

Abstract
Proteomics is a rapidly transforming interdisciplinary field of research that embraces a diverse set of analytical approaches to tackle problems in fundamental and applied biology. This view-point article highlights the benefits of interlaboratory studies and standardization initiatives to enable investigators to address many of the challenges found in proteomics research. Among these initiatives, we discuss our efforts on a comprehensive performance standard for characterizing PTMs by MS that was recently developed by the Association of Biomolecular Resource Facilities (ABRF) Proteomics Standards Research Group (sPRG).
Keywords: LC-MS/MS, Mass spectrometry-based proteomics, Phosphopeptides, Post-translational modifications, Protein identification
Proteomics: advances and challenges
Proteomics has become an indispensable and integral part of biological, clinical, and pharmaceutical research. The use of MS for in-depth profiling of proteomes and biomolecular complexes, along with identification and localization of a vast array of PTMs, has enabled a multitude of discoveries that have been proven essential for resolving important biological questions [1–5]. However, despite rapid progress in development of powerful instrumentation, sample preparation methods, and data analysis strategies, numerous challenges remain. The biological challenges of the extremely wide dynamic range of protein concentrations in biological systems and their differential spatial-temporal agility superimpose the technological challenges, including (i) analytical variability; (ii) comprehensive characterization of trace-level and post-translationally modified proteins; (iii) elucidation of the functional roles of PTMs and their interplay; (iv) elucidation of the dynamics of protein expression, interaction, and localization in biological systems; (v) determination of tertiary structure of proteins and protein complexes and its dynamics; (vi) data processing and interpretation of the increasingly large data sets; (vii) accurate and reproducible quantitative analysis.
Interlaboratory studies and standardization initiatives in proteomics
Proteomics continues to be a multifaceted, interdisciplinary field of research that is rapidly changing. The critical importance of proteomics to biomedical investigations has made it a high priority across many disciplines. Contributions made by the field of proteomics to biological research can be attributed in part to the diversity of its analytical approaches. Such diversity has enabled the field to evolve new areas of study, improve sensitivity, and become more quantitative. The expense and sophisticated nature of the instrumentation, combined with the expertise needed to conduct the analyses, process the data, and interpret the results have been major factors influencing the establishment of proteomics shared facilities all over the world. Since proteomes are spatially, temporally, structurally, and functionally dynamic and complex, the approaches that have been developed for proteomic analysis are also diverse and complex. In view of this tremendous diversity and complexity, there is an urgent need to develop standards, guidelines, protocols, and mechanisms for evaluating lab-to-lab differences. But, the proper balance is needed between standardization and innovation so that proteomics instrumentation and methodology can continue to move forward. Individual researchers are clearly vital to these efforts. However, coordinated interlaboratory studies, where many researchers contribute their unique expertise and insight, are also essential. In general, there is a lack of agreement in the field about the best ways to assess quality control and false discovery rates. This is compounded by lack of uniformity in requirements for presentation of experimental details in publications and for deposition of supporting data files. Issues related to analytical reproducibility, reliability in identification and localization of PTMs, assessment of pre-analytical variability in sample processing, accurate statistical measures of protein inferences, and quantitative differences are additional concerns that are becoming increasingly recognized by scientific journals. Interlaboratory multidisciplinary studies and standardization initiatives are powerful ways to make advances in this arena, design roadmaps to tackle proteome complexity, and provide invaluable mechanisms for self-assessment.
Multisite collaborative efforts aimed at standardization in proteomics can be divided into four main categories: (i) development of guidelines for experimental procedures and reporting information about performed experiments; (ii) development of standards in data processing, data formats, and data sharing; (iii) development of reference standards and applications of such in assessment of analytical approaches and interlaboratory reproducibility; and (iv) interlaboratory initiatives focused on various topics related to proteomics, including development of standards.
Development of guidelines
These collaborative projects are typically organized by consortia or groups of individual researchers. Recently, the Proteomics Standards Initiative of the Human Proteome Organization (HUPO) (http://www.psidev.info) published recommendations for standardization of collection, integration, storage, and dissemination of proteomics data (“minimum information about a proteomics experiment;” the MIAPE guidelines) [6, 7]. The MIAPE guidelines followed several analogous preceding initiatives in other fields of biosciences, including transcriptomics, clinical trials, toxicology, prognostic studies, and in assessment of accuracy of diagnostic tests [7]. The guidelines were well received by the research community and inspired a variety of publications with more detailed recommendations for reporting information about proteomics experiments utilizing different experimental approaches [8–12]. No doubt this trend in standardization will be carried on with more guidelines and publications to come, while novel proteomics technologies continue to emerge. As proteomics expands from focusing mainly on discovery-driven methodology to knowledge-based and hypothesis-driven experiments that include various targeted analyses, new standardization initiatives will evidently be required. Moreover, potentially high-impact biomedical research, including large-scale clinical, biomarker discovery, and translational studies, involving the use of proteomics technologies, will require coordination of interdisciplinary resources and development of new standards for collection, storage, and processing of both specimens and data [13].
Standards in data handling
One of the most important themes of interlaboratory initiatives in proteomics is standardization of applied informatics technologies. There are several major directions in standardization efforts that can be currently identified, such as bioinformatics frameworks for data sharing, integration, processing, and interpretation. A major roadblock to these efforts is the proprietary data formats and data processing algorithms associated with commercial mass spectrometers, programming interfaces and software platforms for data acquisition and processing [14]. Availability of standardized, platform-independent computational pipelines for analysis and interpretation of proteomics data as well as establishment of common data output formats are essential for data sharing and unbiased benchmarking of technologies. Considerable efforts have been made to generate common data formats [15–20] and data format converters [14, 19, 21, 22]. In our opinion, development of standardized platforms such as ProteoWizard Toolkit will facilitate progress in development of proteomics software tools and enable rigorous assessment of diverse computational approaches to accelerate proteomics research [14].
Reference samples
An essential component for standardization of proteomics is the development of reference standards tailored in their chemical formulations for a specific type of analysis. These standards can be used for assessment of performance of analytical techniques, experimental reproducibility, sources of analytical/pre-analytical variability, quality control as well as benchmarking the power of analytical platforms and sample processing workflows within a laboratory or across multiple laboratories for a given analytical capability [23–26]. The wide variety of experimental approaches and analytical technologies in proteomics requires the development of a battery of specialized reference materials. Notable examples of well-studied complex protein standards that arose from the efforts of an interlaboratory initiative are the Universal Proteomics Standards (UPS1 and UPS2), a mixture of 48 human proteins designed and tested by the Association of Biomolecular Resource Facilities (ABRF) Proteomics Standards Research Group (sPRG) and marketed by Sigma-Aldrich [27]. Other examples include: the HUPO Gold MS Protein Standard, a mixture of 20 human proteins, developed by the joint efforts of HUPO and Invitrogen; the Complex Proteomics Standard marketed by Agilent, a protein extract from Pyrococcus furiosus, an extremophile organism of high evolutionary distance from mammals [28, 29]; and the yeast protein lysate (RM8323), a protein extract from Saccharomyces cerevisiae, developed by the National Institute of Standards and Technology (NIST) [25, 30]. There are additional commercial peptide standards designed for method development and assessing the performance of specific qualitative or quantitative applications, such as the Peptide Retention Standard (Thermo Fisher Scientific) and the MS PhosphoMix Standards (Sigma-Aldrich). Interaction between companies and academic laboratories or scientific organizations in development of specialized reference standards is often seen as more efficient than either entity working independently [27]; these collaborations generally result in more thoroughly characterized products that have been evaluated by multiple laboratories using a comprehensive set of available state-of-the-art analytical technologies. Additionally, the information generated about the quality of the standard and the performance of various workflows becomes publicly available in the form of journal articles, deposition in data repositories, and presentations at conferences, representing substantial, additional advantages over standards purely developed by commercial vendors.
Other noteworthy interlaboratory initiative in proteomics for which reference samples were distributed include the HUPO Plasma Proteome Project, focusing on cataloging the plasma proteome in a specially collected and pooled reference plasma sample [31] and the Clinical Proteomic Tumor Analysis Consortium (CPTAC) Phase II, a coordinated effort to accelerate the understanding of the molecular basis of cancer using proteomic technologies, aligned with the high-profile genomics accomplishments of The Cancer Genome Atlas initiative [32]. Phase I of the CPTAC program made strides toward understanding the scope and magnitude of common reproducibility issues associated with both discovery [30] and targeted studies [32] through self-assessment and development of standard operating procedures using a reference yeast lysate and other reference standards. Phase II CPTAC standards will include human-in-mouse xenograft tumor samples. The HUPO Plasma Proteome Project initiative effectively facilitated assessment of pre-analytical variables, including ones caused by sample type, storage, use of pro-tease inhibitors, donor background, and sample collection, as well as variables associated with the wide range of analytical techniques available for profiling of human plasma [31,33,34].
The ABRF has been sponsoring interlaboratory studies from the various Research Groups since the late 1990s. The studies have often centered around development of reference samples to be tested by multiple volunteer, participant laboratories, and have involved numerous diverse technologies, including affinity interactions, bioinformatics, flow cytometry, genomics, metabolomics, microscopy, protein sequencing and proteomics (see Research Groups at www.abrf.org) [35]. In a manner similar to the ABRF studies, several interlabo-ratory consortia have developed reference samples and distributed them among volunteer laboratories. For example, a noncommercial, technique-independent collaborative initiative called Fixing Proteomics (www.fixingproteomics.org) was established to address experimental challenges leading to cross-laboratory irreproducibility. The project initiated two studies focused on reproducibility of 2D gel electrophoresis-based proteomic profiling and on developing a reference standard. Also, several multisite qualitative and quantitative studies involving specifically designed reference samples were performed by the Spain-based consortium ProteoRed (www.proteored.org) [12, 36].
In light of the main focus of this special issue of Proteomics on PTMs, we discuss a recent ABRF-driven study of high relevance in more detail. The ABRF sPRG recently developed and thoroughly evaluated a comprehensive performance standard for characterizing PTMs by MS [37, 38]. This collection of 70 peptides that are individually modified by acetylation, methylation, nitrosylation, phosphorylation, or sulfation, (along with unmodified analogs) provides a powerful means to assess existing methodology and quality control strategies and is valuable for development of new approaches for detection of these PTMs in a complex matrix.
The impetus for creating this new PTM standard was the increased demand for identification and comprehensive characterization of PTMs in efforts to elucidate their roles in cell biology in health and disease as well in response to specific stimuli (Fig. 1). This defined PTM standard will enable laboratories to optimize their instrumentation and methods to overcome common challenges related to substoichiometry, chemical lability, ionization efficiency, peptide fragmentation, enrichment techniques, and suboptimal separation of modified peptides.
Figure 1.
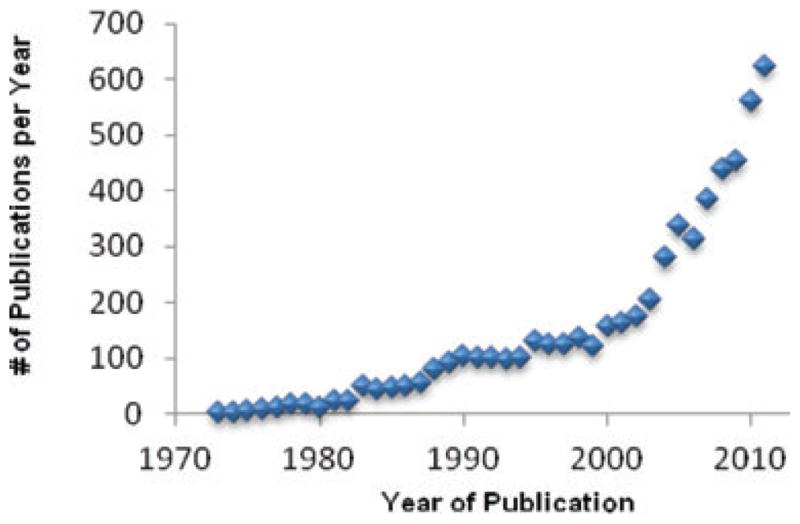
The trend in numbers of PubMed publications containing the following keywords: “PTM”, “post-translational modification”, “post translational protein modification” or “posttranslational modification”.
The synthetic peptides in the sPRG PTM standard were designed and thoroughly evaluated by the research group members using a variety of analytical and bioinformatic approaches. The final preparation was produced in collaboration with Thermo Fisher Scientific and Sigma-Aldrich and was formulated as two mixtures: peptides alone, and peptides mixed with a tryptic digest of six proteins from which the sequences of the synthetic peptides were derived. The sPRG evaluation included a stability study of the sample constituents under different storage conditions over a 3 month period. Additionally, the sPRG assembled a mass spectral library as a public resource for identification of peptides present in the standard. The standard was used in two recent ABRF-sponsored studies by the sPRG [38, 39] and the Informatics Research Group (iPRG) [40]. The sPRG study supplied the standard to volunteer participants in sufficient quantities to permit evaluation using diverse analytical approaches. A new spectral library has been generated from all of the submitted data to be used in conjunction with the standards (manuscript in preparation). The iPRG distributed a dataset produced by the sPRG resulting from LC-MS/MS analysis of the sPRG standard mixed with the digested NIST yeast protein lysate (RM8323) (manuscript submitted).
Other collaborative proteomics studies involving development of standards
There are several ongoing, large-scale interlaboratory initiatives aimed at the development of common resources of validated protein–protein interactions and protein-binding molecules for standardized characterization of the human proteome—especially its clinically relevant constituents. These programs include ProteomeBinders, AffinityProteome (both at www.proteomebinders.org), Affinomics (www.affinomics.org), Human Protein Atlas (www.proteinatlas.org), and Human Antibody Initiative (HUPO) [41–43]. A noteworthy aspect of these initiatives is their common focus on developing standard approaches and criteria for characterization of affinity reagents for analysis of the human proteome. Such criteria are virtually nonexistent despite the fact that antibody binding-based techniques are usually considered as the “gold standard” in specific detection of a protein [43, 44].
Two additional HUPO collaborative efforts include the Molecular Interaction workgroup and the Cardiovascular Initiative. Formed under umbrella of the Proteomics Standards Initiative, the Molecular Interaction workgroup is focused on the development of standardized vocabularies, data formats, and databases of protein interactions [45]. The primary focus of the Cardiovascular Initiative is to overcome challenges in cardiovascular proteomics, especially as related to data organization and unification of results obtained from different laboratories. Among other goals, this initiative also aims at crafting standard operating procedures in proteomics and advancing quantitative proteomics and PTM characterization [46]. A HUPO-driven initiative of the new Chromosome-Centric Human Proteome Project involves enhancement of community-driven standards for proteomic databases as well as standards for conducting large-scale MS- and antibody-based comparative protein and PTM analyses in the context of human disease and diversity [47,48]. The overall goals and mechanisms in support of these interlaboratory initiatives are quite different. However, there is substantial overlap in the specific aims and aspirations to further develop standards in these areas of interest. The joint efforts of multiple proteomics laboratories have to be coordinated to meet the proposed goals and to overcome resource limitations and differences in available technologies in a single research group.
Outlook for the future interlaboratory studies and development of new standards
The progress in identifying reliable biomarkers, elucidating the functions of PTMs and revealing molecular mechanisms of disease and biological phenomena has been slow, due mostly to the tremendous complexity of biological systems and the technological challenges of the analyses. The use of interlaboratory collaborative proteomic initiatives aimed at the standardization and enhancement of analytical strategies is an effective mechanism to accelerate progress in this arena, especially, in the current financial climate. Using a collaborative strategy, multiple sites can “divide and conquer” by using a variety of alternative approaches to more effectively tackle the analytical challenges and issues related to bioinformatics analysis. Researchers of vastly different expertise can productively collaborate in reaching common goals while concomitantly establishing “best practices” for proteomics.
We encourage researchers to join in and contribute to the interlaboratory efforts that are aimed at further advancement of proteomics. Participation in such initiatives will assist researchers in fulfilling their scientific interests, improving the performance and capabilities of their laboratories, expanding their collaborative networks, developing a creative climate for synergistic idea exchange, and, ultimately, improving their competitiveness for funding. The outcomes of interlaboratory collaborative initiatives greatly depend on the level of energy, creativity, and expertise of the participants. We hope that scientific journals and funding agencies will continue to recognize and support these interlaboratory efforts and will encourage researchers to participate in multisite studies by publishing results of these studies and adopting the standards developed in such collaborative initiatives. We expect that interlaboratory efforts such as the ones organized by ABRF, HUPO, CPTAC, and other consortia will ultimately lead to new exciting discoveries and a better understanding of the molecular mechanisms of biology and disease. In our opinion, in addition to past and ongoing studies discussed above, there is a strong need for future collaborative efforts that focus on development of new standards for remaining challenges in proteomics such as: quantitative profiling of physiological fluids, tissues, and organelles; characterization samples with limited protein availability (including single-cell analysis); elucidation of dynamic changes in molecular profiles (including the interplay and combinatorial pattern of PTMs); analysis of functional and interaction pathways; systemic molecular profiling, combining “metaomics” data sets (including genomics, metabolomics, proteomics, and transcriptomics). The benefits of future interlaboratory proteomics initiatives and further standardization of various aspects of proteomics include enhanced technology dissemination, improved inter-laboratory reproducibility, and of progress in fundamental and applied research.
Abbreviations
- ABRF
Association of Biomolecular Resource Facilities
- CPTAC
Clinical Proteomic Tumor Analysis Consortium
- HUPO
Human Proteome Organization
- sPRG
Proteomics Standards Research Group
Footnotes
The authors have declared no conflict of interest.
References
- 1.Domon B, Aebersold R. Mass spectrometry and protein analysis. Science. 2006;312:212–217. doi: 10.1126/science.1124619. [DOI] [PubMed] [Google Scholar]
- 2.Hood LE, Omenn GS, Moritz RL, Aebersold R, et al. New and improved proteomics technologies for understanding complex biological systems: addressing a grand challenge in the life sciences. Proteomics. 2012;12:2773–2783. doi: 10.1002/pmic.201270086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choudhary C, Mann M. Decoding signalling networks by mass spectrometry-based proteomics. Nat Rev Mol Cell Biol. 2010;11:427–439. doi: 10.1038/nrm2900. [DOI] [PubMed] [Google Scholar]
- 4.Cravatt BF, Simon GM, Yates JR. 3rd, The biological impact of mass-spectrometry-based proteomics. Nature. 2007;450:991–1000. doi: 10.1038/nature06525. [DOI] [PubMed] [Google Scholar]
- 5.Dix MM, Simon GM, Wang C, Okerberg E, et al. Functional interplay between caspase cleavage and phosphorylation sculpts the apoptotic proteome. Cell. 2012;150:426–440. doi: 10.1016/j.cell.2012.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orchard S, Hermjakob H, Julian RK, Jr, Runte K, et al. Common interchange standards for proteomics data: public availability of tools and schema. Proteomics. 2004;4:490–491. doi: 10.1002/pmic.200300694. [DOI] [PubMed] [Google Scholar]
- 7.Taylor CF, Paton NW, Lilley KS, Binz PA, et al. The minimum information about a proteomics experiment (MI-APE) Nat Biotechnol. 2007;25:887–893. doi: 10.1038/nbt1329. [DOI] [PubMed] [Google Scholar]
- 8.Robin X, Hoogland C, Appel RD, Lisacek F. MIA-PEGelDB, a web-based submission tool and public repository for MIAPE gel electrophoresis documents. J Proteomics. 2008;71:249–251. doi: 10.1016/j.jprot.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 9.Domann PJ, Akashi S, Barbas C, Huang L, et al. Guidelines for reporting the use of capillary electrophoresis in proteomics. Nat Biotechnol. 2010;28:654–655. doi: 10.1038/nbt0710-654b. [DOI] [PubMed] [Google Scholar]
- 10.Jones AR, Carroll K, Knight D, Maclellan K, et al. Guidelines for reporting the use of column chromatography in proteomics. Nat Biotechnol. 2010;28:654. doi: 10.1038/nbt0710-654a. [DOI] [PubMed] [Google Scholar]
- 11.Hoogland C, O’Gorman M, Bogard P, Gibson F, et al. Guidelines for reporting the use of gel image informatics in proteomics. Nat Biotechnol. 2010;28:655–656. doi: 10.1038/nbt0710-655. [DOI] [PubMed] [Google Scholar]
- 12.Medina-Aunon JA, Martinez-Bartolome S, Lopez-Garcia MA, Salazar E, et al. The ProteoRed MIAPE web toolkit: a user-friendly framework to connect and share proteomics standards. Mol Cell Proteomics. 2011;10:M111 008334. doi: 10.1074/mcp.M111.008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poste G. Bring on the biomarkers. Nature. 2011;469:156–157. doi: 10.1038/469156a. [DOI] [PubMed] [Google Scholar]
- 14.Chambers MC, Maclean B, Burke R, Amodei D, et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat Biotechnol. 2012;30:918–920. doi: 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deutsch E. mzML: a single, unifying data format for mass spectrometer output. Proteomics. 2008;8:2776–2777. doi: 10.1002/pmic.200890049. [DOI] [PubMed] [Google Scholar]
- 16.Cote RG, Reisinger F, Martens L. jmzML, an open-source Java API for mzML, the PSI standard for MS data. Proteomics. 2010;10:1332–1335. doi: 10.1002/pmic.200900719. [DOI] [PubMed] [Google Scholar]
- 17.Martens L, Chambers M, Sturm M, Kessner D, et al. mzML–a community standard for mass spectrometry data. Mol Cell Proteomics. 2011;10:R110 000133. doi: 10.1074/mcp.R110.000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pedrioli PG, Eng JK, Hubley R, Vogelzang M, et al. A common open representation of mass spectrometry data and its application to proteomics research. Nat Biotechnol. 2004;22:1459–1466. doi: 10.1038/nbt1031. [DOI] [PubMed] [Google Scholar]
- 19.Keller A, Eng J, Zhang N, Li X-j, Aebersold R. A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol Syst Biol. 2005;1:2005.0017. doi: 10.1038/msb4100024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schramm T, Hester A, Klinkert I, Both JP, et al. imzML–a common data format for the flexible exchange and processing of mass spectrometry imaging data. J Proteomics. 2012;75:5106–5110. doi: 10.1016/j.jprot.2012.07.026. [DOI] [PubMed] [Google Scholar]
- 21.Kessner D, Chambers M, Burke R, Agus D, Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24:2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sturm M, Bertsch A, Gropl C, Hildebrandt A, et al. OpenMS – an open-source software framework for mass spectrometry. BMC Bioinformatics. 2008;9:163. doi: 10.1186/1471-2105-9-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kocher T, Pichler P, Swart R, Mechtler K. Quality control in LC-MS/MS. Proteomics. 2011;11:1026–1030. doi: 10.1002/pmic.201000578. [DOI] [PubMed] [Google Scholar]
- 24.Pichler P, Mazanek M, Dusberger F, Weilnbock L, et al. SIMPATIQCO: a server-based software suite which facilitates monitoring the time course of LC-MS performance metrics on Orbitrap instruments. J Proteome Res. 2012;11:5540–5547. doi: 10.1021/pr300163u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rudnick PA, Clauser KR, Kilpatrick LE, Tchekhovskoi DV, et al. Performance metrics for liquid chromatography-tandem mass spectrometry systems in proteomics analyses. Mol Cell Proteomics. 2010;9:225–241. doi: 10.1074/mcp.M900223-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lilley KS, Deery MJ, Gatto L. Challenges for proteomics core facilities. Proteomics. 2011;11:1017–1025. doi: 10.1002/pmic.201000693. [DOI] [PubMed] [Google Scholar]
- 27.Andrews PC, Arnott DP, Gawinowicz MA, Kowalak JA, et al. ABRF 2006. Long Beach, CA: 2006. [Google Scholar]
- 28.Vaudel M, Burkhart JM, Breiter D, Zahedi RP, et al. A complex standard for protein identification, designed by evolution. J Proteome Res. 2012;11:5065–5071. doi: 10.1021/pr300055q. [DOI] [PubMed] [Google Scholar]
- 29.Vaudel M, Burkhart JM, Radau S, Zahedi RP, et al. Integral quantification accuracy estimation for reporter ion-based quantitative proteomics (iQuARI) J Proteome Res. 2012;11:5072–5080. doi: 10.1021/pr300247u. [DOI] [PubMed] [Google Scholar]
- 30.Paulovich AG, Billheimer D, Ham AJ, Vega-Montoto L, et al. Interlaboratory study characterizing a yeast performance standard for benchmarking LC-MS platform performance. Mol Cell Proteomics. 2010;9:242–254. doi: 10.1074/mcp.M900222-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Omenn GS. International collaboration in clinical chemistry and laboratory medicine: the Human Proteome Organization (HUPO) Plasma Proteome Project. Clin Chem Lab Med. 2004;42:1–2. doi: 10.1515/CCLM.2004.001. [DOI] [PubMed] [Google Scholar]
- 32.Addona TA, Abbatiello SE, Schilling B, Skates SJ, et al. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat Biotechnol. 2009;27:633–641. doi: 10.1038/nbt.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rai AJ, Gelfand CA, Haywood BC, Warunek DJ, et al. HUPO Plasma Proteome Project specimen collection and handling: towards the standardization of parameters for plasma proteome samples. Proteomics. 2005;5:3262–3277. doi: 10.1002/pmic.200401245. [DOI] [PubMed] [Google Scholar]
- 34.Gelfand CA, Omenn GS. In: Sample Preparation in Biological Mass Spectrometry. Lazarev AV, Ivanov AR, editors. Springer–Verlag; Heidelberg: 2011. pp. 269–289. [Google Scholar]
- 35.Friedman DB, Andacht TM, Bunger MK, Chien AS, et al. The ABRF Proteomics Research Group studies: educational exercises for qualitative and quantitative proteomic analyses. Proteomics. 2011;11:1371–1381. doi: 10.1002/pmic.201000736. [DOI] [PubMed] [Google Scholar]
- 36.Paradela A, Escuredo PR, Albar JP. Geographical focus. Proteomics initiatives in Spain: ProteoRed. Proteomics. 2006;6(Suppl 2):73–76. doi: 10.1002/pmic.200600487. [DOI] [PubMed] [Google Scholar]
- 37.Ivanov AR, Colangelo C, Dufresne C, Farmar J, et al. 59th ASMS Conference on Mass Spectrometry and Allied Topics; Elsevier; 2011. [Google Scholar]
- 38.Ivanov AR, Colangelo C, Dufresne C, Farmar J, et al. ABRF 2011: Technologies to Enable Personalized Medicine, ABRF; San Antonio, TX. 2011. p. 54. [Google Scholar]
- 39.Ivanov AR, Colangelo C, Dufresne CP, Friedman DB, et al. The 60th ASMS Conference on Mass Spectrometry and Allied Topics; Vancouver, BC, Canada: Springer; 2012. [Google Scholar]
- 40.Cottrell J, Clauser KR, Chalkley RG, Sun R, et al. ABRF 2012; Learning from Molecules. The Technology behind the Story; Orlando, FL: ABRF; 2012. [Google Scholar]
- 41.Stoevesandt O, Taussig MJ. European and international collaboration in affinity proteomics. N Biotechnol. 2012;29:511–514. doi: 10.1016/j.nbt.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 42.Gloriam DE, Orchard S, Bertinetti D, Bjorling E, et al. A community standard format for the representation of protein affinity reagents. Mol Cell Proteomics. 2010;9:1–10. doi: 10.1074/mcp.M900185-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taussig MJ, Stoevesandt O, Borrebaeck CA, Bradbury AR, et al. ProteomeBinders: planning a European resource of affinity reagents for analysis of the human proteome. Nat Methods. 2007;4:13–17. doi: 10.1038/nmeth0107-13. [DOI] [PubMed] [Google Scholar]
- 44.Uhlen M, Ponten F. Antibody-based proteomics for human tissue profiling. Mol Cell Proteomics. 2005;4:384–393. doi: 10.1074/mcp.R500009-MCP200. [DOI] [PubMed] [Google Scholar]
- 45.Orchard S. Molecular interaction databases. Proteomics. 2012;12:1656–1662. doi: 10.1002/pmic.201100484. [DOI] [PubMed] [Google Scholar]
- 46.Lam MP, Vivanco F, Scholten A, Hermjakob H, et al. HUPO 2011: The new Cardiovascular Initiative–integrating proteomics and cardiovascular biology in health and disease. Proteomics. 2012;12:749–751. doi: 10.1002/pmic.201270015. [DOI] [PubMed] [Google Scholar]
- 47.Paik YK, Omenn GS, Uhlen M, Hanash S, et al. Standard guidelines for the chromosome-centric human proteome project. J Proteome Res. 2012;11:2005–2013. doi: 10.1021/pr200824a. [DOI] [PubMed] [Google Scholar]
- 48.Paik YK, Jeong SK, Omenn GS, Uhlen M, et al. The Chromosome-Centric Human Proteome Project for cataloging proteins encoded in the genome. Nat Biotechnol. 2012;30:221–223. doi: 10.1038/nbt.2152. [DOI] [PubMed] [Google Scholar]