 6 after conditioning and BMT. WT: wild-type; TBI: total body irradiation; TLI: total lymphoid irradiation; ATS: anti-thymocyte serum; BM: 50 × 106 WT C57BL/6 donor bone marrow cells; SPL: 60 × 106 WT C57BL/6 donor spleen cells; TBI800, TBI400: cGy cumulative doses of myeloablative (TBI800) or non-myeloablative (TBI400) TBI. (C) Representative FACS plots of CD4 and Foxp3 staining of gated H-2Kb+TCRαβ+CD4+ cells in spleen (top row) and MLN (bottom row) of recipients at day 6 after conditioning and BMT from WT C57BL/6 donors. Percentage of cells in each gate is given adjacent to the gate. TBI800, TBI400: cGy cumulative doses of TBI. (D) Mean ± SEM absolute number (log 10) H-2Kb+TCRαβ+CD4+Foxp3+ cells in spleen (top panel) and MLN (bottom panel) of recipients at day 6 after TBI 800/ATS, TBI 400/ATS, or TLI/ATS conditioning followed by BM + SPL infusion from WT C57BL/6 donors (n = 7 per group). (E) Representative proliferation histograms for H-2Kb+TCRαβ+CD4+Foxp3+ cells harvested from spleens of recipient mice of the groups indicated at day 6 after TLI/ATS conditioning and infusion of BM and SPL. SPL were labeled with proliferation dye eFluor™450 at day 0. Percent proliferation by eFluor™450 dye dilution is indicated above the bar within each histogram. Controls include eFluor™450 dye-labeled WT C57BL/6 BM + SPL fixed at day 0 (PRE-BMT) and splenocytes at day 6 from WT BALB/c recipients receiving TLI/ATS followed by infusion of BM + SPL from CD45-congenic (CD45.1+) BALB/c donors (SYN). (F) Representative FACS plots of Foxp3 expression in proliferating contours of gated H-2Kb+TCRαβ+CD4+Foxp3+ cells from spleens of WT hosts receiving WT and STAT6−/− donor BM + SPL. (G) Representative FACS plots of Helios expression on gated H-2Kb+CD4+Foxp3neg (left panel) and H-2Kb+CD4+Foxp3+ (right panel) cells at day 6 from spleens of WT hosts receiving TLI/ATS + WT C57BL/6 BMT. White open histogram: isotype control. Black filled histogram: anti-Helios. Percent positive is indicated above the gate. WT: wild-type host; TBI: total body irradiation; TLI: total lymphoid irradiation; ATS: anti-thymocyte serum; BM: 50 × 106 donor bone marrow cells; SPL: 60 × 106 donor spleen cells; 800 and 400 represent the cGy cumulative dose of TBI (cGy); e450: 450 nm wavelength emission proliferation dye.
Both the percentage amongst total donor CD4+ T cells (Figure 1C) and the absolute number (Figure 1D) of H-2Kb+TCRαβ+CD4+Foxp3+ donor Treg recovered from recipient spleen and MLN at day 6 were significantly increased after TLI/ATS + BMT versus after 800 cGy TBI/ATS or 400 cGy TBI/ATS conditioning. The same trend was seen for donor Treg accumulation in the recipient colon and liver at day 6 after BMT (data not shown).
Donor CD4+Foxp3+ nTreg proliferation after TLI/ATS + allogeneic BMT is not directly driven by recipient IL-4
Analysis of T cell subset proliferation at day 6 demonstrated no loss of Treg proliferation (Fig. 1E) and stable Foxp3 protein expression with ongoing Treg proliferation in vivo (Fig. 1F) and low donor CD8+ T effector cell proliferation (data not shown) when WT BALB/c recipients received TLI/ATS and BMT with 50 × 106 bone marrow cells and 60 × 106 eFluor® 450-labeled splenocytes from STAT6−/− C57BL/6 donors as compared to BMT from WT donors. Notably, there was no increase in GVHD at day 6 after BMT from STAT6−/− donors as compared to WT donor BMT (data not shown). The donor Tregs were confirmed to be naturally occurring thymically derived Treg (nTregs) by their uniform (>90% gated H-2Kb+CD4+Foxp3+ cells) expression of Helios (Figure 1G), a surface marker shown to differentiate thymically-derived murine nTregs from Foxp3+ Tregs induced in the periphery (iTregs) (19).
Since not only nTreg but all donor cells were IL-4 signaling incompetent in BMT from STAT6−/− donors, this data generated the hypothesis that recipient iNKT cell-derived IL-4 effects on donor nTreg proliferation are not direct, and that an IL-4 signaling-dependent recipient cell population is responsible for inducing donor nTreg expansion in vivo after TLI/ATS + BMT.
Three distinct subsets of recipient CD11b+ cells (Gr-1high, Gr-1int, and Gr-1low) are dominant in WT recipients early after TLI/ATS + BMT
After examining B- and T-lymphocyte, monocyte, macrophage, and dendritic cell (DC) sub-populations including plasmacytoid DC (pDC) and myeloid DC (mDC) in spleen, liver, MLN, and colon, we identified three H-2Kb-negB220negCD11b+ populations consistently increased at both days 0 pre-BMT (data not shown) and day 6 after BMT (Figure 2) in TLI/ATS as compared to TBI/ATS conditioned recipients: CD11b+Gr-1high, CD11b+Gr-1int and CD11b+Gr CD11b+Gr-1low. Figure 2A shows representative FACS histograms of gated H-2Kb-negB220neg cells in recipient spleens at day 6. Although these cells are rare in untreated WT BALB/c mice (Fig. 2A, left panel), their recovery is markedly increased both in relative fraction (Fig. 2A, right panel) and in absolute number in the spleen (Fig. 2B) in WT recipients of TLI/ATS versus 800 cGy or 400 cGy TBI/ATS + WT C57BL/6 donor BMT. Similar quantitative comparisons were obtained for MLN, liver, and colon (Fig. 2C).
Figure 2. TLI/ATS + BMT induces generation of distinct subsets of CD11b+ myeloid recipient populations in major GVHD target organs.
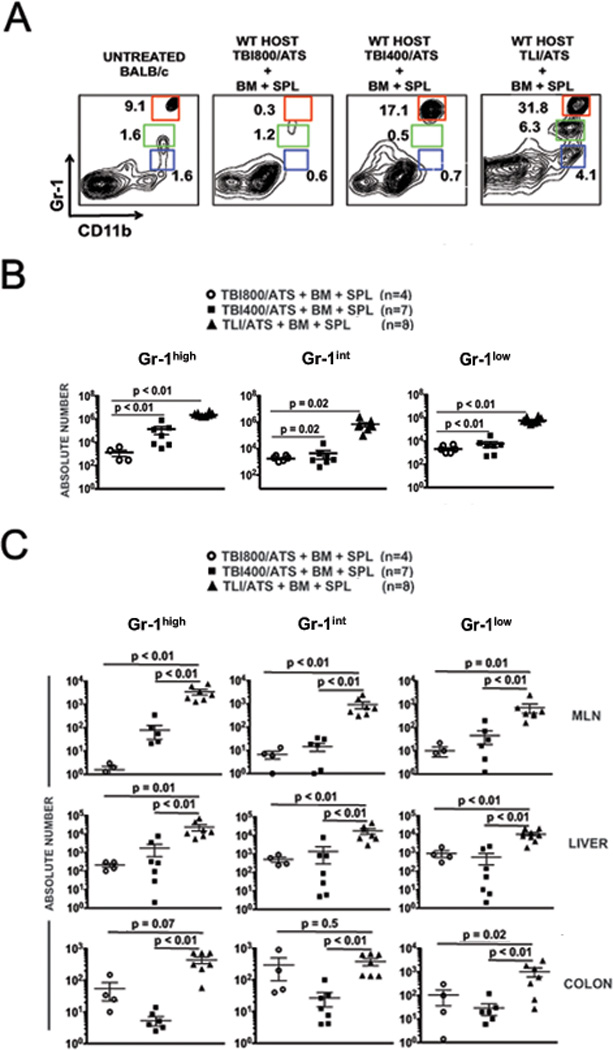
(A) Representative FACS plots of CD11b and Gr-1 staining of gated H-2Kb-negB220neg cells in spleens of recipients at day 6 after conditioning and BMT from WT C57BL/6 donors. Three distinct CD11b+ populations are indicated by colored gates. Percentage of cells is given adjacent to each gate. red gate: H-2Kb-negB220negCD11b+Gr-1high (Gr-1high); green gate: H-2Kb-negB220negCD11b+Gr-1int (Gr-1int); blue gate: H-2Kb-negB220negCD11b+Gr-1low (Gr-1low). (B) Mean ± SEM absolute number of Gr-1high (left panel), Gr-1int (middle panel), and Gr-1low (right panel) cells in spleen of recipients receiving TBI800/ATS (n = 4), TBI400/ATS (n = 7) or TLI/ATS (n = 8) followed by BM + SPL from WT C57BL/6 donors. (C) Mean ± SEM absolute number Gr-1high (left column panels), Gr-1int (middle column panels), and Gr-1low (right column panels) cells in MLN (top row), liver (middle row), and colon (bottom row) of WT BALB/c recipients from B. TBI: total body irradiation; TLI: total lymphoid irradiation; ATS: anti-thymocyte serum; BM: 50 × 106 WT C57BL/6 donor bone marrow cells; SPL: 60 × 106 WT C57BL/6 donor spleen cells; TBI800, TBI400: cGy cumulative doses of TBI.
Recipient B220negCD11b+Gr-1low cells can be specifically delineated and sorted by their expression of CD11c
At day 6 after TLI/ATS + BMT in recipient spleen, the gated H-2Kb-negB220negCD11b+Gr-1low but not the CD11b+Gr-1high or CD11b+Gr-1int subpopulation expressed CD11c (Fig. 3A). The mean ± SEM percentage CD11c+ cells among gated CD11b+Gr-1low cells was 70.0% ± 5.6% (range 60.3 – 79.9%; n = 5 experiments, n = 8 mice). After CD11b enrichment by MACS® selection, both the CD11b+Gr-1int and the CD11b+Gr-1low populations overlap in mean fluorescence intensity for Gr-1 expression by FACS analysis (Figure 4B, left panel). The CD11b+Gr-1low and the CD11b+Gr-1int populations could be differentiated based upon surface CD11c expression (Figure 3B, right panel), resulting in 3 distinguishable populations at sort: H-2Kb-negB220negCD11b+Gr-1highCD11cneg (Gr-1high), H-2Kb-negB220negCD11b+Gr-1intCD11cneg (Gr-1int) and H-2Kb-negB220negCD11b+Gr-1lowCD11c+ (Gr-1lowCD11c+).
Figure 4. Recipient Gr-1lowCD11c+ myeloid-derived immunomodulatory cells after TLI/ATS + BMT is dependent on both recipient STAT6 signaling and iNKT cells.

(A) Mean ± SEM absolute number (log 10) H-2Kb-negB220negCD11b+Gr-1highCD11cneg (Gr-1high) (left panel), H-2Kb-negB220negCD11b+Gr-1intCD11cneg (Gr-1intCD11cneg) (middle panel), and H-2Kb-negB220negCD11b+Gr-1lowCD11c+ cells (Gr-1lowCD11c+) (right panel) from WT (n = 5) and IL-4/IL-13 (Th2) signaling deficient STAT6−/− (n = 4) BALB/c recipient mice at day 6 after TLI/ATS and infusion of BM + SPL from WT C57BL/6 donors. (B) Mean ± SEM absolute number (log 10) Gr-1high (left panel), Gr-1intCD11cneg (middle panel), and Gr-1lowCD11c+ cells (right panel) from WT (n = 10) and iNKT-deficient Jα18−/− (n = 9) BALB/c recipient mice at day 6 after TLI/ATS and infusion of BM + SPL from WT C57BL/6 donors. (C) Mean ± SEM cumulative GVHD scores (left panel) and mean ± SEM colon GVHD scores (right panel) of WT, STAT6−/− (n = 15), and Jα18−/− BALB/c (n = 13) recipient mice at day 6 after TLI/ATS and WT C57BL/6 BMT. Data represent n = 4 experiments. (D) Representative photomicrographs of the hematoxylin/eosin-stained colon sections (200×) of recipient mice shown in C.
Recipient Gr-1lowCD11c+ cells express MHC Class I and II, CD1d, Ly6C, and IL-4Rα and have dendritic cell morphology
CD11b+ populations with variable Gr-1 expression and immunoregulatory properties have been described, including myeloid derived suppressor cells (MDSC) and regulatory dendritic cell subsets (20–25). We analyzed the three CD11b+ populations for surface markers associated with these and other regulatory myeloid cell subsets (Fig. 3C). All three populations expressed MHC class I (H-2d), but only the Gr-1lowCD11c+ population demonstrated significant MHC class II (I-Ad) expression. Surface expression of CD1d, the canonical MHC Class I-like antigen-presenting ligand for iNKT cells, was significantly increased on Gr-1lowCD11c+ as compared to the Gr-1high or Gr-1intCD11cneg cells. CD80 was highly expressed on Gr-1high cells, while CD86 expression was highest on the Gr-1lowCD11c+ population. CD40 but not OX40L was expressed on the Gr-1lowCD11c+ population. CD40 has previously been reported to mediate MDSC- and DC- induced contact dependent expansion of Foxp3+ nTreg via activation of CD40L (CD154) on nTreg (26, 27). Gr-1lowCD11c+ cells did not express CD8α but did express F4/80, supporting their derivation from macrophages. All three populations also expressed the M-CSF receptor (CD115), but lacked CD103 expression, indicating that these cells derive from monocyte progenitors and not lamina propria DCs (28). Ly6C specific antibody (clone AL-21, BD Biosciences) significantly stained all three CD11b+ subsets. ICAM-1 (CD54), a critical adhesion molecule (29), was highly expressed on the Gr-1intCD11cneg and Gr-1lowCD11c+ subsets. Notably, all three CD11b+ subsets uniformly expressed IL-4Rα (CD124).
By light microscopy, the Gr-1high, Gr-1int, and Gr-1lowCD11c+population demonstrated neutrophil, monocyte/macrophage, and dendritic cell morphology respectively (Figure 3D).
Recipient Gr-1lowCD11c+ cells at day 6 after TLI/ATS and BMT up-regulate intracellular iNOS and TNF-α after TLR4 stimulation, and secrete Th1 but not Th2 cytokines
Serbina et al (30) were the first to describe a population of Ly6C+CD11b+CD11c+ monocytes that egress from the bone marrow after L. monocytogenes infection and produce Tumor Necrosis Factor-α (TNF-α) and iNOS measurable by intracellular protein staining upon specific antigen re-stimulation. These cells, termed “TNF and iNOS producing DC” (Tip-DC) have since been described in other settings (31). When stimulated in vitro with LPS for 12 hrs with Brefeldin A, the Gr-1lowCD11c+ cells (Fig. 3E) but not the Gr-1high or Gr1intCD11cneg cells showed a dramatic increase in intracellular iNOS and TNF-α expression.
Gr-1high, Gr-1intCD11cneg, and Gr-1lowCD11c+ populations were sorted from gated H-2Kb-negB220neg cells among pooled recipient splenocytes prepared on day 6 after TLI/ATS + WT C57BL/6 BMT. The Gr-1intCD11cneg and Gr-1lowCD11c+ were differentially sorted as indicated in Figure 3B. TLR4 stimulation (E. coli LPS) was provided for 72 hrs prior to supernatant harvest, and supernatant cytokine and chemokine levels were assessed by 22-plex Luminex® assay. Gr-1high and Gr-1intCD11cneg populations secreted insignificant levels of all cytokines tested after LPS stimulation, whereas Gr-1lowCD11c+ cells stimulated with LPS secreted significant amounts of IFN-γ (500 pg/ml), CCL3 (500 pg/ml), and CCL5 (500 pg/ml), and TNF-α (200 pg/ml) (Fig. 3F). Notably, none of these subsets secreted IL-4, IL-10 (Figure 3F), IL-5, or IL-13 (data not shown). Thus, none of these CD11b+ populations secreted Th2 cytokines, which we and others have shown derive mainly from recipient Th2-polarized relatively radio-resistant invariant natural killer T (iNKT) cells enriched by TLI conditioning and required for graft-versus-host tolerance after TLI/ATS + BMT (7–11).
Recipient iNKT cells and STAT6 signaling are required for induction of Gr-1lowCD11c+ myeloid-derived immunomodulatory cells after TLI/ATS + BMT
As prior work has defined that IL-4 induced Th2 polarization and recipient iNKT cells are both critical to durable transplantation tolerance after non-myeloablative TLI/ATS and allogeneic BMT (7–11), we investigated whether the induction of these recipient CD11b+ subsets is Th2- or iNKT-dependent. We assessed the absolute number of cells of each induced CD11b+ population recovered from the colon of WT, STAT6−/−, and Jα18−/− BALB/c recipients of TLI/ATS + WT C57BL/6 BMT. In each mouse, the entire colon was isolated from 1 cm proximal to the anal verge to the ileocecal valve as previously described (9,11).
As shown in Figure 4A, there was a significant reduction in the Gr-1lowCD11c+ (P < .05) but no difference in the Gr-1high (P = 0.5) and Gr-1intCD11cneg (P = 1.0) subsets in STAT6−/− recipients as compared to WT recipients (Figure 4A), supporting a central role for Th2 (specifically IL-4 or IL-13) signaling in generating these regulatory DCs.
As in STAT6−/− recipients, the absolute number of Gr-1lowCD11c+ cells at day 6 was dramatically reduced in iNKT-deficient Jα18−/− BALB/c recipients of TLI/ATS + WT C57BL/6 donor BMT as compared to WT BALB/c recipients (Gr-1lowCD11c+: P < .01), indicating a requirement for recipient iNKT cells in the enhanced recovery of Gr-1lowCD11c+ cells (Fig. 4B).
Figures 4C and 4D show mean cumulative and colonic GVHD scores and representative photomicrographs of colon (200×) at day 6 after BMT for the 2 groups of knockout recipients of WT C57BL/6 BMT. Loss of GVHD protection was seen in both STAT6−/− and Jα18−/− as compared to WT BALB/c recipients of TLI/ATS + BMT (Figure 4C). There was no significant difference in GVHD severity between STAT6−/− and Jα18−/− BALB/c recipients.
Depletion of recipient Ly6+ cells results in loss of donor nTreg accumulation and development of acute GVHD after TLI/ATS + BMT
The antibody clone RB6-8C5 recognizes both Ly6C and Ly6G epitopes and has been used to deplete Ly6-expressing CD11b+ populations in vivo (18). Due to lack of suitable CD11b-diphtheria toxin receptor (DTR) or CD11c-DTR murine models on BALB/c background, we performed experiments using depletive antibody treatment of WT BALB/c recipients of TLI/ATS + BMT. At day 6, there was a dramatic and significant increase in histopathologic acute GVHD between recipients of RB6-8C5 depletive versus isotype control antibody (cumulative GVHD score, P < 0.01; colon GVHD score, P = 0.01) (Figure 5A). Figure 5B shows representative micrographs of colon sections (200×) obtained at day 6.
Figure 5. Depletion of recipient Gr-1+ cells before TLI/ATS + BMT results in acute GVHD.

(A) Mean ± SEM cumulative GVHD scores (left panel) and mean ± SEM colon GVHD scores (right panel) of recipient mice at day 6 after TLI/ATS and BMT treated with rat IgG2a isotype control antibody (n = 9) or the anti-Ly6C/Ly6G antibody RB6-8C5 (n = 5). (B) Representative photomicrographs of the hematoxylin/eosin-stained colon sections (200×) of recipient mice and treated with isotype control (top panel) or RB6-8C5 (bottom panel) during conditioning with TLI/ATS, followed by BMT from WT C57BL/6 donors. (C) Mean ± SEM percentage CD4+Foxp3+ nTreg amongst total gated H-2Kb+TCRαβ+CD4+ cells in colon of recipients at day 6 after TLI/ATS and treatment with isotype antibody (n = 9) or RB6-8C5 (n = 13). Data represent n = 4 separate experiments. (D) Representative FACS plots of Foxp3 and CD4 staining of gated H-2Kb+TCRαβ+CD4+ cells in colonic mononuclear cells isolated from recipients at day 6 from the experiments shown in Figure 3C. (E) Method of WT Gr-1lowCD11c+ cell adoptive transfer to iNKT-deficient Jα18−/− recipients of TLI/ATS. (1–3), 1 × 105 H-2Kb-negB220negCD11b+Gr-1lowCD11c+ cells were sorted from pooled spleens of WT BALB/c mice at day 6 following TLI/ATS + BMT and adoptively transferred (4) to Jα18−/− recipients of TLI/ATS (5) 4 hours prior to BMT consisting of e450-labeled CD45.1 C57BL/6 splenocytes and unlabeled WT C57BL/6 BM (6). Control Jα18−/− recipients of TLI/ATS received vehicle (PBS) prior to BMT. (7) At day 6, GVHD target organs were harvested and prepared for FACS and histopathologic analysis. Absolute numbers of cell subsets were calculated for individual mice. Percent proliferation and division index were calculated on analyses for n = 2–3 pooled spleens per experiment, n = 2–3 experiments. (F) Mean ± SEM absolute number (log 10) H-2Kb+CD4+Foxp3+ nTreg (top panel) and H-2Kb+TCRαβ+CD8+ effector cells (bottom panel) in spleens of Jα18−/− recipients shown in E receiving vehicle (n = 5) or sorted H-2Kb-negB220negCD11b+Gr-1lowCD11c+ cells (n = 5). Data represent n = 2–3 separate experiments. (G) Mean ± SEM absolute number (log 10) of gated H-2Kb+CD4+Foxp3+ nTreg (top panel) and H-2Kb+TCRαβ+CD8+ effector cells (bottom panel) in colons of Jα18−/− recipients shown in E. (H) Mean ± SEM percent proliferation of gated H-2Kb+CD4+Foxp3+ nTreg (top panel) and H-2Kb+TCRαβ+CD8+ effector cells (bottom panel) in spleens of Jα18−/− recipients shown in E. *: < 1% proliferation. (I) Mean ± SEM division index (DI) of gated H-2Kb+CD4+Foxp3+ nTreg (top panel) and H-2Kb+TCRαβ+CD8+ effector cells (bottom panel) in spleens of Jα18−/− recipients shown in E. *: DI < 0.1. WT: wild-type host; TBI: total body irradiation; TLI: total lymphoid irradiation; ATS: anti-thymocyte serum; BM: 50 × 106 donor bone marrow cells; SPL: 60 × 106 donor spleen cells; 800TBI, 400TBI: cumulative dose of TBI (cGy).
Notably, there was also a significant (P < 0.01) reduction in the percentage of donor CD4+Foxp3+ nTreg recovered amongst total donor CD4+ T cells in the colon at day 6 in RB6-8C5 depletive antibody-treated (mean ± SEM 4.7% ± 2.2%, n = 13) versus isotype control-treated recipients (mean ± SEM 13.4% ± 3.7%, n = 9) (Figure 5C). Figure 5D shows representative FACS plots of percentage Foxp3 expressing cells amongst gated H-2Kb+CD4+ cells from recipient spleen at day 6 in RB6-8C5 antibody- versus isotype antibody-treated recipients of TLI/ATS + BMT.
Adoptive transfer of Gr-1lowCD11c+ myeloid-derived immunomodulatory cells to iNKT-deficient Jα18−/− recipients induces donor nTreg accumulation and proliferation and loss of donor CD8 effector T cell accumulation after TLI/ATS + BMT
Figure 5E details the adoptive transfer strategy utilized to study the direct effect of Gr-1lowCD11c+ cells on nTreg and effector CD8+ T cell recovery in GVHD target organs in iNKT-deficient recipients of TLI/ATS + BMT. At day 6 following adoptive transfer, there was a dramatic increase in accumulation of donor CD4+Foxp3+ nTreg (P < 0.01) and a correlative significant decrease in effector CD8+ T cell accumulation (P < 0.05) in spleen (Figure 5F) and colon (P < 0.05) (Fig. 5G) of Jα18−/− BALB/c recipients of WT Gr-1lowCD11c+ myeloid-derived immunomodulatory cells as compared to vehicle treated controls. Notably, there was also a robust (P < 0.01) increase in donor CD4+Foxp3+ nTreg and concomitant reduction in donor CD8+ effector T cell proliferation (P < 0.01) (Fig. 5H) and calculated division index (DI) (P < 0.01) (Fig. 5I) in pooled spleens at day 6 in recipients of Gr-1lowCD11c+ cells as compared to recipients of vehicle control. This data confirms a mechanistic role for recipient Gr-1lowCD11c+ myeloid-derived immunomodulatory cells in linking iNKT cell-secreted IL-4 and MHC-mismatched donor nTreg proliferation and subsequent donor-recipient immune tolerance after TLI/ATS + BMT.
Gr-1lowCD11c+ recipient myeloid-derived immunomodulatory cells induce contact dependent donor-type nTreg but not CD4+Foxp3neg effector T cell proliferation in vitro
CD11b+ cell subsets sorted to > 98% purity based on Gr-1 and CD11c expression were co-cultured for 72 hours with eFluor® 450 proliferation dye-labeled Foxp3-IRES-mRFP (FIR) C57BL/6 splenocytes. Robust proliferation of gated H-2Kb+CD4+Foxp3+ splenic nTregs was observed in co-culture with sorted Gr-1lowCD11c+ cells (mean ± SEM proliferation 24.1% ± 4.5, n = 5), but not in co-cultures of responders with Gr-1high (mean ± SEM proliferation 4.6% ± 0.6, n = 6) or Gr-1intCD11cneg cells (mean ± SEM proliferation 7.8% ± 1.5, n = 6) (n = 6 experiments) (Figures 6A, top panel, and 6B). Proliferation in co-culture with Gr-1lowCD11c+ cells was restricted to the CD4+Foxp3+ subset and was not seen in gated CD4+Foxp3neg effector cells (Figure 6A, bottom panel), supporting that the induced proliferation is specific to nTreg rather than a pan-T cell stimulatory function of Gr-1lowCD11c+ cells. Of note, stability of expression of Foxp3 was seen with ongoing cycles of nTreg proliferation (Figure 6A, middle panels), suggesting that these cells may have a physiologic role in maintenance of nTreg in specific settings. Whereas the Gr-1high and Gr-1intCD11cneg populations did not induce nTreg proliferation in vitro as compared to responders alone (Gr-1high: P = 0.5 and Gr-1intCD11cneg: P = 0.3), co-culture with the Gr-1lowCD11c+ population of cells dramatically increased nTreg proliferation from either baseline nTreg culture or control nTreg co-culture with Gr-1intCD11cneg cells (P = 0.02) (Figure 6B). nTreg proliferation was abrogated when the responder and stimulator populations were separated in Transwell assays (Figure 6B) (mean ± SEM proliferation 5.2% ± 0.7, n = 5; P = 0.03), supporting dependence upon myeloid-derived immunomodulatory cell-nTreg direct contact for induction of nTreg proliferation. Of note, the induction of proliferation of nTregs in this setting is independent of MHC-TCR interactions, since nTregs in this assay derive from C57BL/6 (H-2b, I-Ab) and Gr-1lowCD11c+ cells from Class I- and Class II-mismatched BALB/c recipients (H-2d, I-Ad). Notably, when CD45.2+Gr-1lowCD11c+ cells were sorted at day 6 from spleens of CD45.2+ (WT) BALB/c recipients given TLI/ATS + BMT from syngeneic (CD45.1+ congenic) BALB/c donors, their capacity to induce FIR C57BL/6-derived Foxp3+ nTreg proliferation was maintained (data not shown), thus excluding the requirement for prior donor cell exposure in priming recipient Gr-1lowCD11c+ cells to induce donor-type nTreg proliferation across MHC barriers.
Figure 6. Recipient Gr-1lowCD11c+ myeloid-derived immunomodulatory cells induce PD-1 ligand dependent proliferation of donor nTregs in vitro and in vivo after TLI/ATS + BMT.
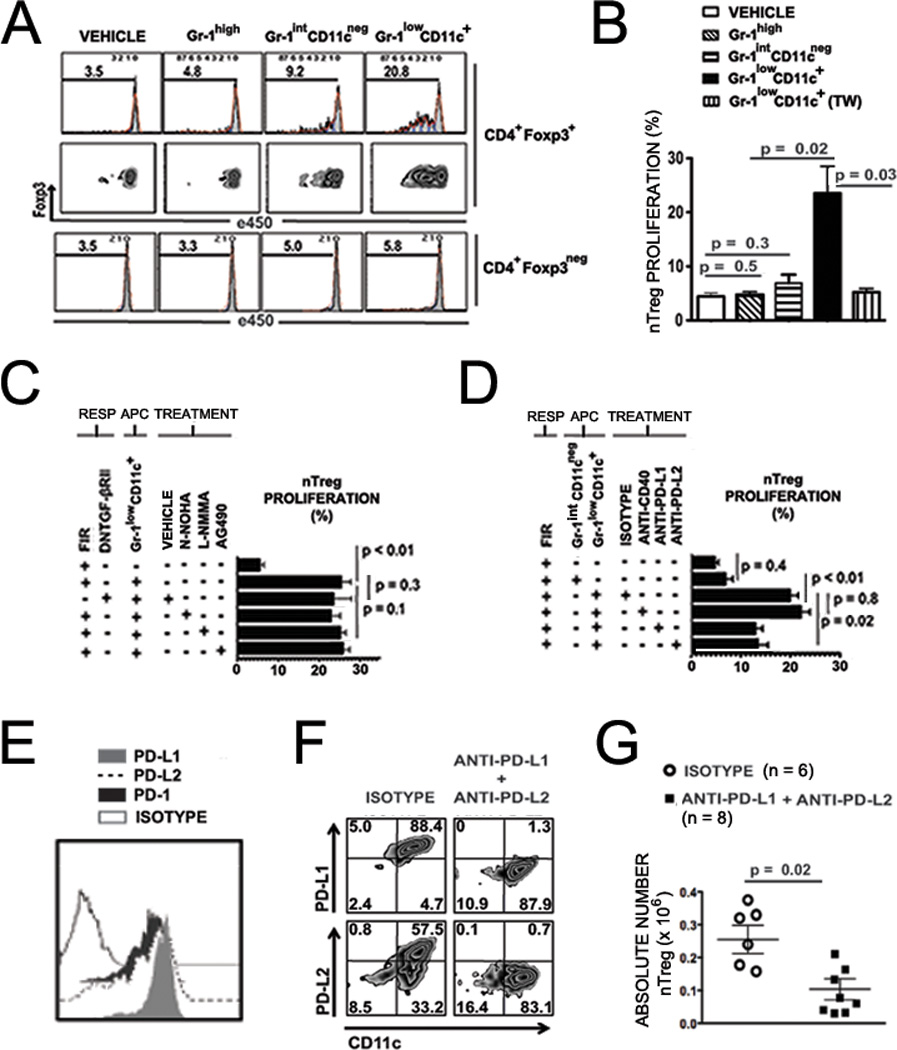
(A) Representative FACS proliferation histograms (top row) and dual axis FACS plots of e450 versus Foxp3 expression (middle row) for H-2Kb+CD4+Foxp3+ cells (CD4+Foxp3+), and representative eFluor®450 (e450) proliferation histograms (bottom row) for H-2Kb+CD4+Foxp3neg cells (CD4+Foxp3neg), both populations gated after 72-hour MLR using responder splenocytes from Foxp3-IRES-mRFP (FIR) C57BL/6 mice labeled with proliferation dye e450. Responders were cultured either alone (VEHICLE) or at 1:1 ratio with H-2Kb-negB220negCD11b+Gr-1highCD11cneg (Gr-1high), H-2Kb-negB220negCD11b+Gr-1intCD11cneg (Gr-1intCD11cneg), and H-2Kb-negB220negCD11b+Gr-1lowCD11c+ (Gr-1lowCD11c+) populations sorted from WT BALB/c hosts at day 6 after TLI/ATS and BMT from WT C57BL/6 donors. (B) Mean ± SEM percent proliferation of e450-labeled, gated H-2Kb+CD4+Foxp3-RFP+ nTreg at 72 hours following co-culture with sorted Gr-1high, Gr-1intCD11cneg, Gr-1lowCD11c+ cells in direct contact, or with recipient-derived Gr-1lowCD11c+ cells in a Transwell chamber (TW). Data represent mean ± SEM for n = 3–8 experiments, n = 6–9 mice per experiment. (C) Mean ± SEM percent proliferation of gated H-2Kb+CD4+Foxp3-RFP+ nTreg among e450-labeled responder splenocytes C57BL/6 mice at 72 hours following co-culture with Gr-1lowCD11c+ cells sorted at day 6 from WT BALB/c hosts given TLI/ATS + BMT from WT C57BL/6 donors. Co-cultures were performed in the presence of vehicle control (VEHICLE) or specific inhibitors, including Arginase 1 inhibitor Nω-Hydroxy-nor-L-arginine (N-NOHA) (50 µM), iNOS inhibitor NG-Monomethyl-L-arginine (L-NMMA) (5 µM) and the Jak2 inhibitor AG490 (25uM). Data represent mean ± SEM of n = 3–5 experiments, n = 4–6 mice per experiment. Responders included splenocytes from Foxp3-IRES-RFP C57BL/6 mice (FIR) and dominant-negative TGF-βRII×Foxp3GFP (DNTGF-βRII) C57BL/6 mice. (D) Mean ± SEM percent proliferation of gated H-2Kb+CD4+Foxp3+ nTreg at 72 hours following co-culture with sorted WT Gr-1lowCD11c+ cells with or without specific treatments. Data represent mean ± SEM for n = 3–6 experiments, n = 5–9 mice per experiment. Responders in all cases are e450-labeled splenocytes from FIR C57BL/6 mice. Treatments beginning at time 0 of co-cultures include rat IgG2a isotype control (ISOTYPE) (5 µg/mL), anti-CD40 (5 µg/ml), anti-PD-L1 (5 µg/ml), and anti-PD-L2 (µg/ml). (E) Representative FACS histograms of mean fluorescence intensity for surface expression of PD-L1, PD-L2 and PD-1 on H-2Kb-negB220negCD11b+Gr-1lowCD11c+ cells sorted from spleens of WT BALB/c recipients at day 6 after TLI/ATS + BMT from WT C57BL/6 donors. (F) Representative FACS plots of PD-L1 and PD-L2 staining using a clone distinct from blocking antibody, among gated H-2Kb-negB220negCD11b+Gr-1lowCD11c+ in spleens at day 6 after TLI/ATS and BMT, in recipients treated with isotype control (left panels) or antibodies blocking PD-L1 and PD-L2 (right panels) during TLI/ATS conditioning and continuing through day +5 post-BMT from WT C57BL/6 donors. (G) Mean ± SEM absolute number H-2Kb+TCRαβ+CD4+Foxp3+ cells in spleen of recipient mice at day 6 after TLI/ATS and BMT and treated with isotype control (ISOTYPE) or antibodies blocking PD-L1 and PD-L2 (anti-PD-L1 + anti-PD-L2). Data represent mean ± SEM for n = 3 experiments using n = 2–3 mice per experiment.
Gr-1lowCD11c+ myeloid-derived immunomodulatory cell-induced donor-type nTreg proliferation is TGF-β, Arg1, iNOS and STAT3/5 –signaling independent
To exclude a requirement for membrane-bound TGF-β in co-cultured Gr-1lowCD11c+ cells with eFluor® 450 proliferation dye labeled responder splenocytes from Foxp3-IRES-GFP (Foxp3GFP) C57BL/6 mice bred with homozygous transgenic C57BL/6 mice expressing a truncated TGFβ type II receptor. This receptor acts as a dominant-negative signaling receptor for TGF-β and serves as a durable model by which to study TGF-β signaling dependence in murine T cell subsets (32). Notably, nTreg proliferation was maintained when the responder cells were obtained from transgenic mice (Fig. 6C).
Expression of the enzyme Arginase 1 (Arg1) has been defined as a major STAT6-dependent regulatory pathway in CD11b+Gr-1+ MDSC (33), and was recently ascribed a role in allo-regulatory function and GVHD protection induced by donor-derived monocytoid MDSC under the influence of the Th2-polarizing cytokine IL-13 (34). MDSC regulation has also been associated with activation of inducible nitric oxide synthase (iNOS) (21). We blocked Arg1 activity in cultures of C57BL/6 CD4+Foxp3+ nTreg and CD11b+Gr-1lowCD11c+ cells using the Arg1 inhibitor, Nω-hydroxy-nor-L-arginine (nor-NOHA) and saw no effect on co-culture induced nTreg proliferation in vitro (Fig. 6C). Treatment of co-cultures with the iNOS inhibitor NG-Monomethyl-L-arginine, (L-NMMA) also did not alter nTreg proliferation induced by Gr-1lowCD11c+ cells (P = 0.1)(Fig. 6C). Granulocytoid MDSC have been shown to suppress T cell responses via reactive oxygen species (ROS), driven by STAT3 and STAT5 signaling (21, 35). We found no alteration in nTreg proliferation induced by Gr-1lowCD11c+ cells with the STAT3/STAT5/Jak2 inhibitor AG490 (35) (P = 0.1) (Fig. 6C), excluding this pathway of nTreg proliferation induction by Gr-1lowCD11c+ cells. Target inhibition by nor-NOHA, L-NMMA, and AG490 were confirmed by functional and phosphoflow assays (data not shown).
Gr-1lowCD11c+ myeloid-derived immunomodulatory cell-mediated induction of CD4+Foxp3+ nTreg proliferation requires PD-1 ligand signaling in vitro
CD40 co-stimulation of CD40L on nTregs has been shown to be critically important for regulatory DC- and MDSC-mediated nTreg expansion (27), and blockade of this axis augments T-effector responses in tumor-bearing mice (26). Gr-1lowCD11c+ cells induced potent proliferation of Foxp3+ nTregs in vitro independent of CD40 blockade (P = 0.8) (Fig. 6D). Programmed Death Ligand 1 and 2 (PD-L1 and PD-L2) signaling are two other previously described MHC-independent, contact-dependent mechanisms by which regulatory APCs including pDC can induce proliferation of nTreg (36–37). The induction of nTreg proliferation was significantly inhibited by addition of either PD-L1 or PD-L2 blocking antibodies (P = 0.02, anti-PD-L1 vs isotype; P = 0.02, anti-PD-L2 vs isotype) (Figure 6D).
Gr-1lowCD11c+ myeloid-derived immunomodulatory cells up-regulate PD-L1 in vivo after TLI/ATS + BMT
At day 6 in the spleens of WT TLI/ATS conditioned BALB/c recipients of WT C57BL/6 BMT, gated H-2Kb-negB220negCD11b+Gr-1lowCD11c+ cells showed significant expression of PD-1, PD-L1, and PD-L2 (Figure 6E). Gated splenic H-2Kb+CD4+Foxp3+ nTreg at day 6 after TLI/ATS + BMT expressed PD-1 and PD-L1 but insignificant levels of PD-L2 (data not shown).
Antibody blockade of PD-L1 and PD-L2 abrogates in vivo donor CD4+Foxp3+ nTreg expansion after TLI/ATS + BMT
Programmed cell death (PD-1) and its ligands (PD-L1 and PD-L2) deliver critical immunomodulatory signals to T cells, thus regulating the balance between T-cell activation and immune tolerance (36). PD-L1 in particular has been found expressed specifically on multiple subsets of tolerogenic DCs (36–40). At day 6 after TLI/ATS and BMT followed by treatment with blocking antibodies, less than 2% PD-L1 and PD-L2 could be detected by counter-staining with non-cross-reactive antibody clones (Fig. 6F, right panels) as compared to counter-staining in isotype antibody-treated control mice (Fig. 6F, left panels), confirming specific therapeutic blockade. Concomitant with this blockade, the absolute numbers of CD4+Foxp3+ nTregs recovered in the spleen at day 6 post-BMT in TLI/ATS and blocking antibody-treated recipients was significantly lower than in isotype control antibody-treated recipients (P = 0.02, mean ± SEM absolute number nTreg 2.6 × 105 ± 0.04 vs. 1 × 105 ± 0.03) (Figure 6G). Significant inhibition of nTreg recovery was not seen when mice were treated with anti-PD-L1 or anti-PD-L2 alone (data not shown; n = 5 mice per experiment, n = 3 experiments), suggesting an overlap of function of these two ligands on Gr-1lowCD11c+ myeloid-derived immunomodulatory cells in inducing nTreg proliferation. The cumulative data indicate that PD-L1 and PD-L2 signaling mediate recipient CD11b+Gr-1lowCD11c+ cell-driven in vivo donor nTreg proliferation after TLI/ATS + BMT.
Discussion
We have previously demonstrated that recipient Th2 polarized iNKT cells induced donor Foxp3+ nTreg cell expansion in vivo early after TLI/ATS and BMT, which results in robust protection against GVHD as compared to myeloablated (TBI800/ATS) controls (11). However, the underlying mechanism by which iNKT cell-derived IL-4 could drive donor nTreg cell proliferation remained unclear, and no data were previously available using non-myeloablated TBI controls. Here, we demonstrate that both recipient iNKT cells and recipient STAT6 signaling were indispensable for the generation of a subset of recipient regulatory APCs (B220negCD11b+Gr-1lowCD11c+) following non-myeloablative TLI/ATS + BMT but not non-myeloablative TBI/ATS + BMT (TBI400/ATS controls). These regulatory APCs induce proliferation of donor nTregs in a PD-1 ligand-dependent manner without requirement for MHC compatibility between donor and recipient. To investigate their role in maintenance of donor nTreg expansion and GVHD protection after TLI/ATS and BMT, we depleted them in vivo and found severe acute GVHD along with significant reduction in donor nTregs in key GVHD target organs. To confirm a cause-and-effect relationship, we adoptively transferred these cells into iNKT deficient Jα18−/− recipients following TLI/ATS but before BMT. We found protection from GVHD and potent induction of nTreg cell proliferation in multiple lymphoid organs of the adoptively transferred recipients as compared to vehicle-treated controls.
Myeloid-Derived Suppressor Cells (MDSCs) are known to suppress effector T cell responses (both allogeneic and syngeneic) via STAT6-dependent expression of Arg1. (21),(40),(41). Recently, Highfill et al also demonstrated that donor-type ex vivo expanded monocytoid MDSCs generated in vitro under the influence of IL-13 can regulate murine experimental GVHD in a C57BL/6 → BALB/c system following TBI-based conditioning, in an Arg1-dependent manner (34). This raised the intriguing possibility that the regulatory nTreg proliferation induced by B220negCD11b+Gr-1lowCD11c+ cells might be Arg1-dependent. Using both nor-NOHA inhibition of Arg1 with WT BALB/c recipient-derived B220negCD11b+Gr-1lowCD11c+ cells or Arg1-deficient B220negCD11b+Gr-1lowCD11c+ cells sorted from ARG1fl/fl × LysM-cre mice (data not shown), we confirmed that B220negCD11b+Gr-1lowCD11c+ cell- induced nTreg proliferation is not Arg1-dependent. Particularly of note since these cells express iNOS upon activation is the finding that selective iNOS inhibition with L-NMMA demonstrated that induction of nTreg proliferation by these APCs is iNOS/STAT1- independent. Cumulatively, this data demonstrates that canonical T cell regulatory pathways defined for MDSCs are not utilized by recipient B220negCD11b+Gr-1lowCD11c+ cells in inducing donor nTreg proliferation after TLI/ATS + BMT.
The regulatory APCs we found in our studies express F4/80, similar to IL-4/IL-13-induced alternatively activated macrophages with regulatory capacity (42). However, alternatively activated macrophages lack surface CD11c (43,44). Although plasmacytoid DC (pDC) have been shown to induce proliferation and/or induction of Foxp3+ nTreg as well as allo-tolerance to vascularized cardiac allografts (45–48), recipient Gr-1lowCD11c+ cells after TLI/ATS + BMT are B220neg, which excludes the possibility as pDC. A recent report describes a CD11c+ murine splenic DC subset capable of MHC class II-independent induction of nTreg proliferation (49). However, these regulatory splenic DC induced nTreg proliferation in a syngeneic system, with a clear requirement for paracrine IL-2 in maintenance of nTreg proliferation, in contrast to the PD-ligand-dependent proliferation of nTreg after MHC Class I and II mismatched allogeneic BMT which we demonstrated here. Consequently, based on phenotypic assessment from our studies and those in the existing literature, recipient B220negCD11b+Gr-1lowCD11c+ cells expanded after TLI/ATS + BMT appear to represent a regulatory monocytoid DC population expressing both CD11b and Gr-1 and secreting TNF-α and iNOS upon activation. This most closely matches the immunophenotype of Tip-DCs (30–31). However, there has been significant debate as to whether Tip-DC represent a distinct DC subset or a myeloid-derived subset with TNF-α and iNOS secretion activated under specific conditions of infectious re-stimulation.. Moreover, the cell population we describe is clearly Gr-1low and Ly6C isoform-expressing, suggesting a divergence from previously described Tip-DCs. Since these cells express iNOS and TNF-α in vitro upon stimulation with the TLR4 agonist LPS (like myeloid immunomodulatory populations including MDSC) but also express CD11c (like Tip-DCs and other immunomodulatory DC subsets) we opted to term refer to them int his manuscript as myeloid-derived immunomodulatory cells.
This is the first data suggesting that myeloid-derived immunomodulatory cells can be generated under the influence of iNKT cells and Th2 polarizing (TLI) but not Th1-polarizing (TBI) non-myeloablative conditioning in a STAT6-dependent manner, and to our knowledge the first description of myeloid-derived cells of the described immunophenotype inducing either nTreg proliferation or immune tolerance in allo-transplantation. The possibility that these B220negCD11b+Gr-1lowCD11c+ cells represent immunomodulatory DC precursors generated from bone marrow myeloid precursors spared in the setting of non-myeloablative lymphoid radiation, the mechanisms of their generation, and why this population is not enriched following non-myeloablative TBI are areas under active investigation.
In summary, B220negCD11b+Gr-1lowCD11c+ myeloid-derived immunomodulatory cellsare regulatory DCs expanded preferentially after non-myeloablative, Th2-polarizing TLI/ATS conditioning but not after non-Th2 polarizing conditioning including non-myeloablative or myeloablative TBI/ATS. We propose that the Th2- and NKT-dependent preservation and enrichment of myeloid-derived immunomodulatory cells after TLI/ATS conditioning explains how recipient Th2-polarizing NKT cells induce the in vivo expansion of donor-type nTreg, which we have previously shown regulates CD8-mediated GVHD and maintains long-term donor-recipient immune tolerance after TLI/ATS + BMT (Figure 7). These data shed critical light on the donor-recipient immunoregulatory networks critical to enhanced development of durable donor-recipient immune tolerance after non-myeloablative TLI/ATS conditioning and BMT as compared to BMT following TBI-based regimens.
Figure 7. Proposed recipient-donor immunoregulatory network regulating GVHD after non-myeloablative TLI/ATS + BMT.

IL-4 (and possibly other growth factors) secreted by recipient invariant natural killer T cells (iNKT CELLS) (blue arrow and text) drives expansion of recipient regulatory Gr-1lowCD11c+ dendritic cells (Gr-1lowCD11c+ REGULATORY DC). Recipient Gr-1lowCD11c+ regulatory DCs generated (likely from radio-protected bone marrow) in the non-myeloablative Th2 polarizing environment of TLI/ATS conditioning induces PD-1/PD-1 ligand contact-dependent, MHC-independent proliferation and in vivo expansion of naturally occurring donor CD4+Foxp3+ regulatory T cells (FOXP3+ nTREG). Donor nTreg then regulate donor effector T cell-mediated GVHD (GVHD EFFECTOR T CELL) (red arrow). Notably, the lack of GVHD T effector accumulation or histopathologic GVHD using STAT6−/− donor BMT after TLI/ATS conditioning of WT BALB/c recipients excludes previously postulated direct role for IL-4-mediated inhibition of GVHD effector T cells in GVHD protection after TLI/ATS + BMT.
Ongoing studies include characterization of molecular mechanisms by which regulatory myeloid-derived immunomodulatory cells may differentially expand under the influence of iNKT cells and Th2-polarizing cytokines, how PD-1/PD-ligand interaction drives Foxp3+ nTreg proliferation in the MHC-mismatched setting, and determination of whether B220negCD11b+Gr-1lowCD11c+ cells play a role in tumor-associated immune suppression via induction of proliferation of autologous nTreg. These studies are expected to delineate targetable pathways of innate immune cell-driven regulation that can be either inhibited to augment cancer immunotherapy or augmented to optimize allogeneic transplant tolerance.
Supplementary Material
Acknowledgments
We thank J. Houston, L. He, S. Schwemberger, S. Perry, and R. Ashmun for cell sorting, Animal Resource Center staff, Drs. V. Frohlich and J. Peters for image acquisition, D. Umetsu (Harvard) for Jα18−/− breeders, P. Murray (St. Jude) for a LysM-cre breeder and primer information, and H. Chi (St. Jude) and D. Flavell (Yale) for Foxp3GFP × DNTGF-βRII breeders. We thank M. Sommers, S. Woolard, V. Morales-Tirado, and W. Luszczek for technical assistance, and Drs. E. Pamer (MSKCC), N. Chao (Duke), and D. Zeng (City of Hope) for their constructive critiques of the manuscript.
Footnotes
Funding support: Sumara Endowed Fellowship in Cellular and Gene Therapy (MvDM), the Lemuel Diggs Endowed Fellowship (HAA), V Foundation for Cancer Research Scholar Award (ABP), grant #1K08-HL088260-05 (NHLBI) (ABP), and the American Lebanese Syrian Association Charities (ALSAC).
Author Contributions: MV and HA designed and performed experiments, analyzed data, and wrote the paper; AS and TO performed experiments; PV reviewed data and wrote the paper; AP conceived and designed the research, performed experiments, analyzed data, and wrote the paper.
Disclosures The authors declare no competing financial interests.
References
- 1.Shlomchik WD. Graft-versus-host disease. Nat Rev Immunol. 2007;7:340–352. doi: 10.1038/nri2000. [DOI] [PubMed] [Google Scholar]
- 2.Welniak LA, Blazar BR, Murphy WJ. Immunobiology of allogeneic hematopoietic stem cell transplantation. Annu Rev Immunol. 2007;25:139–170. doi: 10.1146/annurev.immunol.25.022106.141606. [DOI] [PubMed] [Google Scholar]
- 3.Storb RF, Champlin R, Riddell SR, Murata M, Bryant S, Warren EH. Non-myeloablative transplants for malignant disease. Hematology Am Soc Hematol Educ Program. 2001:375–391. doi: 10.1182/asheducation-2001.1.375. [DOI] [PubMed] [Google Scholar]
- 4.Pillai A, Hartford C, Wang C, Pei D, Yang J, Srinivasan A, Triplett B, Dallas M, Leung W. Favorable preliminary results using TLI/ATG-based immunomodulatory conditioning for matched unrelated donor allogeneic hematopoietic stem cell transplantation in pediatric severe aplastic anemia. Pediatr Transplant. 2011;15:628–634. doi: 10.1111/j.1399-3046.2011.01542.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lowsky R, Takahashi T, Liu YP, Dejbakhsh-Jones S, Grumet FC, Shizuru JA, Laport GG, Stockerl-Goldstein KE, Johnston LJ, Hoppe RT, Bloch DA, Blume KG, Negrin RS, Strober S. Protective conditioning for acute graft-versus-host disease. N Engl J Med. 2005;353:1321–1331. doi: 10.1056/NEJMoa050642. [DOI] [PubMed] [Google Scholar]
- 6.Kohrt HE, Turnbull BB, Heydari K, Shizuru J, Laport G, Miklos D, Johnston L, Arai S, Weng WK, Hoppe RT, Lavori P, Blume KG, Negrin RS, Strober S, Lowsky R. TLI and ATG conditioning with low risk of graft-versus-host disease retains antitumor reactions after allogeneic hematopoietic cell transplantation from related and unrelated donors. Blood. 2009;114(5):1099–1109. doi: 10.1182/blood-2009-03-211441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lan F, Zeng D, Higuchi M, Higgins JP, Strober S. Host conditioning with total lymphoid irradiation and antithymocyte globulin prevents graft-versus-host disease: the role of CD1-reactive natural killer T cells. Biol Blood Marrow Transplant. 2003;9:355–363. doi: 10.1016/s1083-8791(03)00108-3. [DOI] [PubMed] [Google Scholar]
- 8.Rigby SM, Rouse T, Field EH. Total lymphoid irradiation nonmyeloablative preconditioning enriches for IL-4-producing CD4+-TNK cells and skews differentiation of immunocompetent donor CD4+ cells. Blood. 2003;101:2024–2032. doi: 10.1182/blood-2002-05-1513. [DOI] [PubMed] [Google Scholar]
- 9.Pillai AB, George TI, Dutt S, Teo P, Strober S. Host NKT cells can prevent graft-versus-host disease and permit graft antitumor activity after bone marrow transplantation. J Immunol. 2007;178:6242–6251. doi: 10.4049/jimmunol.178.10.6242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yao Z, Liu Y, Jones J, Strober S. Differences in bcl-2 expression by T-cell subsets alter their balance after in vivo radiation to favor CD4+Bcl-2hi NKT cells. Eur J Immuno. 2009;l(39):763–775. doi: 10.1002/eji.200838657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pillai AB, George TI, Dutt S, Strober S. Host natural killer T cells induce an interleukin-4-dependent expansion of donor CD4+CD25+Foxp3+ T regulatory cells that protects against graft-versus-host disease. Blood. 2009;113:4458–4467. doi: 10.1182/blood-2008-06-165506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mantel PY, Kuipers H, Boyman O, Rhyner C, Ouaked N, Ruckert B, Karagiannidis C, Lambrecht BN, Hendriks RW, Crameri R, Akdis CA, Blaser K, Schmidt-Weber CB. GATA3-driven Th2 responses inhibit TGF-beta1-induced FOXP3 expression and the formation of regulatory T cells. PLoS Biol. 2007;5:e329. doi: 10.1371/journal.pbio.0050329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prochazkova J, Fric J, Pokorna K, Neuwirth A, Krulova M, Zajicova A, Holan V. Distinct regulatory roles of transforming growth factor-beta and interleukin-4 in the development and maintenance of natural and induced CD4+ CD25+ Foxp3+ regulatory T cells. Immunology. 2009;128:e670–e678. doi: 10.1111/j.1365-2567.2009.03060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Su MA, Wan YY. An essential role of the transcription factor GATA-3 for the function of regulatory T cells. Immunity. 2011;35:337–348. doi: 10.1016/j.immuni.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campbell DJ. Regulatory T cells GATA have it. Immunity. 2011;35:313–315. doi: 10.1016/j.immuni.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 16.Akbari O, Stock P, Meyer E, Kronenberg M, Sidobre S, Nakayama T, Taniguchi M, Grusby MJ, DeKruyff RH, Umetsu DT. Essential role of NKT cells producing IL-4 and IL-13 in the development of allergen-induced airway hyperreactivity. Nat Med. 2003;9:582–588. doi: 10.1038/nm851. [DOI] [PubMed] [Google Scholar]
- 17.Higuchi M, Zeng D, Shizuru J, Gworek J, Dejbakhsh-Jones S, Taniguchi M, Strober S. Immune tolerance to combined organ and bone marrow transplants after fractionated lymphoid irradiation involves regulatory NK T cells and clonal deletion. J Immunol. 2002;169:5564–5570. doi: 10.4049/jimmunol.169.10.5564. [DOI] [PubMed] [Google Scholar]
- 18.Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol. 2008;83:64–70. doi: 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- 19.Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, Shevach EM. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol. 2010;184:3433–3441. doi: 10.4049/jimmunol.0904028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–5802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. 2010;22:238–244. doi: 10.1016/j.coi.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 23.Gehrie E, Van der Touw W, Bromberg JS, Ochando JC. Plasmacytoid dendritic cells in tolerance. Methods Mol Biol. 2011;677:127–147. doi: 10.1007/978-1-60761-869-0_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Segura E, Wong J, Villadangos JA. Cutting edge: B220+CCR9- dendritic cells are not plasmacytoid dendritic cells but are precursors of conventional dendritic cells. J Immunol. 2009;183:1514–1517. doi: 10.4049/jimmunol.0901524. [DOI] [PubMed] [Google Scholar]
- 25.Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pan PY, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, Divino CM, Chen SH. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010;70:99–108. doi: 10.1158/0008-5472.CAN-09-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guiducci C, Valzasina B, Dislich H, Colombo MP. CD40/CD40L interaction regulates CD4+CD25+ T reg homeostasis through dendritic cell-produced IL-2. Eur J Immunol. 2005;35:557–567. doi: 10.1002/eji.200425810. [DOI] [PubMed] [Google Scholar]
- 28.Varol C, Vallon-Eberhard A, Elinav E, Aychek T, Shapira Y, Luche H, Fehling HJ, Hardt WD, Shakhar G, Jung S. Intestinal lamina propria dendritic cell subsets have different origin and functions. Immunity. 2009;31:502–512. doi: 10.1016/j.immuni.2009.06.025. [DOI] [PubMed] [Google Scholar]
- 29.Herman S, Krenbek D, Klimas M, Bonelli M, Steiner CW, Pietschmann P, Smolen JS, Scheinecker C. Regulatory T cells form stable and long-lasting cell clusters with myeloid dendritic cells (DC) Int Immunol. 2012;24:417–426. doi: 10.1093/intimm/dxs039. [DOI] [PubMed] [Google Scholar]
- 30.Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 2003;19:59–70. doi: 10.1016/s1074-7613(03)00171-7. [DOI] [PubMed] [Google Scholar]
- 31.Bosschaerts T, Guilliams M, Stijlemans B, Morias Y, Engel D, Tacke F, Herin M, De Baetselier P, Beschin A. Tip-DC development during parasitic infection is regulated by IL-10 and requires CCL2/CCR2, IFN-gamma and MyD88 signaling. PLoS Pathog. 2010;6:e1001045. doi: 10.1371/journal.ppat.1001045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gorelik L, Flavell RA. Abrogation of TGF-beta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–181. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- 33.Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 2008;68:5439–5449. doi: 10.1158/0008-5472.CAN-07-6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, Taylor PA, Panoskaltsis-Mortari A, Serody JS, Munn DH, Tolar J, Ochoa AC, Blazar BR. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood. 2010;116:5738–5747. doi: 10.1182/blood-2010-06-287839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang C, Yang G, Jiang T, Huang K, Cao J, Qiu Z. Effects of IL-6 and AG490 on regulation of Stat3 signaling pathway and invasion of human pancreatic cancer cells in vitro. J Exp Clin Cancer Res. 2010;29:51. doi: 10.1186/1756-9966-29-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yi T, Li X, Yao S, Wang L, Chen Y, Zhao D, Johnston HF, Young JS, Liu H, Todorov I, Forman SJ, Chen L, Zeng D. Host APCs augment in vivo expansion of donor natural regulatory T cells via B7H1/B7.1 in allogeneic recipients. J Immunol. 2011;186:2739–2749. doi: 10.4049/jimmunol.1002939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fukaya T, Takagi H, Sato Y, Sato K, Eizumi K, Taya H, Shin T, Chen L, Dong C, Azuma M, Yagita H, Malissen B, Sato K. Crucial roles of B7-H1 and B7-DC on expressed mesenteric lymph node dendritic cells in the generation of antigen-specific CD4+Foxp3+ regulatory T cells in the establishment of oral tolerance. Blood. 2010;116:2266–2276. doi: 10.1182/blood-2009-10-250472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wolfle SJ, Strebovsky J, Bartz H, Sahr A, Arnold C, Kaiser C, Dalpke AH, Heeg K. PD-L1 expression on tolerogenic APCs is controlled by STAT-3. Eur J Immunol. 2011;41:413–424. doi: 10.1002/eji.201040979. [DOI] [PubMed] [Google Scholar]
- 40.Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. 2010;22:238–244. doi: 10.1016/j.coi.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 41.Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5:641–654. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- 42.Karp CL, Murray PJ. Non-canonical alternatives: What a macrophage is 4. J Exp Med. 2012;209:427–431. doi: 10.1084/jem.20120295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hume DA. Macrophages as APC and the dendritic cell myth. J Immunol. 2008;181:5829–5835. doi: 10.4049/jimmunol.181.9.5829. [DOI] [PubMed] [Google Scholar]
- 45.Ouabed A, Hubert HX, Chabannes D, Gautreau L, Heslan M, Josien R. Differential control of T regulatory cell proliferation and suppressive activity by mature plasmacytoid dendritic cells. J Immunol. 2008;180:5862–5870. doi: 10.4049/jimmunol.180.9.5862. [DOI] [PubMed] [Google Scholar]
- 46.Kuwana M. Induction of anergic and regulatory T cells by plasmacytoid dendritic cells and other dendritic cell subsets. Hum Immunol. 2002;63:1156–1163. doi: 10.1016/s0198-8859(02)00754-1. [DOI] [PubMed] [Google Scholar]
- 47.Mahnke K, Enk AH. Dendritic cells: key cells for the induction of regulatory T cells? Curr Top Microbiol Immunol. 2005;293:133–150. doi: 10.1007/3-540-27702-1_7. [DOI] [PubMed] [Google Scholar]
- 48.Ochando JC, Homma C, Yang Y, Hidalgo A, Garin A, Tacke F, Angeli V, Li Y, Boros P, Ding Y, Jessberger R, Trinchieri G, Lira S, Randolph G, Bromberg J. Alloantigen-presenting plasmacytoid dendritic cells mediate tolerance to vascularized grafts. Nature Immunol. 2006;7:652–662. doi: 10.1038/ni1333. [DOI] [PubMed] [Google Scholar]
- 49.Zou T, Caton AJ, Koretzky GA, Kambayashi T. Dendritic cells induce regulatory T cell proliferation through antigen-dependent and -independent interactions. J Immunol. 2010;185:2790–2799. doi: 10.4049/jimmunol.0903740. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.