

Abstract
Hypochlorous acid (HOCl) is generated by myeloperoxidase (MPO), using chloride and hydrogen peroxide as substrate. HOCl and its conjugate base (OCl−) bind to the heme moiety of hemoglobin (Hb) and generate a transient ferric species whose formation and decay kinetics indicate it can participate in protein aggregation and heme destruction along with subsequent free iron release. The oxidation of Hb heme moiety by OCl− was accompanied by marked heme destruction as judged by the decrease and subsequent flattening of the Soret absorbance peak at 405 nm. HOCl-mediated Hb heme depletion was confirmed by HPLC analysis and in-gel heme staining. Exposure of Hb to increasing concentrations of HOCl produced a number of porphyrin degradation products resulting from oxidative cleavage of one or more of the carbon-methene bridges of the tetrapyrrole ring, as identified by their characteristic HPLC fluorescence and LC-MS. A non-reducing denaturing SDS PAGE showed several degrees of protein aggregation. Similar, porphyrin degradation products were identified after exposure of red blood cells to increasing concentration of HOCl indicating biological relevance of this finding. This work provides a direct link between Hb heme destruction and subsequent free iron accumulation, as occurs under inflammatory conditions where HOCl is formed in substantial amounts.
Keywords: Free iron, hypochlorous acid, hemoglobin, inflammation, mammalian peroxidase, RBC, oxidative stress
INTRODUCTION
Red blood cells (RBCs) are the most common cells in the blood which derive their red color from hemoglobin (Hb), a protein that makes up about 92% of the RBC's dry content [1, 2]. Hemoglobin is made up of a tetramer consisting of four protein globin subunits and four heme prosthetic groups [2]. The main function of Hb is to deliver oxygen from lung to peripheral tissue for cellular metabolism and recycle carbondioxide from tissue to the lung [3]. Hemoglobin displays high oxygen affinity with carrying capacity ranging from 1.36 to 1.37 mL O2 per gram of Hb [4].
Within the body, Hb exists mainly in two ferrous forms: oxygenated (oxy-Hb; Hb-Fe(II)-O2) and deoxygenated (Hb-Fe(II). Major interest has been focused on Hb heme destruction and the formation of other higher oxidative states such as a ferryl porphyrin radical cation Hb-Fe(IV)=O+Π● (Compound I) and Hb-Fe(IV)=O (Compound II), and their possible role in the development of several pathophysiological processes [5–7]. Compound I has two oxidizing equivalents above Hb-Fe(III) while Compound II has one oxidizing equivalent above Hb-Fe(III). Most of the heme degradation in biological systems occurs in two different pathways: an enzymatic pathway that requires the heme oxygenase system [5]; or a nonenzymatic pathway that requires the interaction with reactive oxygen species (ROS), reducing agents, or xenobiotics (as reviewed in [5]). In the enzymatic pathway, heme oxygynase catalyzes heme cleavage and subsequently releases the heme iron in the ferrous form, and in a specific manner it eliminates the carbon-methene bridge of heme as CO to form bilivedrin [5, 8]. In the non-enzymatic pathways, ROS such as superoxide (O2−), hydrogen peroxide (H2O2), and hypochlorous acid (HOCl) can mediate heme destruction unselectively at any position of the heme double bonds [5, 9]. Several fluorescence and non-fluorescence heme degradation products have been identified using NMR and/or mass spectrometry (MS) techniques [10, 11]. Furthermore, several studies have demonstrated the ability of OCl− not only to bind to the heme moiety of several hemoproteins, but also cause heme destruction [12], protein modification and protein aggregation [13], as well as mediate the formation of peroxidase-like activity (formation of Compounds I and II) [14]. For example, Chapman et al. have shown that a strong non-covalent protein aggregation occurs when apohemoglobin and apomyoglobin was treated with HOCl [13]. Indeed exposure of RBCs to HOCl causes protein aggregation and as a result, the lysis of these cells [15]. Protein aggregation may also disrupt normal tissue organization when fibronectin is oxidized at sites of inflammation [16].
Hemoprotein heme destruction is extremely toxic to different organs and cells leading to serious pathological consequences [17]. The cellular toxicity is mainly due to the generation of free iron, which displays the capacity to participate in the further generation of ROS that mediates cellular mitochondria poisoning, lipid peroxidation, and uncoupling of oxidative phosphorylation [17–20]. Free iron can damage blood vessels and produce vasodilation with increased vascular permeability, leading to hypotension and metabolic acidosis [21, 22].
Hypochlorous acid is generated enzymatically by myeloperoxidase (MPO), which uses H2O2 to catalyze the two-electron oxidation of chloride (Cl−) [23]. Hypochlorous acid and its conjugate base (OCl−) are potent oxidants that function as powerful antimicrobial agents [24]. The normal plasma MPO concentration ranges from 18–39 ng/mL [25, 26], but it could be increased to 55 ng/mL [26] and 287 ng/mL [27] in myocardial infarction and acute coronary syndrome respectively. Activated neutrophils generate around 150−425 µM HOCl/h [28, 29] whereas at sites of inflammation HOCl level is estimated to reach as high as 5 mM [30]. Under these circumstances, HOCl can damage the host tissue by the same mechanism used to destroy invading pathogens. Indeed, HOCl has been implicated to play a role in a number of pathological conditions such as inflammatory diseases, atherosclerosis, respiratory distress, acute vasculitis, rheumatoid arthritis, glomerulonephritis, and cancer [31–34]. Under these pathological conditions where MPO has been known to play a role, there have been reports of significant free iron accumulation [21, 22, 35–37]. The source of this iron is still unclear, but it is thought to be Hb released from damaged RBCs at sites of vascular turbulence or in hemorrhagic atheromatous plaques [22]. We believe there is a mechanistic link between high HOCl and higher free iron. Our hypothesis is that the source of this iron is HOCl mediated destruction of the heme moiety from Hb. Here we studied the reaction between purified bovine Hb (Hb-Fe(III) and oxy-Hb) as well as isolated human RBCs with increasing HOCl concentrations utilizing a variety of spectroscopic and analytical techniques. Our rapid kinetic measurements demonstrate that HOCl can mediate free iron release through a mechanism that involves formation and destruction of peroxidase-like intermediates, ultimately resulting in oxidative cleavage of heme moiety generating fluorescent and non-fluorescent porphyrin derivatives. Additionally, HOCl treatment also resulted in different levels of protein aggregation. This mechanism may accentuate tissue damage in states of inflammation.
MATERIALS AND METHODS
Materials
All the materials used were of highest purity grade and used without further purification. Sodium hypochlorite (NaOCl), ammonium acetate (CH3COONH3), ferrozine, L-methionine, ascorbic acid, bovine Hb, dimethylformamide, methanol and trifluroacetic acid (TFA) - HPLC grade, were obtained from Sigma Aldrich (St. Louis, MO, USA). HPLC grade acetonitrile (CH3CN) was obtained from EMD Chemicals Inc. (Gibbstown, NJ, USA).
Absorbance Measurements
The absorbance spectra were recorded using a Cary 100 Bio UV–visible spectrophotometer, at 25°C, pH 7.0. Experiments were performed in a 1-mL phosphate buffer solution supplemented with fixed amount of Hb (0.63 µM) and increasing concentration of HOCl (0, 5, 10, 20, 40, 80, 100, 120, 160, 180, and 200 µM). After a 10-minute incubation for reaction completion, methionine (5-fold of the final HOCl concentration) was added to eliminate excess HOCl and absorbance changes were recorded from 300 to 700 nm.
Rapid Kinetic Measurements
The kinetic measurements of HOCl-mediated Hb heme destruction were performed using a dual syringe stopped-flow instrument (Hi-Tech, Ltd. Model SF-61). Measurements were carried out under an aerobic atmosphere at 10°C following rapid mixing of equal volumes of a buffer solution containing a fixed amount of Hb (0.63 µM) and a buffer solution containing increasing concentration of HOCl (0–200 µM) . The time course of the absorbance change was fitted to a single-exponential, (Y = 1 − e-kt), or a double-exponential (Y = Ae-k1t + Be-k2t) function as indicated. Signal-to-noise ratios for all kinetic analyses were improved by averaging at least six to eight individual traces. In some experiments, the stopped-flow instrument was attached to a rapid scanning diode array device (Hi-Tech) designed to collect multiple numbers of complete spectra (200–800 nm) at specific time ranges. The detector was automatically calibrated relative to a holmium oxide filter, as it has spectral peaks at 360.8, 418.5, 446.0, 453.4, 460.4, 536.4, and 637.5 nm, which were used by the software to correctly align pixel positions with wavelength.
High Performance Liquid Chromatography (HPLC) analysis
HPLC analyses was carried out using a Shimadzu HPLC system equipped with a SCL-10A system controller, with a binary pump solvent delivery (LC-10 AD) module and a SIL-10AD auto-injector connected to a SPD-M10A diode array detector (DAD) and a RF-10A XL fluorescence detector. Alltech 5 µm particle size, 4.6 × 150 mm reverse-phase octadecylsilica (C18) HPLC column was used. The photodiode array detector was set at 400 nm and the fluorescent detector was set at excitation 321 nm and emission 465 nm to monitor the chromatogram. The column was eluted at a flow rate of 1.0 mL/min with linear gradients of solvents A and B (A, 0.1% TFA in water; B, 0.1% TFA in 80% acetonitrile). The solvent gradient was as follows: 0 to 10 min, 55–65% B; 10 to 14 min, 65–90% B; then the solvent B composition was dropped down to 55% within 14 to 24 min. After treatment of Hb with HOCl, 500 µL of the reaction mixture was diluted with 500 µL of injection solvent (55% B and 45% A) and 50 µL were injected. At the end of the run the system was equilibrated with 45% solvent A. For fluorescence detection the excitation and emission wavelength was set at 321 nm and 465 nm, respectively. Each sample was analyzed in triplicate. For hemoglobin, after treatment with HOCl the reaction mixture was filtered through Amicon Ultra-15 centrifugal filter unit with Ultracel-10 membrane (from Millipore) with a 3-kDa cut-off by centrifuging at 14000 rcf for 30 min at 4 °C.
Mass spectrometric analysis of heme degradation products
Mass spectrometry (MS) experiments were performed using an Agilent 6410 Triple Quadrupole mass spectrometer coupled with an Agilent 1200 HPLC system (Agilent Technologies, New Castle, DE), equipped with an electrospray source. Waters symmetry C18 column (particle size 3.5 µm; 2.1 × 100mm) (Milford, MA) was used to separate reaction products. Solvent A was H2O with 0.1% formic acid and solvent B was acetonitrile with 0.1% formic acid. The column was equilibrated with 80% solvent A and 20% solvent B. The gradient was: 20–95% solvent B over 10 min; 95% solvent B for 10 min; 95–20% solvent B for 1 min; and 80% solvent A for 14 min. Five µL of the sample was injected at a flow rate of 0.25 mL/min.Liquid chromatography electrospray ionization (LC/ESI) MS in the positive mode was performed using the following parameters: spray voltage 4000 V, drying gas flow 10 L/min, drying gas temperature 325°C, and nebulizer pressure 40 psi. Fragmentor voltage was optimized using Flow injection analysis (FIA) of hematin (St. Louis, MO, USA). Optimal fragmentor voltage was 300 V in MS2 scan mode. Mass range between m/z 200 to 900 was scanned to obtain full scan mass spectra.
Free iron analysis
Free iron release was measured colorimetrically by using ferrozine, following a slight modification of a published method [38]. To 100 µL of the sample (RBC-HOCl or Hb-HOCl reaction mixture) 100 µL of ascorbic acid (100 mM) was added. After 5 minutes of incubation at room temperature, 50 µL of ammonium acetate (16%) and the same volume of ferrozine (16 mM) were added to the mixture and mixed well. Subsequently, the reaction mixture was, incubated for 5-minutes at room temperature and the absorbance was measured at 562 nm. A standard curve prepared by using ammonium Fe (III) sulfate was used for the calculation of free iron concentration. Final concentrations of the additives are as follows, ascorbic acid-33.33 µM, ammonium acetate-5.3 %, and ferrozine-5.3 µM.
In-gel heme staining and reducing SDS-PAGE
8 µg of Hb (from the reaction mixture) was incubated with Laemmli buffer containing 63 mM Tris–HCl (pH 6.8), 2% (w/v) SDS, 10% (w/v) glycerol, and 0.0025% (w/v) bromophenol blue. 2-Mercaptoethanol/dithiothreitol, which interfered with the heme-staining dye, was omitted from the Laemmli buffer. A non-reducing denaturing gel electrophoresis was performed for 4 h at a constant voltage of 60 V on discontinuous 12% SDS gel. Gels and buffers, which had been prepared according to Laemmli [39], were equilibrated at 4 °C (by submerging the gel apparatus in a ice bath) before electrophoresis, and electrophoresis was performed at the same temperature. Gels were then stained either for heme with O-dianisidine/H2O2 following a slight modification of a published method [40]. Gels were washed for 10 min in methanol/sodium acetate (0.25 M, pH 5.0) 3/7 (v/v) and, subsequently, incubated in the dark for 20 min in a freshly prepared solution, containing 7 parts 0.25 M sodium acetate, pH 5.0, and 3 parts 12 mM O-dianisidine in methanol. Gels were developed by adding H2O2 to a final concentration of 1.25 M (bands developed immediately) and washed for 30 min in H2O/methanol/acetic acid 8/1/1 (v/v/v). The gels were scanned. For reducing SDS-PAGE, same procedure was followed except, the Laemmli sample buffer had 10% 2-mercaptoethanol and the samples were boiled for 5 minutes at 100 °C before loading. The gels for reducing SDS-PAGE were run at room temperature and stained with Coomassie blue. Relative amounts of protein or heme were estimated by densitometric analysis of the scanned image using ImageJ software from the NIH [41].
Isolation of human red blood cells
Red blood cells were isolated from heparinized blood collected from healthy individuals according to a previously published method [18]. Briefly, blood sample were centrifuged at 3000 rpm for 5 min to remove the plasma and buffy coat and some of the least-dense cells containing most of the reticulocytes. The red cell pellet was washed two times with 5 volumes of phosphate-buffered saline (PBS), pH 7.4, to make sure that all of the plasma was removed. Cells were diluted to a hematocrit of 50% with PBS, pH 7.4. The Hb concentration was measured using Drabkin’s reagent (Sigma Aldrich, St. Louis, MI, USA) using the manufacturer’s protocol.
HOCl treatment of red blood cells
Isolated human RBCs were diluted in PBS to give a Hb concentration of 12.5 µM. this solution of intact red blood cells were treated with increasing concentrations of HOCl (0– 2000 µM) for 1 hour at room temperature. Following which the reaction was stopped by addition of methionine at 5 times the final HOCl concentration. The viability of the red blood cells after HOCl treatment was evaluated by the trypan blue exclusion method [42]. After HOCl treatment, cells were lysed by mixing with an equal volume of distilled water and incubating at room temperature for 2 minutes and then by sonication. The cell lysates were then centrifuged at 13,000 rpm for 80 minutes to separate the membrane debris from the cytosol. Both the cytosolic fraction and the membrane fraction were extracted with the HPLC injection solvent and filtered through Amicon Ultra-15 centrifugal filter unit with Ultracel-10 membrane (from Millipore) with a 3-kDa cut-off by centrifuging at 14,000 rcf for 30 min at 4 °C. The filtrate was analyzed by HPLC for the presence of heme degradation products. Red blood cell lysate, after HOCl treatment, was also analyzed for free iron by ferrozine.
Solution preparation
HOCl preparation
HOCl was prepared as previously described with some modification [43]. Briefly, a stock solution of HOCl was prepared by adding 1 mL NaOCl solution to 40 mL of 154 mM of NaCl and the pH was adjusted to around 3 by adding HCl. The concentration of active total chlorine species in solution expressed as [HOCl]T (where [HOCl]T = [HOCl] + [Cl2] + [Cl3−] + [OCl−]) in 154 mM NaCl was determined by converting all the active chlorine species to OCl− by adding a bolus of 40 µL 5 M NaOH and measuring the concentration of OCl−. The concentration of OCl− was determined spectrophotometrically at 292 nm (ε = 362 M−1 cm−1). As HOCl is unstable, the stock solution was freshly prepared on a daily basis, stored on ice, and used within one hour of preparation. For further experimentations, dilutions were made from the stock solution using 200 mM phosphate buffer pH 7, to give working solutions of lower HOCl concentration.
Methemoglobin solution
10 mg of lypophilized Hb powder was dissolved in 10 mL of 200 mM phosphate buffer pH 7 to give a solution of concentration 15.5 µM. Methemoglobin solution prepared in this manner was kept on ice and used within one hour.
Oxyhemoglobin solution
10 mg of lyophilized Hb powder was dissolved in 1 ml of 200 mM phosphate buffer pH 7.0. A few crystals of sodium dithionate was added and mixed well. The solution was aspirated by pipette to saturate with oxygen. Excess sodium dithionate was removed by passing the solution through a Sephadex G-25 M column (Amersham Biosciences). The prepared oxyhemoglobin solution was kept on ice and used within an hour of preparation. The concentration of oxy-Hb in the stock solution was determined spectrophotometrically at 577 nm (ε = 14.6 mM− 1 cm−1/heme) [44].
RESULTS
Optical spectroscopy and stopped-flow measurements
We first characterized OCl− binding to Hb-Fe(III) and its subsequent effects on Hb heme destruction. The visible spectrum of met-Hb displayed a Soret absorbance peak centered at 405 nm with absorbance shoulders at 500 and 631 nm, indicative of a ferric heme. Exposure of a fixed amount of Hb (0.6 µM) to increasing concentration of HOCl caused Hb heme destruction, as judged by the loss and flattening of the Soret peak region. Incubation of Hb with 40 µM of HOCl caused 50 % decrease in the Soret band, whereas the addition of 200 µM of HOCl led to a complete flattening of the Soret absorbance peak indicating heme destruction. We next, utilized diode array stopped-flow spectrophotometry to continuously monitor and identify species that formed upon mixing the protein solution with phosphate buffer supplemented with increasing concentration of HOCl. All HOCl concentrations employed were in larger molar excess to Hb to ensure pseudo first order conditions. Figure 1 shows spectra collected over time for the reaction of Hb-Fe(III) with HOCl (200 µM final). As shown in Fig. 1A, the starting spectrum recorded after 0.05s is characteristic of Hb-Fe(III), which displays a Soret absorbance peak centered at 405 nm with absorbance shoulders at 500 and 631 nm, indicative of a ferric heme. This was followed by a buildup of a transient intermediate, whose spectrum was characterized by absorbance peak at 415, 533, and 567 nm, attributable to Hb–Fe(III)-OCl complex formation (traces 2–5). This transition species then decayed to form another transient intermediate whose spectral features match those of authentic Hb ferryl complex (Hb-Fe(IV)=O complex, Compound II) via rapid initial formation of Compound I (Fig. 1B). Compound II possesses a characteristic Soret absorbance peak at 420 nm that is easily distinguished from the Soret absorbance peaks of both Hb-Fe(III) and Hb-Fe(III)-OCl, as shown in Figure 1A and 1B. The oxidation of Compound II by HOCl was accompanied by a remarkable decrease and subsequent flattening in the Soret absorbance region, suggesting heme degradation (Fig. 1C). These spectral changes may also suggest that the majority of Hb was converted to Fe(IV)=O complex before heme destruction. The occurrence of two defined sets of isosbestic points (at 412 and 414 nm) at two different time frames suggest that the Hb heme degradation succeeds through subsequent intermediate products and indicate that at least three phases of the reaction can be distinguished (Fig. 1).
Fig. 1.

Interaction of met-Hb with HOCl leads to the transient formation of Compound II, prior to heme destruction. Absorbance spectra recorded by diode array stopped-flow when a phosphate buffer solution (0.20 M, pH 7.0) containing 1.25 µM met-Hb was rapidly mixed with a buffer solution containing 400 µM HOCl, at 10°C. The upper panel contains spectra collected after 0.05, 0.15, 0.25, 0.95 and 1.5 s of initiating the reaction, which shows the accumulation of compound II. The inset in upper panel shows the accumulation of Compound II as judged by the transition of the Soret absorbance peak from 405 nm to 420 nm. The lower panel contains spectra collected after 1.5, 9.5, 34.5, 89.5, 149.5, 599.5 s of initiating the reaction, which shows Compound II heme destruction. Arrows indicate the direction of spectral change over time. The experiment shown is representative of three.
To examine the kinetics of interaction between HOCl and Hb-Fe(III) and determine rate constant for the various forms of Hb, we monitored the changes in absorbance at two different wavelengths; 405 and 420 nm. These wavelengths were chosen to facilitate the direct examination of HOCl not only as a ligand, but also as a mediator for heme destruction. Formation of Hb-Fe(III)-OCl was monitored by following the decrease in absorbance at 405 nm which also showed the formation of Compound II. The influence of HOCl on Compound II decay was also monitored by spectral changes occurring at the isosbestic point 412 nm. Therefore, the formation of Compound II, if any, following interaction of HOCl with Hb, would not have been seen. The influence of HOCl on the kinetics of Compound II buildup, duration, and its exhaustion were monitored by following the both increase and decrease in absorbance at 420 nm. Experiments were carried out under aerobic conditions following rapid mixing of fixed amount of Hb-Fe(III) solution with various concentration of HOCl (25, 50, 100, 150, and 200 µM, final concentration).
At all of the HOCl concentrations tested, there was a monophasic decrease in absorbance at 405 nm attributed to buildup of Hb-Fe(III)-OCl complex formation, followed by a slower, essentially monophasic decrease attributed to heme destruction. As shown in Figure 2A, the plot of kobs versus HOCl concentration was linear, with a positive intercept indicating that the reaction is reversible and follows a one step mechanism. The kinetic parameters for HOCl binding, kon and koff, estimated from the slope and intercept, respectively, were found to be 0.008 µM−1s−1 and 0.2 s−1. Only the slow phase was observed when the same reaction was monitored at 420 nm. HOCl significantly accelerated the rate of heme destruction in a concentration dependent fashion. A plot of HOCl concentration versus rate of Compound II destruction demonstrated linear kinetics with y-intercept close to zero and yielded a second order rate constant of 6 X 10−5 µM−1s−1 (Fig. 2C). The accelerated rate of Compound II destruction in the presence of HOCl indicates that it also binds and oxidized the porphyrin ring.
Fig. 2.

Plots of observed rate constants of various intermediates that formed upon mixing of met-Hb against increasing concentration of HOCl. The observed rates of Fe(III)-OCl complex formation was monitored by following the decrease in the absorbance at 405 nm (Panel A). The observed rate constant of the buildup of compound II was followed by monitoring the increase in absorbance at 420 nm (Panel B). The observed rate constant of heme destruction was monitored by following the decrease in absorbance at 405 and 420 nm (Panel C). The experiments were carried out as in Fig. 1. The rates plotted are the average of three or four separate experiments.
Based on spectra of Figure 1B and previous work with Hb, we followed the buildup of Hb Compound II and its decay at 420 nm. As shown in Fig. 1B, formation of Hb Compound II appeared rapidly and required 1.5 s to reach completion, whereas its decay was very slow and reach completion in the next 600 s. In both cases, the spectral changes were essentially monophasic and fitted well to a single exponential function. For the rate of Compound II formation, the plot of kobs versus HOCl concentration was linear with an intercept at y-axis close to zero, indicating that reaction is irreversible with a second order rate constant of 0.02 µM−1s−1 (Fig. 2B). The rate of the second step is essentially identical to the slow phase that was observed at 405 nm consistent with only Compound II destruction occurring under this condition. For heme destruction, the plot of the kobs as a function of HOCl concentration was also linear with a y-intercept close to zero, giving a second order rate constant of 6.0 X 10−5 µM−1s−1 (Fig. 2C). Together, these results indicate that HOCl-mediated Hb heme destruction is relatively slow and occurs upon complete conversion of Hb-Fe(III) to Compound II.
In order to facilitate comparison, identical experiments were repeated with oxy-Hb. The absorbance spectrum of oxy-Hb is characterized by its Soret absorbance peak centered at 412 nm and two weaker well-resolved absorption maxima at 541 and 577 nm. Addition of HOCl to oxy-Hb solution initially caused a rapid small decrease in absorbance at 412 nm with time, followed by instant buildup of a transient species, whose spectrum was characterized by absorbance peak at 405, (upper panel of Figure 3). These spectral features clearly showed that the majority of oxy-Hb was oxidized and converted essentially to Hb-Fe(III) in the first 150 s of initiating the reaction. The decrease in absorbance was best fitted to a one exponential function with pseudo first order rate constant of 0.023 s−1 for 10 µM HOCl. At higher concentrations of HOCl the change in absorbance was very small to accurately measure the rates. This transient intermediate was characterized by a Soret peak at 405 nm and a visible band at 537 and 576 nm. These spectral changes suggest that OCl− is coordinated to the ferric heme iron and form a low spin six-coordinate Fe(III)-OCl complex which is associated with the destabilization of the Hb heme moiety after complete conversion to Compound II. This can be determined by the decrease and flatting in the Soret peak region from 350 nm to 450 nm. This interpretation is based on the previously reported spectral changes that showed addition of H2O2 to Hb leads to the accumulation of Compound II via rapid initial formation of Compound I. Monitoring the spectral decrease at 412 nm indicated that the reaction was monophasic at lower concentrations of HOCl (< 150 µM) and at higher concentrations (≥ 150 µM ) the reaction was biphasic and best fitted to a two exponential function with pseudo-first order rate constant of 0.029 and 0.003 s−1 for 150 µM HOCl. About 50% of the total absorbance change at 150 µM HOCl was associated with the slow phase. To determine the kinetic parameters of the reactions, the reactions were repeated at different HOCl concentration. For the first phase, the plot was linear with a positive intercept at the y-axis, indicating that OCl− binding is reversible with kon and koff, estimated from the slope and intercept of 0.044 µM−1s−1 and 1.2 s−1, respectively (Fig. 4A). For the formation of compound II the plot was linear with an intercept at the y-axis close to zero yielding a second order rate constant of 0.024 µM−1s−1 (Fig. 4B). The third phase of the reaction attributed to the process of heme destruction was found to be irreversible as indicated by the linear plot with y-intercept close to zero yielding a second order rate of 2.0 X 10−5 µM−1s−1 (Fig 4C).
Fig. 3.

Oxy-hemoglobin heme oxidation and subsequent heme destruction mediated by HOCl. Absorbance spectra recorded by diode array stopped-flow when a phosphate buffer solution (0.20 M, pH 7.0) containing 1.25 µM oxy-Hb was rapidly mixed with a buffer solution containing increasing concentration of HOCl, at 10°C. The upper panel shows heme oxidation when the protein solution was rapidly mixed with a buffer solution supplemented with 20 µM, HOCl, spectra were collected after 0.5, 1.5, 24.5 and 599.5 s of initiating the reaction. The inset shows the transition of the Soret absorbance peak from 415 nm (oxy-Hb) to 405 nm (met-Hb). The lower panel shows Hb heme destruction when the oxy-Hb solution was rapidly mixed with 400 µM HOCl, spectra were collected after 0.5, 1.5, 6.5, 19.5, 44.5, 89.5, 199.5, 599.5 s of initiating the reaction. Arrows indicate the direction of spectral change over time. The data are representative of three independent experiments.
Fig. 4.

Plots of observed rate constants of various intermediates that formed upon mixing of oxy-Hb against increasing concentrations of HOCl. The observed rate constant of the Fe(III)-OCl complex formation, as in Fig. 3, was monitored by following the decrease in absorbance at 405 nm (Panel A). The observed rate constant of the buildup of compound II was monitored by the following the increase in absorbance at 420 nm (Panel B). The observed rate constant of heme destruction was monitored by following the decrease in absorbance at both 405 and 420 nm (panel C). The rates plotted are the average of three or four separate experiments.
HOCl-mediated heme destruction, protein aggregation and free iron release in hemoglobin
As HOCl is thought to oxidize the heme moiety of Hb, we examined whether these spectral transformations that are apparent from our UV–visible spectral analysis may represent oxidation, free iron release, and subsequent protein aggregation. To investigate how the flattening in the Soret absorbance peak at 405 nm, in HOCl treated samples, is related to Hb heme depletion, we analyzed the heme content in Hb (6.25 µM) after treatment with increasing molar excess of HOCl, by in-gel heme staining. Fig. 5A shows the heme content following HOCl treatment compared to untreated control. By comparing the band intensity of the untreated control it can be said that the treatment with HOCl leads to appreciable loss in heme content in Hb confirming our previous spectrophotometric studies. It was also observed that Hb undergoes aggregation as evidenced by the higher molecular weight bands observed in the coomassie staining (Fig. 5B). To study this phenomenon in more detail we performed reducing SDS-PAGE (Fig. 6). The Hb monomer band intensity decreases slightly until quite near the 1:32 Hb:HOCl ratio, followed by a steep plunge until 1:160 Hb:HOCl ratio after which it saturates. In contrary, the monomer:oligomer band intensity showed a steady increase till 1:160 Hb:HOCl ratio after which it saturates. Higher amounts of dimers were formed at 1:32 and 1:64 ratios of Hb:HOCl. At concentration higher than that (1:32 or 1:64) dimer content decreased but high molecular weight oligomers were formed. With the largest amounts of HOCl, the distinct pattern of aggregation disappeared, producing smears over the entire length of the lanes (as visualized with coomassie stain). This could be attributed either to the formation of aggregates that were too large to enter the separating gel, or to fragmentation. Kettle and coworker obtained similar aggregation patterns when apo-Hb and apomyoglobin were treated with HOCl [13]. Strong, non-covalent interactions between protein chains has been assumed, which could apply to this work. Thus, we conclude that HOCl-mediated protein aggregation of Hb occur independent of heme presence. Free iron accumulation when assayed by ferrozine method showed a linear increase as a function of HOCl concentration (Fig. 7).
Fig. 5.

In-gel heme staining confirms that HOCl treatment causes heme destruction from hemoglobin. Hb (6.25 µM) was treated with 160 fold of HOCl for 10 minutes at room temperature and in-gel heme staining was performed by o-dianisidine-H2O2 method, as detailed in Materials and Methods section (Panel A). Non-reducing SDS PAGE analysis of the same samples as in stained with coomassie blue for protein (Panel B). The data are representative of three independent experiments.
Fig. 6.

HOCl concentration dependent depletion of hemoglobin monomer and subsequent formation of high molecular weight aggregates. 6.25 µM of Hb dissolved in phosphate buffer was treated with different molar ratios of HOCl (0 to 256 fold HOCl) for 10 minutes at room temperature. Panel A shows the SDS-PAGE of HOCl treated Hb. After HOCl treatment, samples were run in 12% SDS-PAGE under reducing condition. The first lane on the left has the standard molecular weight markers, the masses (Kd) of the marker proteins closest to hemoglobin monomer, dimer and tetramer have been shown. The arrows indicate the expected position of the monomer, dimer, trimer and tetramer bands of hemoglobin. The molar ratio of HOCl:Hb with which the sample was treated is shown at the top of each well. The figure shown is a representative of three independent experiments. Panel B shows densitiometric scanning of the gel image was performed as mentioned in the Materials and Methods section. The percentage of monomer (as calculated by the monomer band intensity) after HOCl treatment (solid line) and the ratio between oligomer/monomer band intensity (represented in the graph as oligomer/monomer, as dotted line) is plotted as a function of HOCl concentration. The results shown are an average of three independent experiments and the error bars represents the standard error of measurement.
Fig. 7.

Release of free iron from hemoglobin following treatment with HOCl. 6.25 µM of Hb was treated with different molar ratios of HOCl (0 to 320 fold) for 10 minutes at room temperature. Free iron was assayed colorimetrically using ferrozine (for details see Materials and Methods section). The above result is an average of three independent experiments and the error bars represents the standard error of measurement.
HPLC analysis of heme degradation products from Hb
Heme by itself does not have any intrinsic fluorescence, but porphyrin derivatives generated due to oxidative fragmentation of heme have an intrinsic fluorescence. We exploited this property to analyze the heme fragmentation pattern after HOCl treatment of Hb. Fig. 8 shows the chromatograms when Hb was treated with different molar ratios of HOCl. We incubated a fixed amount of Hb (6.3 µM) with increasing molar ratios of HOCl (1:64, 1:160 and 1:256). When Hb was reacted with HOCl there was a progressive accumulation of new heme degradation products (as a function of HOCl concentration) eluting at earlier time (Fig. 8). HOCl treatment led to the formation of four different fluorescent degradation products with retention times of 1.8, 3.09, 5 and 8 minutes respectively. The appearance of newer earlier eluting peaks in the chromatograms could be due to the formation of degradation products with decreasing hydrophobicity generated by fragmentation of the tetrapyrrole ring of the heme.
Fig. 8.

Treatment of hemoglobin with HOCl leads to the generation of fluorescent heme degradation products. Hb (6.25 µM) was treated with a range of HOCl:Hb molar ratios (0 to 256 fold HOCl) and analyzed by HPLC, as detailed in the Materials and Methods section. The fluorescent detector was set at excitation 321 nm and emission 465 nm. The molar ratio of the HOCl:Hb used for treatment is mentioned in inset of each panel. The above set of chromatograms is a representative of three independent experiments.
HOCl treatment significantly reduce viability of RBC senescence
Human RBC were treated with various molar ratios of HOCl:Hb for 1 hour at room temperature and their viability was evaluated using the trypan blue exclusion technique (as specified in the Materials and Methods section). Our results show that the RBC viability did not significantly change up to 20:1 HOCl:Hb ratio, but above that there was remarkable decrease in RBC viability, with complete loss of viability occurring at 120:1 HOCl:Hb ratio (Fig. S1).
Free iron release and accumulation of heme degradation products from RBC after HOCl treatment
Human RBC were treated with different molar ratios of HOCl:Hb and the formation of fluorescent heme degradation products were studied using HPLC both in the membrane (data not shown) as well as the cytosolic fraction (Fig. 9). HPLC chromatograms show the presence of mainly three distinct fluorescent peaks in the membrane and as well as the cytosol. These products were not observed in the HOCl untreated control but were only seen in HOCl treated samples. Comparison of the retention times of the fluorescent products observed from RBC with those obtained when Hb was treated with HOCl reveals that they were very similar. Thus it can be concluded that these fluorescent products were generated by HOCl mediated heme destruction of Hb in RBC. Free iron accumulation as assayed by ferrozine method showed a similar trend as observed for Hb-HOCl i.e. a linear increase as a function of HOCl concentration (Fig. 10).
Fig. 9.

Treatment of human RBC with HOCl leads to the generation of fluorescent heme degradation products in the RBC cytosol. Isolated human RBC adjusted to Hb concentration of 12.5 µM was treated with a range of HOCl:Hb molar ratios (0 to 160 fold HOCl) and analyzed by HPLC, as detailed in the Materials and Methods section. The fluorescent detector was set at excitation 321 nm and emission 465 nm. The molar ratio of the HOCl:Hb used for treatment is mentioned in inset of each panel. The above set of chromatograms is a representative of three independent experiments.
Fig. 10.

Release of free iron from isolated human RBC following treatment with HOCl. Isolated human RBC adjusted to Hb concentration of 12.5 µM was treated with different molar ratios of HOCl (0 to 160 fold) for 1 hour at room temperature. Free iron was assayed colorimetrically using ferrozine (for details see Materials and Methods section). The above result is an average of three independent experiments and the error bars represents the standard error of measurement.
LC-ESI-MS of heme fragmentation products
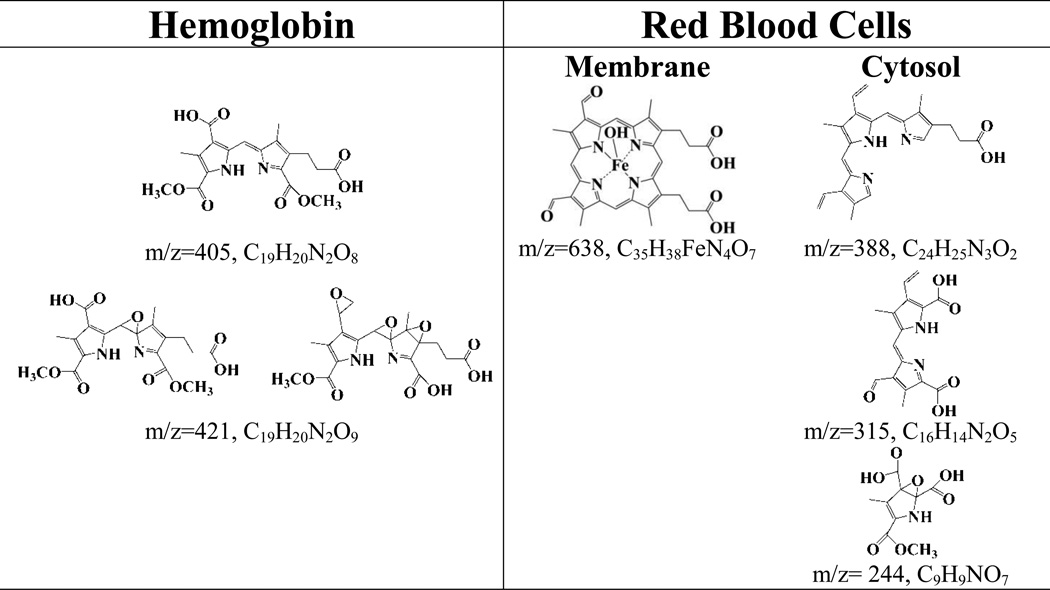
LC/ESI/MS in the positive mode was utilized to elucidate HOCl-mediated reaction products of hemoglobin. We subjected the reaction products of varying concentrations HOCl with Hb and with RBCs (membrane and cytosolic fractions). The majority of the products were tentatively identified by detecting the molecular ions [M + H]+ from the most abundant peaks following LC/MS. The molecular weights were then compared with the proposed structures with the previously identified products and/or depending on the chemical reactivity of HOCl with carbon-carbon double bonds [10, 11, 23]. These cleavage products were classified into four major categories according to the degree of oxidation and their cleavage position within the heme molecule. Group 1 consists of compound with the intact tetrapyrrole ring system with the iron but with oxidative modification in the terminal carbons. m/z 638 (Fig. S2) eluting at 8 min identified from Hb:HOCl 1:120 reaction mixture (identified from RBC membrane fraction) is an example of this class of compound. Group 2 consists of tripyrrole derivatives, which resulted by oxidative cleavge of the tetrapyrrole ring of heme by HOCl. Compounds identified that fall in group 2 include m/z 388, from RBC cytosolic fraction (Hb:HOCl and 1:20 and 1:160), eluting at 7.8 min. Group 3 consists of dipyrrole derivatives of heme. There are three compounds that fall under this category m/z 421 (Fig. S3) eluting at 11.1 min (identified from Hb:HOCl 1:128, 1:288, 1:320) , m/z 315 (Fig. S4) identified from RBC cytosol fraction (Hb:HOCl 1:40) and m/z 405 identified from Hb. Group 4 consists of mono-pyrrole derivatives; m/z 244 identified from RBC cytosol is an example of this class of compound. Group 4, consists of compounds that have resulted from extensive oxidation and cleavage of the tetrapyrrole ring, example of compounds in this group include m/z 449, and m/z 594. Table 1 shows, the structures of all the heme degradation compounds tentatively identified by LC-MS.
TABLE 1.
Structures of different heme degradation products as tentatively identified by LC-MS following treatment of Hb and isolated human RBC with HOCl.
 |
DISCUSSION
Hb is the most abundant hemeprotein in humans with an average of 750 g in adult [45]. RBC develops in bone marrow, which is rich in Hb (20 mM). RBCs have a limited life span (~120 days) before their components are recycled by macrophages [5]. Using a combination of biochemical, physiological, and kinetic approaches, we showed that HOCl oxidizes oxyHb, binds to the heme iron, mediates Hb heme destruction thereby generating more toxic free iron, and heme degradation products, resulting in protein aggregation. In order to perform its physiological function, Hb has to tightly bind molecular oxygen (in the oxygen-rich atmosphere of the lungs) and release it in the relatively oxygen-poor environment of the tissues. Therefore, we propose that the oxidation of oxy-Hb/met-Hb by HOCl may play an important role in altering the protein function by destabilizing Fe-O2 complex, causing heme destruction, or forming Hb Compounds I and II. Methemoglobin and Hb ferryl intermediates are inactive form of Hb, which cannot bind oxygen. Accumulation of these species in the blood could result in tissue hypoxia, impeding the normal function of Hb. Oxy-Hb and met-Hb also react with other ROS such as O2− and H2O2 to produce Compounds I and II [14]. Similarly, a molar excess of H2O2 has been reported to degrade the heme moiety of Hb and other related hemoproteins such as cytochrome c, releasing iron from heme [46–48]. The dissociation and the destruction of heme moiety may also be enhanced by damage to the globin, which binds the heme [49]. Thus, increased HOCl levels or the deficiency of potent scavengers of HOCl such as taurine, glutathione, and lycopene [23, 50, 51] may contribute to inflammation, cancers and other related disorders by increasing catalytically active free iron levels.
A general kinetic Scheme of how HOCl interacts with Hb intermediates incorporated into the classic peroxidase-like cycle is illustrated in Scheme 1. The capacity of HOCl to influence both the oxidation state transition of Hb heme iron and the stability of heme moiety supports the notion that HOCl displays multiple functions over the time course of the reaction. The ability of OCl− to serve as a ligand for met-Hb was directly demonstrated employing diode array stopped-flow methods. It has been thought that the heterolytic cleavage of the O-Cl bond in an Fe(III)-OCl intermediate preferentially occur at neutral conditions to degrade HOCl and form a ferryl porphyrin radical cation, Compound I [52]. Compound I is highly unstable intermediate, which immediately decayed to form Compound II. Because Compound I formation is slower than its decay to Compound II, Compound I cannot be detected. Thus, the observed absorbance changes should reflect the alteration in Compound II accumulation, duration, and decay. Such a species has been proposed to be formed in catalase and LPO during reaction with HOCl, as well as, during inactivation of horseradish peroxidase by cyclopropanone hydrate [53–55]. Our kinetic measurements indicated that Compound II is the predominant species formed prior to heme destruction, providing direct evidence for involvement of Compound II in heme destruction. This conclusion is in full agreement with Nagababu and Rifkind who showed that substances that react with Hb Compound II, such as sodium sulfide and peroxidase substrate prevent H2O2-mediated heme degradation [56]. Our results show that HOCl not only accelerates the formation of Compound II, but also enhances Hb heme destruction. Hb heme destruction is an irreversible process, relatively slow and associated with free iron release and protein aggregation. Heme degradation process is spectrally distinguishable by the flattening in the heme Soret region which can be explained by the release of the heme prosthetic group, the heme oxidation and destruction, or combination of both. Spontaneous breakage or weakening of the natural imidazole nitrogen ligand provided by the distal histidine (His58) can be explained by the negative trans effect of OCl−, which in this case, favors cleavage of the iron-nitrogen bond located on the opposite side of heme iron or through the formation of the Hb-Fe(III)-OO•− radical that is formed through the formation of Compound II [5]. Our HPLC and mass spectrometric results clearly show a direct correlation between Hb oxidation and the formation of fluorescent and non-fluorescent degradation products as well as free iron accumulation.
Scheme 1.

A general kinetic scheme showing the interaction of Hb with HOCl leading to heme destruction and liberation of free iron.
The direct reaction between oxy-Hb complex and HOCl first involves oxidation of heme iron from ferrous to ferric state. Thus, Hb can no longer bind oxygen but it can bind an OCl− molecule. Because significant Fe(III)-OCl complex formation can be seen immediately after oxy-Hb oxidation, it seems likely that OCl− molecule that mediated heme oxidation is still retained in the heme pocket and binds to the ferric heme iron immediately after oxygen release. This may contribute to the binding rate constant enhancement which in turn influences the alteration in the intermediate distributions during the reaction progression, but otherwise the kinetic parameters of Compound II formation, duration, and heme destruction were almost similar to those obtained for met-Hb with HOCl (Figs. 2A & 4A).
Heme depletion was confirmed by in-gel heme staining, which clearly showed loss of heme from Hb following HOCl treatment. HOCl mediated heme destruction was also associated with increased free iron release and a buildup of three new fluorescent products. The excitation and emission wavelengths of these fluorescent products are similar to those observed when H2O2 reacted with Hb, and indistinguishable from those obtained when isolated human RBC were treated with HOCl. It is interesting to note that these fluorescent bands are observed when oxy-Hb, hematin, as well as protoporphyrin IX was treated with increasing concentrations of HOCl (data not shown). Thus, the cleavage of porphyrin ring is occurring independent of the metal center. The destruction of heme molecule leads to the liberation of heme iron as determined by the ferrozine method. In this respect, Hb may serve as a source of free iron at sites of inflammation when HOCl is elevated. The free iron generated by Hb destruction may induce oxidative stress and make it highly toxic, as it can rapidly react with H2O2 and molecular oxygen to produce free radicals [17]. Toxicity of free iron can damage blood vessels and produce vasodilation with increased vascular permeability, leading to hypotension and metabolic acidosis [20, 21]. Recent studies have shown that catalytically active MPO, the major generator of HOCl, and its oxidative species are present in human atherosclerotic lesions [57], promote lipid peroxidation, DNA strand breaks, and modification or degradation of biomolecules, eventually leading to cell death [58]. It has also been shown that iron accumulates in atherosclerotic lesions in a catalytically active form [57].
HPLC and LC-MS analysis indicate that multiple fluorescent and non-fluorescent cleavage products were generated when heme was degraded by HOCl. A total of seven cleavage products have been tentatively identified based on their mass signals and comparison with the previously identified products and/or depending on the chemical reactivity of HOCl with carbon-carbon double bonds from Hb and RBC treated with different concentrations of HOCl [10, 11, 23]. The degree of degradation of heme depends mainly on the ratio of HOCl to heme, suggesting that multiple molecules of HOCl are required per molecule of heme. The proposed chemical mechanism for the HOCl mediated cleavage of heme moiety is shown in Scheme 2A. In this model, HOCl can randomly attack any of the four carbon methyne bridges between the adjacent pyrrole ring and forms a chlorinated adduct, which by releasing of chloride, forms an epoxide or aminal. The epoxide gives rise to a hydroxylated compound with the hydroxyl group being attached to the carbon-methyne bridge of the tetrapyrrole moiety, where the initial attack by HOCl occurred. Attack by another hydroxyl group to this carbon leads to the formation of a vicinyl diol (two hydroxyl groups attached to two adjacent carbon atoms). Cleavage of the vicinyl diol leads to the formation of two carbonyl compounds. The cleavage of the vicinyl diol can either occur through a homolytic cleavage forming two carbonyl compounds, or in presence of iron, through the formation of dioxetane intermediate by a heterolytic two electron process (shown in Scheme 2A in red) [59]. The aldehydes that are generated may further be oxidized by HOCl to form carboxylic acid, through a mechanism described previously [23]. Cleavage of the C=C bond is not only limited to the carbon-methyne bridge, but it can also occur at the terminal C=C bond, leading to the formation of formaldehyde (Scheme 2B). This single carbon aldehyde can be oxidized to formic acid by the electrophilic addition of HOCl (24). The formaldehyde thus generated is used for the formation of methyl esters as seen in some of the structures (Scheme 2B).
Scheme 2.

Proposed chemical mechanism for the HOCl mediated cleavage and subsequent methylation of the heme moiety.
Taken together, the results of our studies suggest that the HOCl-mediated cleavage reaction of heme and the degree of oxidation could occur non-selectively at any position of the heme double bonds independent of HOCl concentration. Many of the metabolites in Table 1 have been reported, as their metabolic pathways were investigated, whereas other cleavage products and their functions are either not yet fully explained or not mentioned before in the literature [10, 11]. H2O2 have been shown to be a major player in non-enzymatic heme destruction. For example, Schaefer et al. have identified six major products generated by the reaction of hemin with H2O2 by using mass spectroscopy and NMR, four dipyrrolic propentdyopents, hematic acid, and methylvinylmaleimide [10]. Groves et al. demonstrated the formation of a formal perferryl species from the reaction of heme and H2O2 [60]. This species has been implicated in the random cleavage of the porphyrin ring to form dipyrroles and monopyrroles. Nagababu and Rifkind have shown that the reaction of heme and hemin with H2O2 produces two fluorescent products similar to that observed when Fe(II)-hemoglobin reacts with H2O2 [46] . They have concluded that the metal center is essential to form these fluorescent products [46]. From these results, one may naturally come to the conclusion that HOCl behaves as a stronger oxidant with a higher oxidative potential than H2O2 and other oxidants for heme destruction.
Reducing denaturing SDS-PAGE revealed a different profile of protein aggregation as a function of HOCl concentration. Previously, Chapman et. al. [13] have studied aggregation of apo-Hb (heme free Hb) upon exposure to HOCl. From extensive biochemical and mass-spectrometric studies they concluded that apo-Hb on exposure to HOCl undergoes aggregation and produces a regular series of high molecular weight oligomers [13]. This change in the protein structure in Hb was facilitated by the formation of protein carbonyls and possibly chloramines, along with methionine oxidation, which altered the protein folding and subsequently the secondary/tertiary structure of the protein. The process of aggregation was due to non-covalent interaction between the exposed hydrophobic areas on neighboring molecules that associate to form dimers and higher-molecular mass aggregates [13]. Our results with intact Hb shows similar pattern of protein aggregation in reducing SDS PAGE. The only difference being that we observed a distinct dimer band, even at higher HOCl-Hb ratios, while Chapman et. al. reported that a higher HOCl-Hb ratios the dimer was not observed [13]. This difference could be attributed to the presence of heme in our reaction system. As shown in our results, Hb with intact heme when treated with HOCl undergoes heme destruction which releases free iron. Thus, we conclude that HOCl-mediated protein aggregation of Hb occur independent of heme presence. Previous, studies by Lucana et. al. [61] has shown that free iron can mediated protein cross linking by dityrosine formation. Additionally, Nagy et. al. [62] have shown that Hb isolated from atheromatous lesions have a higher dityrosine content. Thus it can be presumed that HOCl treatment of intact Hb causes free iron release, which mediates dityrosine formation, as judged by the presence of dimer bands even at higher HOCl-Hb ratios. This process could lead to the formation of aggregated proteins at sites of inflammation where MPO activity and subsequently HOCl generation is enhanced and may contribute to tissue injury. HOCl-mediated aggregation is not specific for Hb, but it can occur in any protein depending on the susceptibility of the constituent amino acids towards HOCl [63, 64]. Aggregated proteins are formed during aging [65], in diabetes [66] and in neurodegenerative diseases, including Creutzfeldt–Jacob disease, Huntington's disease, Alzheimer's disease and Parkinson's disease [66–68].
Several studies have shown that RBCs with abnormal Hb are usually exposed to sustained oxidative stress from both exogenous and endogenous sources of ROS. Exposure to oxidative stress is associated with enhanced Hb autoxidation, accumulation of iron in membranes, and increased membrane damage, which in sum results in shorter life span for pathological RBCs. It is worth noting that the mechanisms of these changes have not been explored yet. Previous studies by Vissers et. al. [69] have shown that HOCl exposure of human RBC caused a dose dependent depletion cellular GSH content. Thus destruction of GSH in RBC after HOCl treatment makes them more vulnerable towards oxidative insults since glutathione peroxidase-glutathione system cannot function efficiently. Our results showed that HOCl treatment of isolated intact human RBC caused a HOCl dose dependent senescence, formation of fluorescent heme degradation products along with a concomitant increase in free iron level. Our HPLC analysis revealed that at least three fluorescent heme degradation products are found in the isolated cytosolic and membrane fractions with overwhelming majority of those found in cytosol fraction. Similar studies with H2O2 have shown that 90% of the fluorescent heme degradation products are found to be localized on the RBC membrane [70]. Additionally these products are not transferred from the cytosol to the membrane. A plausible explanation for this finding is that cytosolic H2O2 is easily scavenged by the cytosolic antioxidant system. On the contrary, H2O2 generated by the membrane associated Hb is not, leading to increased heme destruction in the membrane. HOCl treatment have been reported to cause a dose dependent depletion of the cytosolic glutathione level in RBC [69] thereby inactivating the glutathione peroxidase-glutathione cycle. This makes HOCl a stronger oxidant (to destroy Hb) in the RBC cytosol. Previous studies by Nagababu et al. have shown that H2O2 formed in association with Hb autoxidation reacts with Hb and initiates a cascade of reactions that results in heme degradation with the formation of two fluorescent emission bands and the release of iron [46, 56]. A 5.6 fold increase in fluorescence was found in red cells from sickle transgenic mice that expressed exclusively human globins when compared to red cells from control mice [71]. Heme degradation was also increased 3.5 fold in β-thalassemic mice generated by deletion of murine β major [71].
Therefore, inhibiting MPO and/or eliminating its final products may play a beneficiary role in biological system in reducing the free iron release mediated by HOCl. Recently we have shown that melatonin, tryptophan, and tryptophan analogs display the potential capacity in inhibiting MPO, the major sources of HOCl [72–75]. More recently we have shown that lycopene can function as a potent scavenger of HOCl at a wide range of concentrations that span various physiological and supplemental ranges [23]. In related studies, we have also shown that peroxynitrite in combination of H2O2 in a low chloride concentration environment inhibit MPO through a mechanism that involves heme destruction and iron release [41].
In summary, the reaction between HOCl with different forms of Hb occurs through a complex pathway involving the sequential generation and decay of oxoferryl intermediates. HOCl reacts with the heme moiety of Hb and causes heme degradation and the release of free iron. HOCl could also mediate different degrees of protein aggregation in the globin molecule.
Supplementary Material
Isolated human RBC adjusted to Hb concentration of 12.5 µM was treated with different molar ratios of HOCl (0 to 160 fold) for 1 hour at room temperature. The viability of the cells was assayed by the trypan blue exclusion technique. The results are expressed as percent of cells alive as a function of molar ratio of HOCl:Hb. For details see Materials and Methods section. The above result is an average of three independent experiments and the error bars represents the standard error of measurement.
The reaction mixture from the membrane fraction was separated by reverse-phase HPLC and subjected to ESI/MS in the positive mode as described under Materials and methods. The major reaction product produced an intense peak at 8 min. Examination of the MS spectrum revealed that the [M + H]+ ion had a m/z 638. (A) The extracted ion chromatogram (EIC) and (B) the MS spectrum of this peak are depicted.
The reaction mixture was separated by reverse-phase HPLC and subjected to ESI/MS in the positive mode as described under Materials and methods. The major reaction product produced an intense peak at 11 min. Examination of the MS spectrum revealed that the [M + H]+ ion had a m/z 421. (A) The extracted ion chromatogram (EIC) and (B) the MS spectrum of this peak are depicted.
The reaction mixture was separated by reverse-phase HPLC and subjected to ESI/MS in the positive mode as described under Materials and methods. The major reaction product produced an intense peak at 8 min. Examination of the MS spectrum revealed that the [M + H]+ ion had a m/z 315. (A) The extracted ion chromatogram (EIC) and (B) the MS spectrum of this peak are depicted.
List of Abbreviations
- MPO
myeloperoxidase
- H2O2
hydrogen peroxidase
- HOCl
hypochlorus acid
- Hb
hemoglobin
- met-Hb
methemoglobin
- oxy-Hb
oxyhemoglobin
- HPLC
High Pressure Liquid chromatography
- RBC
Red Blood Cells
- LC-ESI-MS
Liquid Chromatography-Electro Spray Ionization-Mass Spectrometry
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ACKNOWLEDGEMENT
This work was supported by the National Institutes of Health grant RO1 HL066367 (to H. M. A.-S.), a grant from the Children’s Hospital of Michigan. SP is supported by the the Doris Duke Foundation Clinical Scientist Development Award the American College of Rheumatology Within our Reach Award, and by the Molecular Phenotyping Core of the Michigan Nutrition and Obesity Research Center (DK089503).
References
- 1.Stadler AM, Digel I, Artmann GM, Embs JP, Zaccai G, Büldt G. Hemoglobin Dynamics in Red Blood Cells: Correlation to Body Temperature. Biophysical Journal. 2008;95:5449–5461. doi: 10.1529/biophysj.108.138040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maton A. Human biology and health. Englewood Cliffs, N.J.: Prentice Hall; 1993. [Google Scholar]
- 3.Schechter AN. Hemoglobin research and the origins of molecular medicine. Blood. 2008;112:3927–3938. doi: 10.1182/blood-2008-04-078188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dominguez de Villota ED, Ruiz Carmona MT, Rubio JJ, de Andres S. Equality of the in vivo and in vitro oxygen-binding capacity of haemoglobin in patients with severe respiratory disease. Br J Anaesth. 1981;53:1325–1328. doi: 10.1093/bja/53.12.1325. [DOI] [PubMed] [Google Scholar]
- 5.Nagababu E, Rifkind JM. Heme degradation by reactive oxygen species. Antioxid Redox Signal. 2004;6:967–978. doi: 10.1089/ars.2004.6.967. [DOI] [PubMed] [Google Scholar]
- 6.Alayash AI, Patel RP, Cashon RE. Redox reactions of hemoglobin and myoglobin: biological and toxicological implications. Antioxid Redox Signal. 2001;3:313–327. doi: 10.1089/152308601300185250. [DOI] [PubMed] [Google Scholar]
- 7.Winterbourn CC. Oxidative reactions of hemoglobin. Methods Enzymol. 1990;186:265–272. doi: 10.1016/0076-6879(90)86118-f. [DOI] [PubMed] [Google Scholar]
- 8.Kikuchi G, Yoshida T, Noguchi M. Heme oxygenase and heme degradation. Biochemical and Biophysical Research Communications. 2005;338:558–567. doi: 10.1016/j.bbrc.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 9.Iakutova E, Osipov AN, Kostenko OV, Arnkhol'd I, Arnol'd K, Vladimirov Iu A. Interaction of hypochlorite with oxyhemoglobin leads to liberation of iron in a catalytically active form. Biofizika. 1992;37:1021–1028. [PubMed] [Google Scholar]
- 10.Schaefer WH, Harris TM, Guengerich FP. Characterization of the enzymatic and nonenzymatic peroxidative degradation of iron porphyrins and cytochrome P-450 heme. Biochemistry. 1985;24:3254–3263. doi: 10.1021/bi00334a027. [DOI] [PubMed] [Google Scholar]
- 11.He K, Bornheim LM, Falick AM, Maltby D, Yin H, Correia MA. Identification of the heme-modified peptides from cumene hydroperoxide-inactivated cytochrome P450 3A4. Biochemistry. 1998;37:17448–17457. doi: 10.1021/bi9808464. [DOI] [PubMed] [Google Scholar]
- 12.Mashino T, Fridovich I. Reactions of hypochlorite with catalase. Biochim Biophys Acta. 1988;956:63–69. doi: 10.1016/0167-4838(88)90298-1. [DOI] [PubMed] [Google Scholar]
- 13.Chapman AL, Winterbourn CC, Brennan SO, Jordan TW, Kettle AJ. Characterization of non-covalent oligomers of proteins treated with hypochlorous acid. Biochem J. 2003;375:33–40. doi: 10.1042/BJ20030685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Jesus-Bonilla W, Cortes-Figueroa JE, Souto-Bachiller FA, Rodriguez L, Lopez-Garriga J. Formation of compound I and compound II ferryl species in the reaction of hemoglobin I from Lucina pectinata with hydrogen peroxide. Arch Biochem Biophys. 2001;390:304–308. doi: 10.1006/abbi.2001.2392. [DOI] [PubMed] [Google Scholar]
- 15.Vissers MC, Carr AC, Chapman AL. Comparison of human red cell lysis by hypochlorous and hypobromous acids: insights into the mechanism of lysis. Biochem J. 1998;330(Pt 1):131–138. doi: 10.1042/bj3300131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vissers MC, Winterbourn CC. Oxidative damage to fibronectin. I. The effects of the neutrophil myeloperoxidase system and HOCl. Arch Biochem Biophys. 1991;285:53–59. doi: 10.1016/0003-9861(91)90327-f. [DOI] [PubMed] [Google Scholar]
- 17.Kumar S, Bandyopadhyay U. Free heme toxicity and its detoxification systems in human. Toxicology Letters. 2005;157:175–188. doi: 10.1016/j.toxlet.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 18.Nagababu E, Chrest FJ, Rifkind JM. Hydrogen-peroxide-induced heme degradation in red blood cells: the protective roles of catalase and glutathione peroxidase. Biochim Biophys Acta. 2003;1620:211–217. doi: 10.1016/s0304-4165(02)00537-8. [DOI] [PubMed] [Google Scholar]
- 19.Clark RA. Oxidative stress and "senescent" fibroblasts in non-healing wounds as potential therapeutic targets. J Invest Dermatol. 2008;128:2361–2364. doi: 10.1038/jid.2008.257. [DOI] [PubMed] [Google Scholar]
- 20.Crichton RR, Wilmet S, Legssyer R, Ward RJ. Molecular and cellular mechanisms of iron homeostasis and toxicity in mammalian cells. Journal of Inorganic Biochemistry. 2002;91:9–18. doi: 10.1016/s0162-0134(02)00461-0. [DOI] [PubMed] [Google Scholar]
- 21.Ong W-Y, Halliwell B. Iron, Atherosclerosis, and Neurodegeneration: A Key Role for Cholesterol in Promoting Iron-Dependent Oxidative Damage? Annals of the New York Academy of Sciences. 2004;1012:51–64. doi: 10.1196/annals.1306.005. [DOI] [PubMed] [Google Scholar]
- 22.Trinder D, Fox C, Vautier G, Olynyk JK. Molecular pathogenesis of iron overload. Gut. 2002;51:290–295. doi: 10.1136/gut.51.2.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pennathur S, Maitra D, Byun J, Sliskovic I, Abdulhamid I, Saed GM, Diamond MP, Abu-Soud HM. Potent antioxidative activity of lycopene: A potential role in scavenging hypochlorous acid. Free Radical Biology and Medicine. 2010;49:205–213. doi: 10.1016/j.freeradbiomed.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pullar JM, Vissers MCMM, Winterbourn CC. Living with a killer: The effects of hypochlorous acid on mammalian cells. IUBMB Life. 2000;50:259–266. doi: 10.1080/713803731. [DOI] [PubMed] [Google Scholar]
- 25.Brennan ML, Penn MS, Van Lente F, Nambi V, Shishehbor MH, Aviles RJ, Goormastic M, Pepoy ML, McErlean ES, Topol EJ, Nissen SE, Hazen SL. Prognostic value of myeloperoxidase in patients with chest pain. N Engl J Med. 2003;349:1595–1604. doi: 10.1056/NEJMoa035003. [DOI] [PubMed] [Google Scholar]
- 26.Mocatta TJ, Pilbrow AP, Cameron VA, Senthilmohan R, Frampton CM, Richards AM, Winterbourn CC. Plasma concentrations of myeloperoxidase predict mortality after myocardial infarction. J Am Coll Cardiol. 2007;49:1993–2000. doi: 10.1016/j.jacc.2007.02.040. [DOI] [PubMed] [Google Scholar]
- 27.Baldus S, Heeschen C, Meinertz T, Zeiher AM, Eiserich JP, Munzel T, Simoons ML, Hamm CW. Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation. 2003;108:1440–1445. doi: 10.1161/01.CIR.0000090690.67322.51. [DOI] [PubMed] [Google Scholar]
- 28.Kettle AJ, Winterbourn CC. Assays for the chlorination activity of myeloperoxidase. Methods Enzymol. 1994;233:502–512. doi: 10.1016/s0076-6879(94)33056-5. [DOI] [PubMed] [Google Scholar]
- 29.Weiss SJ, Klein R, Slivka A, Wei M. Chlorination of taurine by human neutrophils. Evidence for hypochlorous acid generation. J Clin Invest. 1982;70:598–607. doi: 10.1172/JCI110652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weiss SJ. Tissue destruction by neutrophils. N Engl J Med. 1989;320:365–376. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- 31.Malech HL, Gallin JI. Current concepts: Immunology - Neutrophils in human diseases. New England Journal of Medicine. 1987;317:687–694. doi: 10.1056/NEJM198709103171107. [DOI] [PubMed] [Google Scholar]
- 32.Malle E, Buch T, Grone HJ. Myeloperoxidase in kidney disease. Kidney Int. 2003;64:1956–1967. doi: 10.1046/j.1523-1755.2003.00336.x. [DOI] [PubMed] [Google Scholar]
- 33.Ohshima H, Tatemichi M, Sawa T. Chemical basis of inflammation-induced carcinogenesis. Archives of Biochemistry and Biophysics. 2003;417:3–11. doi: 10.1016/s0003-9861(03)00283-2. [DOI] [PubMed] [Google Scholar]
- 34.Schiller J, Fuchs B, Arnhold J, Arnold K. Contribution of reactive oxygen species to cartilage degradation in rheumatic diseases: Molecular pathways, diagnosis and potential therapeutic strategies. Current Medicinal Chemistry. 2003;10:2123–2145. doi: 10.2174/0929867033456828. [DOI] [PubMed] [Google Scholar]
- 35.Defrère S, Lousse JC, González-Ramos R, Colette S, Donnez J, Van Langendonckt A. Potential involvement of iron in the pathogenesis of peritoneal endometriosis. Molecular Human Reproduction. 2008;14:377–385. doi: 10.1093/molehr/gan033. [DOI] [PubMed] [Google Scholar]
- 36.Chau LY. Iron and atherosclerosis. Proc Natl Sci Counc Repub China B. 2000;24:151–155. [PubMed] [Google Scholar]
- 37.Yamaguchi K, Mandai M, Toyokuni S, Hamanishi J, Higuchi T, Takakura K, Fujii S. Contents of Endometriotic Cysts, Especially the High Concentration of Free Iron, Are a Possible Cause of Carcinogenesis in the Cysts through the Iron-Induced Persistent Oxidative Stress. Clinical Cancer Research. 2008;14:32–40. doi: 10.1158/1078-0432.CCR-07-1614. [DOI] [PubMed] [Google Scholar]
- 38.Carter P. Spectrophotometric determination of serum iron at the submicrogram level with a new reagent (ferrozine) Anal Biochem. 1971;40:450–458. doi: 10.1016/0003-2697(71)90405-2. [DOI] [PubMed] [Google Scholar]
- 39.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 40.Thomas PE, Ryan D, Levin W. An improved staining procedure for the detection of the peroxidase activity of cytochrome P-450 on sodium dodecyl sulfate polyacrylamide gels. Anal Biochem. 1976;75:168–176. doi: 10.1016/0003-2697(76)90067-1. [DOI] [PubMed] [Google Scholar]
- 41.Galijasevic S, Maitra D, Lu T, Sliskovic I, Abdulhamid I, Abu-Soud HM. Myeloperoxidase interaction with peroxynitrite: chloride deficiency and heme depletion. Free Radic Biol Med. 2009;47:431–439. doi: 10.1016/j.freeradbiomed.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fonseca AM, Porto G, Uchida K, Arosa FA. Red blood cells inhibit activation-induced cell death and oxidative stress in human peripheral blood T lymphocytes. Blood. 2001;97:3152–3160. doi: 10.1182/blood.v97.10.3152. [DOI] [PubMed] [Google Scholar]
- 43.Wang L, Bassiri M, Najafi R, Najafi K, Yang J, Khosrovi B, Hwong W, Barati E, Belisle B, Celeri C, Robson MC. Hypochlorous acid as a potential wound care agent: part I. Stabilized hypochlorous acid: a component of the inorganic armamentarium of innate immunity. J Burns Wounds. 2007;6:e5. [PMC free article] [PubMed] [Google Scholar]
- 44.Spagnuolo C, Rinelli P, Coletta M, Chiancone E, Ascoli F. Oxidation reaction of human oxyhemoglobin with nitrite: a reexamination. Biochim Biophys Acta. 1987;911:59–65. doi: 10.1016/0167-4838(87)90270-6. [DOI] [PubMed] [Google Scholar]
- 45.Ames BN, Cathcart R, Schwiers E, Hochstein P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc Natl Acad Sci U S A. 1981;78:6858–6862. doi: 10.1073/pnas.78.11.6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagababu E, Rifkind JM. Formation of fluorescent heme degradation products during the oxidation of hemoglobin by hydrogen peroxide. Biochem Biophys Res Commun. 1998;247:592–596. doi: 10.1006/bbrc.1998.8846. [DOI] [PubMed] [Google Scholar]
- 47.Harel S, Salan MA, Kanner J. Iron release from metmyoglobin, methaemoglobin and cytochrome c by a system generating hydrogen peroxide. Free Radic Res Commun. 1988;5:11–19. doi: 10.3109/10715768809068554. [DOI] [PubMed] [Google Scholar]
- 48.Florence TM. The degradation of cytochrome c by hydrogen peroxide. J Inorg Biochem. 1985;23:131–141. doi: 10.1016/0162-0134(85)83017-8. [DOI] [PubMed] [Google Scholar]
- 49.Bunn HF, Jandl JH. Exchange of heme among hemoglobins and between hemoglobin and albumin. J Biol Chem. 1968;243:465–475. [PubMed] [Google Scholar]
- 50.Schaffer SW, Azuma J, Mozaffari M. Role of antioxidant activity of taurine in diabetes. Canadian Journal of Physiology and Pharmacology. 2009;87:91–99. doi: 10.1139/Y08-110. [DOI] [PubMed] [Google Scholar]
- 51.Bouckenooghe T, Remacle C, Reusens B. Is taurine a functional nutrient? Current Opinion in Clinical Nutrition and Metabolic Care. 2006;9:728–733. doi: 10.1097/01.mco.0000247469.26414.55. [DOI] [PubMed] [Google Scholar]
- 52.Mori H, Nishida K, Ozaki T, Inoue H, Nakanishi T. Isolation of a mRNA Preferentially Expressed in Synoviocytes from Rheumatoid Arthritis That is Identical with Lumican, which Encodes a Collagen Binding, Extracellular Matrix Protein. Journal of Hard Tissue Biology. 2008;17:125–130. [Google Scholar]
- 53.Wiseman JS, Nichols JS, Kolpak MX. Mechanism of inhibition of horseradish peroxidase by cyclopropanone hydrate. J Biol Chem. 1982;257:6328–6332. [PubMed] [Google Scholar]
- 54.Furtmüller PG, Jantschko W, Regelsberger Gn, Jakopitsch C, Arnhold Jr, Obinger C. Reaction of Lactoperoxidase Compound I with Halides and Thiocyanateâ€. Biochemistry. 2002;41:11895–11900. doi: 10.1021/bi026326x. [DOI] [PubMed] [Google Scholar]
- 55.Bonini MG, Siraki AG, Atanassov BS, Mason RP. Immunolocalization of hypochlorite-induced, catalase-bound free radical formation in mouse hepatocytes. Free Radical Biology and Medicine. 2007;42:530–540. doi: 10.1016/j.freeradbiomed.2006.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nagababu E, Rifkind JM. Reaction of hydrogen peroxide with ferrylhemoglobin: superoxide production and heme degradation. Biochemistry. 2000;39:12503–12511. doi: 10.1021/bi992170y. [DOI] [PubMed] [Google Scholar]
- 57.Nicholls SJ, Hazen SL. Myeloperoxidase and Cardiovascular Disease. Arterioscler Thromb Vasc Biol. 2005;25:1102–1111. doi: 10.1161/01.ATV.0000163262.83456.6d. [DOI] [PubMed] [Google Scholar]
- 58.Kohgo Y, Ikuta K, Ohtake T, Torimoto Y, Kato J. Body iron metabolism and pathophysiology of iron overload. Int J Hematol. 2008;88:7–15. doi: 10.1007/s12185-008-0120-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sugimoto H, Spencer L, Sawyer DT. Ferric chloride-catalyzed activation of hydrogen-peroxide for the demethylation of N,N-Dimethylaniline, the epoxidation of olefins, and the oxidative cleavage of vicinal diols in acetonitrile - A reaction mimic for cytochrome-P-450. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:1731–1733. doi: 10.1073/pnas.84.7.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Groves JT, Haushalter RC, Nakamura M, Nemo TE, Evans BJ. High-valent iron-porphyrin complexes related to peroxidase and cytochrome P-450. Journal of the American Chemical Society. 1981;103:2884–2886. [Google Scholar]
- 61.Ortiz de Orue Lucana D, Roscher M, Honigmann A, Schwarz J. Iron-mediated oxidation induces conformational changes within the redox-sensing protein HbpS. J Biol Chem. 285:28086–28096. doi: 10.1074/jbc.M110.127506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nagy E, Eaton JW, Jeney V, Soares MP, Varga Z, Galajda Z, Szentmiklosi J, Mehes G, Csonka T, Smith A, Vercellotti GM, Balla G, Balla J. Red cells, hemoglobin, heme, iron, and atherogenesis. Arterioscler Thromb Vasc Biol. 30:1347–1353. doi: 10.1161/ATVBAHA.110.206433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bonini MG, Gabel SA, Ranguelova K, Stadler K, Derose EF, London RE, Mason RP. Direct magnetic resonance evidence for peroxymonocarbonate involvement in the cu,zn-superoxide dismutase peroxidase catalytic cycle. J Biol Chem. 2009;284:14618–14627. doi: 10.1074/jbc.M804644200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chapman ALP, Winterbourn CC, Brennan SO, Jordan TW, Kettle AJ. Characterization of non-covalent oligomers of proteins treated with hypochlorous acid. Biochemical Journal. 2003;375:33–40. doi: 10.1042/BJ20030685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stadtman ER, Berlett BS. Reactive Oxygen-Mediated Protein Oxidation in Aging and Disease. Chemical Research in Toxicology. 1997;10:485–494. doi: 10.1021/tx960133r. [DOI] [PubMed] [Google Scholar]
- 66.Horwich A. Protein aggregation in disease: a role for folding intermediates forming specific multimeric interactions. The Journal of Clinical Investigation. 2002;110:1221–1232. doi: 10.1172/JCI16781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of Huntingtin in Neuronal Intranuclear Inclusions and Dystrophic Neurites in Brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 68.Koo EH, Lansbury PT, Kelly JW. Amyloid diseases: Abnormal protein aggregation in neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:9989–9990. doi: 10.1073/pnas.96.18.9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vissers MC, Winterbourn CC. Oxidation of intracellular glutathione after exposure of human red blood cells to hypochlorous acid. Biochem J. 1995;307(Pt 1):57–62. doi: 10.1042/bj3070057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nagababu E, Mohanty JG, Bhamidipaty S, Ostera GR, Rifkind JM. Role of the membrane in the formation of heme degradation products in red blood cells. Life Sci. 86:133–138. doi: 10.1016/j.lfs.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nagababu E, Fabry ME, Nagel RL, Rifkind JM. Heme degradation and oxidative stress in murine models for hemoglobinopathies: thalassemia, sickle cell disease and hemoglobin C disease. Blood Cells Mol Dis. 2008;41:60–66. doi: 10.1016/j.bcmd.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sliskovic I, Abdulhamid I, Sharma M, Abu-Soud HM. Analysis of the mechanism by which tryptophan analogs inhibit human myeloperoxidase. Free Radic Biol Med. 2009;47:1005–1013. doi: 10.1016/j.freeradbiomed.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 73.Lu T, Galijasevic S, Abdulhamid I, Abu-Soud HM. Analysis of the mechanism by which melatonin inhibits human eosinophil peroxidase. Br J Pharmacol. 2008;154:1308–1317. doi: 10.1038/bjp.2008.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Galijasevic S, Abdulhamid I, Abu-Soud HM. Melatonin is a potent inhibitor for myeloperoxidase. Biochemistry. 2008;47:2668–2677. doi: 10.1021/bi702016q. [DOI] [PubMed] [Google Scholar]
- 75.Galijasevic S, Abdulhamid I, Abu-Soud HM. Potential role of tryptophan and chloride in the inhibition of human myeloperoxidase. Free Radic Biol Med. 2008;44:1570–1577. doi: 10.1016/j.freeradbiomed.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Isolated human RBC adjusted to Hb concentration of 12.5 µM was treated with different molar ratios of HOCl (0 to 160 fold) for 1 hour at room temperature. The viability of the cells was assayed by the trypan blue exclusion technique. The results are expressed as percent of cells alive as a function of molar ratio of HOCl:Hb. For details see Materials and Methods section. The above result is an average of three independent experiments and the error bars represents the standard error of measurement.
The reaction mixture from the membrane fraction was separated by reverse-phase HPLC and subjected to ESI/MS in the positive mode as described under Materials and methods. The major reaction product produced an intense peak at 8 min. Examination of the MS spectrum revealed that the [M + H]+ ion had a m/z 638. (A) The extracted ion chromatogram (EIC) and (B) the MS spectrum of this peak are depicted.
The reaction mixture was separated by reverse-phase HPLC and subjected to ESI/MS in the positive mode as described under Materials and methods. The major reaction product produced an intense peak at 11 min. Examination of the MS spectrum revealed that the [M + H]+ ion had a m/z 421. (A) The extracted ion chromatogram (EIC) and (B) the MS spectrum of this peak are depicted.
The reaction mixture was separated by reverse-phase HPLC and subjected to ESI/MS in the positive mode as described under Materials and methods. The major reaction product produced an intense peak at 8 min. Examination of the MS spectrum revealed that the [M + H]+ ion had a m/z 315. (A) The extracted ion chromatogram (EIC) and (B) the MS spectrum of this peak are depicted.