

Abstract
Cellular production of such cytokines as interferon (IFN)-γ and tumor necrosis factor (TNF)-α is used to determine disease-specific immune responses and may be used to diagnose infectious diseases such as tuberculosis. In this paper, we describe the development of micropatterned electrodes functionalized with electroactive aptamers for multiplexed detection of immune-cell-produced cytokines. A sequence of electrode deprotection and aptamer incubation steps were used to assemble anti–IFN–γ DNA aptamers and anti-TNF-α RNA aptamers on individually addressable half-ring electrodes. Aptamer molecules were thiolated for assembly on gold and were functionalized with methylene blue redox reporter for electrochemical signal transduction. Specificity of individual sensors to the correct cytokine species was confirmed by exposure to recombinant cytokines. For cell detection experiments, electrode arrays were integrated into microfluidic devices and incubated with immune cells. Design of the surface was such that a small group of ~400 cells attached in the circular adhesion sites surrounded by half-ring electrodes sensing IFN-γ and TNF-α. The microdevice consisted of two parallel microfluidic channels, each channel containing four cell capture/sensing sites. Upon mitogenic activation, secreted IFN-γ and TNF-α molecules were monitored by performing square wave voltammetry (SWV) at different time points at individually addressable electrodes. This biosensing platform was used to analyze the quantity and rate of cytokine release from primary T cells and a monocyte cell line. Upon further development of this platform may be enhanced to enable detection of larger number of cytokines and used to correlate the levels and dynamics of cytokine release in immune cells to diagnosis and treatment of infectious diseases.
Keywords: Cytokine biosensors, Aptasensors, Blood analysis, Surface modification, Micropatterning
1. Introduction
Cytokines are signaling proteins often secreted by immune cells in order to regulate immune response, wound healing, and tissue regeneration. In immunology, IFN-γ production by T helper cells (CD4+) upon stimulation with antigens is used to determine prior exposure to infectious diseases. In fact, IFN-γ release by T cells is employed clinically to diagnose latent tuberculosis and is used in basic immunology to determine presence of disease-specific T cells [1,2]. TNF-α is another important inflammatory cytokine produced by immune and non-immune cells [3–5]. In an effort to determine blood-based biomarkers of infectious diseases, increasing attention is focused on assessing how cytokine profiles correlate with disease diagnosis, progression or regression. There is therefore a strong impetus to develop new technologies for cytokine detection.
At the present time, antibody-based immunoassays such as ELISA/ELISpot represent the gold standard in cytokine detection [6–8]. While these immunoassays are sensitive and specific to cytokines, they are often cumbersome, expensive, and time-consuming, involving multiple washing steps and requiring large sample volumes for analysis. Recently, miniaturization and microfluidics have been used to multiplex immunoassays, scale down volume requirements, and enable novel features such as single cell analysis [9–13]. However, these miniature assays rely on antibodies for cytokine detection, require secondary labeling to develop the signal, and are somewhat difficult to adapt for continuous monitoring of cytokine release.
Aptamers are emerging as viable alternatives to antibodies, offering the advantages of high thermal/chemical stability, regenerability, and ease of modification [14–16]. Composed of DNA or RNA molecules, aptamers have been selected for binding of targets ranging from small molecules to proteins [17–23] and have been used as biorecognition elements in electrochemical, optical, and mass-sensitive biosensors [24–27]. Previously, our lab has developed a DNA hairpin aptamer for IFN-γ detection. The hairpin was thiolated, conjugated with methylene blue (MB) (a redox reporter), and immobilized on a gold electrode [28]. Binding of IFN-γ caused a conformational change in the aptamer, resulting in a decrease of the redox current. Most recently, these electrochemical aptamer beacons were immobilized onto micropatterned gold electrodes and were used for continuous monitoring of IFN-γ release from primary T cells captured on the same surface [29]. The present paper builds on these previous studies by describing a strategy for co-immobilizing aptamers against IFN-γ and TNF-α on micropatterned electrodes and then deploying these sensing surfaces for cell analysis (see Fig. 1). Surface modification, biosensor characterization, and continuous monitoring of cytokine release from living cells are described.
Fig. 1.
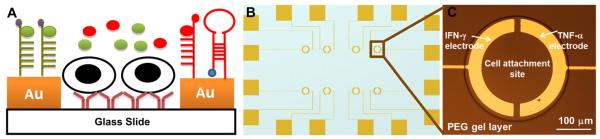
(A) Schematic illustration depicting selective modification of gold electrodes with different cytokine-binding aptamers. A pair of half ring-shaped Au electrodes fabricated on glass slides are embedded inside one PEG hydrogel and incubated with antibodies for immune cell capture. Upon injection of cells, T cells and human monocytes are bound on Abmodified glass regions. Two cytokine-binding aptamers are respectively modified on individually addressable electrodes for detecting cytokine release in real time (B) Electrode layout, where the overall device size is half of a glass slide (25 mm × 37.5 mm) (C) 300 μm diameter of PEG wells are used to capture approximately 400 cells inside one well.
2. Materials and methods
2.1. Materials
10× phosphate-buffered saline (PBS) without calcium and magnesium, Na4EDTA, KHCO3, NH4Cl, anhydrous toluene (99.9%), 11-Mercaptoundecanoic acid (MUA), 200-proof ethanol, poly(ethylene glycol) diacrylate (PEG-DA, MW 575), and 2-hydroxy-2-methyl-propiophenone (photoinitiator) were purchased from Sigma–Aldrich (St. Louis, MO). Chromium etchant (CR-4S) and gold etchant (Au-5) were purchased from Cyantek Corporation (Fremont, CA). Positive photoresist (S1813) and developer solution (MIF-318) were purchased from Shipley (Marlborough, MA). 3-Acryloxypropyl trichlorosilane, was purchased from Gelest, Inc. (Morrisville, PA). Monoclonal purified mouse anti-human CD4 Abs (13B8.2) from Beckman-Coulter (Fullerton, CA). Anti-human CD3–FITC (UCHT1) and anti-CD4-PE (L120) were used for immunostaining of surface-bound cells and were purchased from BD Pharmingen. Human recombinant IFN-γ, TNF-α, Interleukin-2, Interleukin-12, Interleukin-17 were from R&D Systems (Minneapolis, MN). T cell and monocyte activation reagents: phorbol 12-myristate 13-acetate (PMA) and ionomycin were purchased from Sigma–Aldrich (St. Louis, MO). Cell culture medium RPMI 1640 with l-glutamine was purchased from VWR. Glass slides (25 mm × 75 mm × 1 mm) were obtained from VWR (West Chester, PA). Sodium bicarbonate (NaHCO3), 6-mercapto-1-hexanol (MCH), tris-(2-carboxyethyl) phosphine hydrochloride (TCEP) were purchased from Sigma–Aldrich (St. Louis, MO). Methylene Blue (MB), carboxylic acid, succinimidyl ester (MB-NHS, Biosearch Technologies, INC, Novato, CA). All chemicals were used without further purification. The 34-mer IFN-γ-binding aptamer sequence (IDT Technologies, San Diego, CA) was as follows: 5′-NH2-C6-GGGGTTGGTTGTGTTGGGTGTTGTGTCCAACCCCC3 SH-3′. The 28-mer TNF-α-binding aptamer sequence (also from IDT Technologies) was as follows: 5′/5AmMC6/rG*rG*rA*rG*rU*rA*rU*rC*rU*rG*rA*rU*rG*rA*rC*rA*rA*rU*rU*rC*rG*rG*rA*rG*rC*rU*rC*rC/3ThioMC3-D/-3′. Phosphorothioates (or S-oligos, marked with *) were used to stabilize RNA against RNase degradation. Both TNF-α and IFN-γ aptamers were modified at the 3-terminus with a C6-disulfide [HO(CH2)6–S–S–(CH2)6–] linker and at the 5-end with an amine group for redox probe (MB) conjugation. The aptamers were dissolved in 1 × PBS buffer (pH 7.4).
2.2. Attachment of redox labels to aptamers
The methylene blue (MB) redox reporter was conjugated to IFN-γ and TNF-α aptamers using previously reported protocols [29,30]. Briefly, NHS-labeled MB was conjugated to the 3′-end of an amino-modified DNA/RNA aptamer through succinimide ester coupling. First, the MB-NHS solution was dissolved in 10 μL of DMF and 50 μL of 0.5 m NaHCO3, and then added to 50 μL of 100 μm aptamer solution. Upon stirring, the compounds were allowed to react for 4 h at room temperature in the dark. To concentrate and purify the MB-modified aptamers, the resulting sample was filtered using a centrifugal filter (Millipore, Amicon Ultra 3 K 0.5 mL). Prior to use, stock aptamer solution of 50 μm was kept at −20 °C.
2.3. Fabrication of Au electrode arrays
The electrode array layout was designed in AutoCAD and converted into plastic transparencies by CAD/Art Services (Portland, OR). The electrodes were fabricated using standard photolithography and metal etching processes described previously [29]. Glass slides (75 mm × 25 mm) were sputter-coated with 15 nm Cr adhesion layer and 100 nm Au layer (Lance Goddard Associates, Santa Clara, CA). The Au/Cr layers were etched to create eight pairs of half-ring shaped (100 μm wide with 300 μm inner diameter) micropatterned electrodes that were individually connected to 2 mm × 2 mm contact pad via 15 μm wide leads. Wires were soldered to contact pads to connect electrode arrays to a home-built multiplexing setup capable of automated data acquisition. It should be noted that the photoresist layer was not removed immediately after metal etching but was kept for protection of the underlying Au regions during the silane modification protocol.
2.4. Packaging Au electrodes in PEG hydrogel
The glass substrates with photoresist-covered Au electrodes were modified with an acrylated silane coupling agent [29] to ensure covalent anchoring of hydrogel structures onto glass substrates. After silane modification, substrates were sonicated in acetone for 2 min to remove photoresist and then placed in an oven for 3 h at 100 °C to fully crosslink the silane layer. To create non-fouling PEG hydrogel micropatterns, a prepolymer solution containing PEG-diacrylate (PEG-DA) (MW 575) and 2% (v/v) photoinitiator was spin-coated at 800 rpm for 4 s onto the patterned substrate. A photomask was aligned to fiduciary marks using a mask aligner, followed by 1 s UV exposure at 60 mJ/cm2, using an OmniCure series 1000 light source or a Canon PLA-501F mask aligner. The regions exposed to UV light underwent radical polymerization and became cross-linked, while the unexposed regions were dissolved in DI water after 5 min of development. In this way, 400 μm diameter glass/Au regions were formed within the non-fouling hydrogel layer. This micropatterning strategy allowed for targeted attachment of cells next to electrodes.
2.5. Characterization of sensitivity and specificity of aptasensors
Electrochemical measurements were carried out with a CHI 842B potentiostat (CH Instruments, Austin, TX) operating inside a homemade Faraday cage. A three-electrode electrochemical cell was constructed based on a microfluidic device, with a flow-through Ag/AgCl (3 m KCl) reference electrode in the outlet, a Pt wire counter electrode in the inlet and aptamer-modified electrodes serving as working electrodes. Measurements were performed using SWV with the following parameters: 40 mV amplitude, 60 Hz frequency, and a voltage range of −0.10 to −0.50 V. An automated switching system was used to sample individual electrodes at predefined time intervals.
For IFN-γ aptamer immobilization, Au electrodes were kept in an aqueous solution for 2 h in the dark. Following incubation, the electrodes were rinsed with copious amounts of DI water and then immersed in an aqueous solution of 3 mm 6-mercapto-1-hexanol solution (MCH) for 1 h to displace non-specifically adsorbed aptamer molecules and to passivate the electrode surface. Aptamer-coated electrodes were allowed to equilibrate in PBS buffer (pH 7.4) for 30 min, as determined by stable faradaic current, and were then challenged with human recombinant IFN-γ dissolved in PBS buffer. Before conducting voltammetric measurements, the sensor was allowed to react with the analyte for 15 min. After cytokine detection experiments, aptasensors could be regenerated by immersion in 7 m urea buffer for 1 min followed by copious rinsing with DI water.
Prior to modification of the electrodes, the TNF-α aptamer stock solution (50 μm) was reduced in 10 mm TCEP for 1 h to cleave disulfide bonds. Electrodes were then immersed in an aqueous TNF-α binding aptamer solution at a diluted concentration of 0.5 μm, followed by rinsing with of DI water. Finally, the electrodes were immersed in an aqueous solution of 3 mm 6-mercapto-1-hexanol solution (MCH) for 1 h same as described above for IFN-γ aptasensor immobilization. The sensor was allowed to equilibrate as determined by stable faradaic current obtained via SWV, and was then challenged with human recombinant TNF-α dissolved in RPMI media solution.
Calibration of the TNF-α aptasensor was performed by challenging the sensor with known concentrations of TNF-α (ranging from 10 to 500 ng/mL) dissolved in RPMI 1640 media. During detection of recombinant TNF-α, the microchip was placed onto a custom-made heating stage to maintain temperature of 37 °C. These steps were taken to mimic conditions used in cell detection experiments described below.
To demonstrate specificity, TNF-α aptasensor was challenged with 50 ng/mL of non-specific cytokines dissolved in PBS. The cytokines tested included IL-12, IL-2, IL-17 and IFN-γ, along with the analyte of interest, TNF-α also at 50 ng/mL and 100 ng/mL.
2.6. Assembly of aptasensors for simultaneous detection of IFN-γ and TNF-α
The micropatterned gold electrodes were passivated with a monolayer of 11-Mercaptoundecanoic acid (MUA) formed by immersing the electrodes in a 3 mm ethanolic solution of MUA for 1 h. All electrochemical reductive desorption experiments were performed in 1× PBS using a standard three-electrode system. A negative potential (−1.2 V vs Ag/AgCl) was applied to the desired electrodes for 1 min in order to remove the MUA monolayer from these individually addressable electrodes. Following the reductive desorption of the MUA sacrificial layer, the Au electrodes were kept in an aqueous solution of IFN-γ aptamer (0.5 μm) for 1 h in the dark, followed by passivation with MCH. This sequence of steps resulted in selective assembly of IFN-γ aptamers on specific electrodes. Next, the gold electrodes still passivated with MUA were deprotected by applying a negative reductive potential, incubated with 0.5 μms aqueous solution of TNF-α aptamer for 1 h, and finally passivated by exposure to 3 mm MCH in water.
To test for cross-contamination, electrodes were challenged with either IFN-γ or TNF-α at 50 ng/mL concentration. Responses of electrodes to “correct” and “incorrect” cytokine molecules were measured using SWV as described above.
2.7. Integration of aptasensors with microfluidics and capture of immune cells
Microfluidic channels were fabricated out of poly (dimethyl siloxane) (PDMS) using standard soft lithography approaches. These devices were secured on micropatterned substrates using vacuum suction as described previously [31,32]. The microfluidic device contained two flow chambers with width-length-height dimensions of 3 × 10 × 0.1 mm as well as a web of auxiliary channels for applying negative pressure.
Following Au electrode modification with IFN-γ and TNF-α aptamers, micropatterned surfaces were incubated with either anti-CD4 or anti-CD14 Ab (0.1 mg/mL in 1 × PBS) to capturing T cells or monocytes respectively. IFN-γ/TNF-α detection from primary T cells was performed using red blood cell (RBC)-depleted whole blood. Blood was collected from healthy adult donors through venipuncture under sterile conditions with informed consent and approval of the Institutional Review Board of the University of California at Davis (protocol number 200311635–6). RBCs were lysed using an ammonia chloride-based erythrocyte lysis solution (89.9 g NH4Cl, 10.0 g KHCO3, and 370.0 mg tetrasodium EDTA in 10 L of deionized water) as described previously [31].
In other experiments, a human monocyte U937 cell line was used. Human monocytic cell line, CRL-1593.2/U-937 (American Type Culture Collection, ATCC), were cultured in suspension in 25 or 75 cm2 tissue culture flasks and incubated in RPMI 1640 supplemented with 10% FBS and 100 U/mL penicillin/streptomycin and l-glutamine. Cells were seeded at 1 × 106/mL, passaged three times per week, and cultured under a 5% CO2/95% air humidified atmosphere at 37 °C.
The cell suspension was introduced into fluidic chambers at flow rate of 20 μL/min. Given the dimensions of the channel this flow rate translates into shear stress of ~0.8 dyn/cm2. Upon filling the chamber with cells, the flow was stopped for 15 min to allow for cell capture. In order to wash away non-specific cells, the flow rate was raised to 100 μL/min for ~15 min.
A human T cell line, MOLT-3 (American Type Culture Collection, ATCC) was used as a negative control in cytokine detection experiments. Cells were cultured in suspension in 25 or 75 cm2 tissue culture flasks and incubated in RPMI 1640 supplemented with 10% FBS and 100 U/mL penicillin/streptomycin and l-glutamine. Cells were seeded at 1 × 106/mL, passaged three times per week, and cultured under a 5% CO2/95% air humidified atmosphere at 37 °C.
2.8. Electrochemical detection of IFN-γ and TNF-α release from captured cells
To analyze IFN-γ/TNF-α release from cells, microfluidic devices were incubated with either RBC-depleted blood or a monocyte cell population. After capturing cells, microfluidic devices were infused with PMA (50 ng/mL) and ionomycin (2 nm) dissolved in phenol-red-free RPMI 1640 media without serum. A surgical clamp was secured around the outlet tubing to eliminate convective mixing. The microdevice was then placed on top of a heating stage (37 °C) and aptasensor electrodes were connected to a potentiostat via a multiplexer (NI-ER16, National Instruments, TX) (a layout of the experimental setup is shown in Figure S1). Our microchips contained eight addressable electrode pairs, with four pairs per microfluidic channel. One channel was used to quantify IFN-γ/TNF-α release from mitogenically stimulated cells whereas the other channel was used to monitor cytokine production in unactivated cells. In these experiments, SWV measurements were made every 15 min for up to 4 h. The microsystems were placed on top of the heating stage to maintain temperature at 37 °C for the duration of experiment.
2.9. Determining cytokine production rates
IFN-γ and TNF-α production rates per cell were calculated using COMSOL Multiphysics (COMSOL, Inc., Burlington, MA). A three-dimensional model was constructed based on the 3 × 10 × 0.1 mm microfluidic chamber. After applying a flux boundary condition on the cell capture area and insulating boundary conditions on all other surfaces, the average profile of measured IFN-γ/TNF-α concentrations at each electrode were then fit against the predicted values from the numerical simulation. Using data from the first 60 min of production, a scaled least-squares regression was used to determine the best-fit production rate to 0.0001 pg/cell/h precision. Complete details are available in the Supporting Information.
3. Results and discussion
This paper describes aptasensing surfaces integrated with microfluidics to enable detection of IFN-γ and TNF-α release from immune cells (see Fig. 1). The novelty of this work lies in creating bi-functional aptasensors for simultaneous, local detection of two cytokines produced by a small number of cells.
3.1. Fabrication of micropatterned surfaces
The micropatterned sensing surfaces were designed to enable detection of IFN-γ and TNF-α molecules appearing in the immediate vicinity of cells. We chose to label aptamers with the same redox reporter molecule, MB, and then resolve the signal by placing each aptamer type on its own electrode (Fig. 1 A). The surface modification process began by microfabrication of Au electrodes onto glass slides. A layout of individual electrodes within the arrays is shown in Fig. 1 B. These slides were then modified with acrylate-terminated silane to promote attachment of PEG hydrogel microstructures to glass regions. As shown in Fig. 1 C, after patterning of the hydrogel each cell culture/sensing region contained a 400 μm diameter PEG well, inside the well were Au half-ring electrodes 100 μm wide and a 300 μm diameter cell attachment site.
3.2. Characterization of specificity and sensitivity of TNF-α sensor
We previously reported the development and characterization an electrochemical aptasensor for IFN-γ detection. Our IFN-γ aptasensor was based on a DNA aptamer hairpin labeled with MB redox probe and immobilized on a gold surface via thiol-gold chemistry. The limit of detection for optimized IFN-γ aptasensor was 0.06 nm with linear response extending to 10 nm [33]. The goal of the present paper was to develop an aptasensor for TNF-α detection and to combine this novel sensor with the existing IFN-γ sensor for dual cytokine detection. While the sequence of RNA aptamer against TNF-α was reported in the literature [34], to the best of our knowledge, an aptasensor for detection of this cytokine has not been demonstrated to date. The susceptibility of wild-type RNA aptamers to degradation by endogenous ribonucleases may compromise sensor performance [18,35]. One strategy to stabilize oligonucleotides is to replace nonbridging oxygens in the backbone with sulfur, producing phosphorothioates (or S-oligos) [36,37]. This modification was employed in the present study to improve the stability of the RNA TNF-α aptamers.
The response of aptamer-modified electrodes to varying concentrations of TNF-α was characterized by SWV and is presented in Fig. 2A. The binding of the cytokine molecules causes aptamer conformation to change and may also insulate the electrode. This explains the decrease in cathodic peak current when capture of TNF-α. In these experiments, solutions of recombinant TNF-α with increasing concentrations were infused into the microfluidic device successively. Surfaces were regenerated by exposure to urea between infusions of different cytokine concentrations. Taking values for cathodic peak current at different cytokine concentrations, we constructed a calibration curve for TNF-α. These data, presented in Fig. 2 B, reveal that the sensor had limit of detection of 10 ng/mL (0.58 nm) with linear range extending to 100 ng/mL (5.8 nm). The results were presented as the difference in faradaic current before and after target binding divided by the initial faradaic current. Because binding of IFN-γ and TNF-α caused a decrease in current, the sensitivity of the biosensor was reported in terms of signal loss or suppression. The signal suppression was defined as final cathodic peak current divided by initial peak current.
Fig. 2.

Characterization of sensor response to TNF-α. (A) SWV showed that a decrease in faradaic current was proportional to the solution concentration of TNF-α. These results were obtained using gold electrodes modified with 0.5 μm aptamer. Detection limit of recombinant TNF-α was 10 ng/mL using this RNA aptasensor. (B) Calibration curve of current vs TNF-α concentration for aptasensor prepared using 0.5 μm aptamer concentration. The calibration curve was fitted to a line with the equation of y = 0.3806x–0.1454 and R2 = 0.9924. The data points and error bars represent average and standard deviations of measurements from three different aptamer-modified electrodes (n = 3).
Specificity is an important characterisitic of a biosensor. In our experiments, specificity was tested by challenging aptasensors with multiple non-specific proteins including cytokines IL-2, IL-12, IL-17 and IFN-γ, all at concentrations of 50 ng/mL, and 1% BSA. Fig. 3 demonstrates 20-fold higher sensor response to 50 ng/mL TNF-α compared to non-specific proteins and therefore points to the high specificity of TNF-α biosensor.
Fig. 3.

Specificity of aptasensor response to TNF-α. SWV measurements of aptasensor response to 50 ng/mL concentration of non-specific cytokines (IL-2, IL-12, IL-17 and IFN-γ) and 1% BSA as well as 50 ng/mL and 100 ng/mL of TNF-α. These results show that biosensor did not respond to non-specific proteins but did respond to TNF-α.
3.3. Multiplex aptasensors for simultaneous detection of IFN-γ and TNF-α
The ability to functionalize specific electrodes with desired sensing molecules is an important part of developing a multi-analyte biosensor. In this paper, selective attachment of different aptamers onto adjacent miniature Au electrodes was controlled using a sequence of alkanethiol removal and aptamer incubation steps. As shown in Fig. 4, all electrodes were modified with MUA, creating a passivation/sacrificial layer. Subsequently, reductive voltage was applied to the desired set of electrodes, removing the alkanethiol and making the underlying Au available for aptamer assembly. After incubating and washing away unbound aptamer, the other set of electrodes was activated and exposed to a different population of aptamer molecules. This functionalization method allowed us to place IFN-γ and TNF-α aptamers on neighboring electrodes and to employ the same redox reporter (MB) to resolve signals from these aptamers.
Fig. 4.

A process flow diagram for the surface modification of dual cytokine-binding aptasensors on adjacent gold electrodes. Electrochemical stripping method was utilized to selectively immobilize different aptamers on a desired gold surface. A negative potential (−1.2 V) was applied for 60 s in order to facilitate complete desorption of thiol SAMs layers from the electrodes prior to further modification.
3.4. Characterizing cross-contamination of dual cytokine aptasensor
In order to validate that the correct aptamer population assembled on the correct electrode and that cross-contamination did not occur, aptasensors were challenged with either IFN-γ or TNF-α individually. Results from a typical experiment are shown in Fig. 5 A–D. As seen from this experiment, challenging dual aptasensors to 50 ng/mL IFN-γ resulted in 30% signal suppression of IFN-γ sensing electrode and no response from TNF-α electrode. When reversing the experiment and infusing 50 ng/mL TNF-α, only the TNF-α sensing electrode responded. Fig. 5 E summarizes responses from several electrode pairs (n = 3) and highlights that the IFN-γ aptasensor had a 28-fold higher response to correct cytokine and that TNF-α sensor had showed a 22-fold higher signal when challenged with the correct vs. incorrect cytokine. These results suggest that the desired aptamer type was assembled on the desired electrode with minimal cross-contamination. In fact, ratios of specific vs. non-specific cytokine responses reported for TNF-α aptasensor are comparable in multiplex and singleplex formats. The same is true for limit of detection and linear range for IFN-γ and TNF-α—these parameters were found to be similar in multiplex and singleplex aptasensors.
Fig. 5.

Cross-contamination test with dual aptasensor responding to either IFN-γ or TNF-α. (A–D) responses of a typical pair of half-ring electrodes challenged with cytokines. (A) IFN-γ sensing electrode challenged with 50 ng/mL IFN-γ shows a significant change in signal whereas (B) TNF-α sensing electrode exposed to 50 ng/mL IFN-γ did not respond. In contrast, (C) IFN-γ sensing half-ring electrode did not respond to 50 ng/mL TNF-α but (D) TNF-α sensing electrode showed a strong signal to this cytokine. (E) Responses of two electrodes to their specific target cytokine. The data points and error bars represent average and standard deviations of measurements from three different aptasensors (n = 3).
3.5. Modifying micropatterned sensing surfaces to capture cells
After characterization of the dual cytokine aptasensor, we proceeded to design surfaces where cells could be captured in well-defined areas adjacent to the sensing electrodes. There have been a number of reports utilizing antibody-coated microfabricated surfaces for cell capture [31,38–40]. In the present study anti-CD4 and anti-CD14 were used to bind primary human T cells and U937 monocytes respectively. Packaging surfaces in a non-fouling PEG hydrogel was used to define the areas of attachment so as to control the number of cells to be captured and analyzed. While the biosensor functions without this packaging layer, confining cell attachment to specific regions and spacing these regions far enough apart allows for the creation of multiple independent sensing sites within the same channel and provides redundancy to the measurements. Fig. 6A shows an example of selective attachment of fluorescently labeled anti-CD4 Ab on glass attachment sites defined by the PEG gel. RBC-depleted whole blood was introduced to a microfluidic device and T cells were captured on anti-CD4 absorbed regions as shown in Fig. 6 B. Staining of captured cells with PE-labeled anti-CD4 antibodies and FITC-labeled anti-CD3 demonstrated the prevalence of CD3+ and CD4+ T cells (see Fig. 6 C). These data are in line with our previous results pointing to high purity of T cell isolation (>95%) on antibody-modified surfaces enclosed inside our microfluidic device [31]. U937 monocytes were captured next to sensing electrodes in a similar manner (data not shown).
Fig. 6.
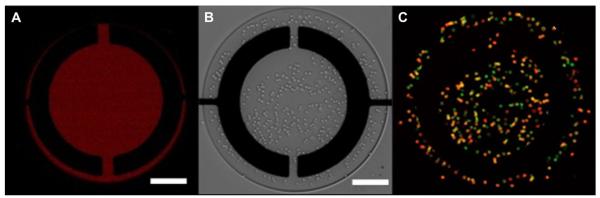
Cell capture on anti-CD4 modified surfaces. (A) Fluorescently labeled CD4 antibody localized on the glass regions but not on the hydrogel layer defining the microwell. (B) Brightfield images after leukocyte attachment on the surface. (C) Staining with anti-CD3-FITC (green) and anti-CD4-PE (red) reveals that majority of captured cells express both of these markers, confirming CD4 T cell phenotype. Scale bars in each panel are 100 μm. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.6. Simultaneous detection of IFN-γ and TNF-α release from immune cells
As the final step in validating the sensing device, release of IFN-γ and TNF-α from primary human T cells and a monocyte cell line was demonstrated. Activated primary T cells are known for robust production of both IFN-γ and TNF-α, whereas monocytes are known as a source of TNF-α but not IFN-γ. By choosing these two different immune cell types, we wanted to demonstrate that our biosensor will reflect differences in function of these cells. T cells or monocytes were captured on antibody-modified region in the proximity of aptamer-modified electrodes. The dimensions of individual attachment sites (300 μm diameter) allowed for the capture of ~400 cells. While in the device, the cells were exposed to PMA/Ionomycin (a mitogen) to induce IFN-γ/TNF-α production and cytokine release was monitored concurrently with mitogenic activation. Fig. 7 summarizes cytokine release data from primary T cell and U937 monocytes. After 4 h stimulation, T cell production was 24.7 ± 2.3 ng/mL for IFN-γ and 53.4 ± 4.2 ng/mL for TNF-α, whereas monocyte production was 9.9 ± 0.7 ng/mL and 77.1 ± 5.2 ng/mL for IFN-γ and TNF-α respectively.
Fig. 7.

Continuous monitoring of IFN-γ/TNF-α release using dual cytokine-binding aptamers modified electrodes from captured cells. (A) Analysis of primary T cells for production of TNF-α and IFN-γ. (B) Cytokine production from activated U937 monocytes. Error bars are one standard deviation from the mean. Number of electrodes used for each data point is n = 4.
Several trends may be noted from these data. The fact that primary T cells produce significantly more TNF-α has been observed by us in previous experiments employing antibody-based detection [40]. In fact the ratio of TNF-α to IFN-γ signal from T cell observed in our previous studies with antibodies is similar to the present work with aptamers (2.20:1 with antibodies and 2.16:1 with aptamers). Also, as expected, monocytes do not produce appreciable quantities of IFN-γ.
It is important to note that multiple control experiments were carried out to ensure validity of the detected signal. 1) In the absence of mitogenic stimulation, T cells and monocytes produced several fold lower levels of IFN-γ (4.1 ± 0.6 ng/mL and 3.1 ± 0.5 ng/mL) and TNF-α (6.5 ± 3.3 ng/mL and 7.4 ± 3.6 ng/mL). 2) Molt3 cells expressing CD4 antigen but known to lack the ability to produce cytokines upon mitogenic activation were tested in our microsystem and did not produce appreciable levels of either IFN-γ or TNF-α (Figure S2).
One may note from Fig. 7 that the signal begins to saturate after 3–4 h. According to our calibration studies, signal suppression associated with saturation of the surface is 60% for TNF-α and 80% for IFN-γ. Therefore, leveling off of the signal seen in our data should be attributed to decrease in cell activity, more specifically to the attenuation in cellular production of cytokines.
Cytokine production rates could be computed based on the release data presented in Fig. 7. Taking into consideration the positions of the captured cells and the electrodes, we modeled the diffusion of secreted cytokines inside the microfluidic device using a numerical approach (COMSOL Multiphysics, Figure S3). In our previous study, we determined that for a given electrode geometry and number of cells, the diffusion field of adjacent cell culture/sensing sites do not overlap appreciably (<2%) on the timescale of 1 h [29]. In the present study, cytokine release data for the first hour was used in calculating production rates.
The production rate of TNF-α and IFN-γ for U937 monocytes were determined to be 0.0160 ± 0.0003 pg/cell/hr and 0.0030 ± 0.0001 pg/cell/hr respectively. For primary T cells, the production rates were 0.0100 ± 0.001 pg/cell/hr for TNF-α and 0.0074 ± 0.0002 pg/cell/hr for IFN-γ. Production rate of IFN-γ detected in a dual cytokine sensor is similar to that reported by us previous for single aptasensor configuration [29]. TNF-α production rates detected in our system are well aligned with production rates of 0.0025–0.009 pg/cell/hr and 0.005–0.012 pg/cell/hr reported in the literature for T cells and monocyte respectively [14–16,41].
4. Conclusions
This paper describes the development of a microsystem for simultaneously quantifying two cell-secreted inflammatory cytokines, IFN-γ and TNF-α. The microsystem consisted of several Au electrode pairs microfabricated on glass and packaged with a non-fouling PEG hydrogel. A sequence of alkanethiol desorption and aptamer immobilization steps was used to assemble MB-functionalized aptamers specific to either TNF-α or IFN-γ onto individual members of electrode pairs. The resultant dual cytokine biosensor had detection limits of 0.06 nm IFN-γ and 0.58 nm TNF-α. Importantly, the sensitivity was similar in dual cytokine and single cytokine biosensor, suggesting that the multi-step assembly process did not compromise sensor performance. The utility of this dual cytokine aptasensors was demonstrated by detecting IFN-γ and TNF-α concurrently from activated T cells and U937 monocytes. Production of these two important cytokines was monitored continuously for 4 h and rates of cytokine release were determined. While demonstrating detection of IFN-γ and TNF-α in the present format, this microsystem may be enhanced in the future to detect a larger array of cytokines or biomarkers and will have applications in blood analysis and infectious disease diagnostics.
Supplementary Material
Acknowledgments
This work was supported by NSF EFRI (gs1) Grant No. 0937997 awarded to AR. TK was supported by NIH Fellowship T32 (gs2) - NIBIB 5T32EB003827. We also thank Qing Zhou, James Enomoto, Dr. Dongsik Shin, Tam Vu for their technical help.
Footnotes
Appendix A. Supplementary material Supplementary data related to this article can be found online at http://dx.doi.org/10.1016/j.biomaterials.2012.06.089.
References
- [1].Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An essential role for interferon-gamma in resistance to mycobacterium-tuberculosis infection. J Exp Med. 1993;178:2249–54. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Harari A, Vallelian F, Pantaleo G. Phenotype heterogeneity of antigen-specific CD4 T-cells under different conditions of antigen persistence and antigen load. Euro J Immun. 2004;34:3525–33. doi: 10.1002/eji.200425324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Moller DE. Potential role of TNF-alpha in the pathogenesis of insulin resistance and type 2 diabetes. Trends Endocrinol Metab. 2000;11:212–7. doi: 10.1016/s1043-2760(00)00272-1. [DOI] [PubMed] [Google Scholar]
- [4].Winthrop KL. Risk and prevention of tuberculosis and other serious opportunistic infections associated with the inhibition of tumor necrosis factor. Nat Rev Rheuatol. 2006;2:602–10. doi: 10.1038/ncprheum0336. [DOI] [PubMed] [Google Scholar]
- [5].Popa C, Netea MG, van Riel PLCM, van der Meer JWM, Stalenhoef AFH. The role of TNF-alpha in chronic inflammatory conditions, intermediary metabolism, and cardiovascular risk. J Lipid Res. 2007;48:751–62. doi: 10.1194/jlr.R600021-JLR200. [DOI] [PubMed] [Google Scholar]
- [6].Makitalo B, Andersson M, Arestrom I, Karlen K, Villinger F, Ansari A, et al. ELISpot and ELISA analysis of spontaneous, mitogen-induced and antigen-specific cytokine production in cynomolgus and rhesus macaques. J Immunol Methods. 2002;270:85–97. doi: 10.1016/s0022-1759(02)00274-0. [DOI] [PubMed] [Google Scholar]
- [7].Steffen MJ, Ebersole JL. Sequential ELISA for cytokine levels in limited volumes of biological fluids. BioTechniques. 1996;21:504–9. doi: 10.2144/96213rr04. [DOI] [PubMed] [Google Scholar]
- [8].Siawaya JFD, Roberts T, Babb C, Black G, Golakai HJ, Stanley K, et al. An evaluation of commercial fluorescent bead-based luminex cytokine assays. Plos One. 2008;3:e2535. doi: 10.1371/journal.pone.0002535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bradshaw EM, Kent SC, Tripuraneni V, Orban T, Ploegh HL, Hafler DA, et al. Concurrent detection of secreted products from human lymphocytes by microengraving: cytokines and antigen-reactive antibodies. Clin Immunol. 2008;129:10–8. doi: 10.1016/j.clim.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fan R, Vermesh O, Srivastava A, Yen BKH, Qin LD, Ahmad H, et al. Integrated barcode chips for rapid, multiplexed analysis of proteins in microliter quantities of blood. Nat Biotechnol. 2008;26:1373–8. doi: 10.1038/nbt.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dishinger JF, Reid KR, Kennedy RT. Quantitative monitoring of insulin secretion from single islets of langerhans in parallel on a microfluidic chip. Anal Chem. 2009;81:3119–27. doi: 10.1021/ac900109t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ma C, Fan R, Ahmad H, Shi QH, Comin-Anduix B, Chodon T, et al. A clinical microchip for evaluation of single immune cells reveals high functional heterogeneity in phenotypically similar T cells. Nat Med. 2011;17:738–43. doi: 10.1038/nm.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Stybayeva G, Kairova M, Ramanculov E, Simonian AL, Revzin A. Detecting interferon-gamma release from human CD4 T-cells using surface plasmon resonance. Colloid Surface B. 2010;80:251–5. doi: 10.1016/j.colsurfb.2010.06.015. [DOI] [PubMed] [Google Scholar]
- [14].North ME, Ivory K, Funauchi M, Webster ADB, Lane AC, Farrant J. Intra-cellular cytokine production by human CD4(+) and CD8(+) T cells from normal and immunodeficient donors using directly conjugated anti-cytokine antibodies and three-colour flow cytometry. Clin Exp Immunol. 1996;105:517–22. doi: 10.1046/j.1365-2249.1996.d01-795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sullivan KE, Cutilli J, Piliero LM, Ghavimi-Alagha D, Starr SE, Campbell DE, et al. Measurement of cytokine secretion, intracellular protein expression, and mRNA in resting and stimulated peripheral blood mononuclear cells. Clin Diagn Lab Immunol. 2000;7:920–4. doi: 10.1128/cdli.7.6.920-924.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jansky L, Reymanova P, Kopecky J. Dynamics of cytokine production in human peripheral blood mononuclear cells stimulated by LPS or infected by Borrelia. Physiol Res. 2003;52:593–8. [PubMed] [Google Scholar]
- [17].Bini A, Minunni M, Tombelli S, Centi S, Mascini M. Analytical performances of aptamer-based sensing for thrombin detection. Anal Chem. 2007;79:3016–9. doi: 10.1021/ac070096g. [DOI] [PubMed] [Google Scholar]
- [18].Ferapontova EE, Olsen EM, Gothelf KV. An RNA aptamer-based electro-chemical biosensor for detection of theophylline in serum. J Am Chem Soc. 2008;130:4256–8. doi: 10.1021/ja711326b. [DOI] [PubMed] [Google Scholar]
- [19].Li Y, Qi HL, Peng Y, Yang J, Zhang CX. Electrogenerated chemiluminescence aptamer-based biosensor for the determination of cocaine. Electrochem Commun. 2007;9:2571–5. [Google Scholar]
- [20].Liu CW, Huang CC, Chang HT. Highly selective DNA-based sensor for lead(II) and mercury(II) ions. Anal Chem. 2009;81:2383–7. doi: 10.1021/ac8022185. [DOI] [PubMed] [Google Scholar]
- [21].McCauley TG, Hamaguchi N, Stanton M. Aptamer-based biosensor arrays for detection and quantification of biological macromolecules. Anal Biochem. 2003;319:244–50. doi: 10.1016/s0003-2697(03)00297-5. [DOI] [PubMed] [Google Scholar]
- [22].Tombelli S, Minunni A, Luzi E, Mascini M. Aptamer-based biosensors for the detection of HIV-1 tat protein. Bioelectrochemistry. 2005;67:135–41. doi: 10.1016/j.bioelechem.2004.04.011. [DOI] [PubMed] [Google Scholar]
- [23].Wang J, Jiang YX, Zhou CS, Fang XH. Aptamer-based ATP assay using a luminescent light switching complex. Anal Chem. 2005;77:3542–6. doi: 10.1021/ac050165w. [DOI] [PubMed] [Google Scholar]
- [24].Xiao Y, Lubin AA, Heeger AJ, Plaxco KW. Label-free electronic detection of thrombin in blood serum by using an aptamer-based sensor. Angew Chem Int Ed. 2005;44:5456–9. doi: 10.1002/anie.200500989. [DOI] [PubMed] [Google Scholar]
- [25].Hansen JA, Wang J, Kawde AN, Xiang Y, Gothelf KV, Collins G. Quantum-dot/aptamer-based ultrasensitive multi-analyte electrochemical biosensor. J Am Chem Soc. 2006;128:2228–9. doi: 10.1021/ja060005h. [DOI] [PubMed] [Google Scholar]
- [26].Liss M, Petersen B, Wolf H, Prohaska E. An aptamer-based quartz crystal protein biosensor. Anal Chem. 2002;74:4488–95. doi: 10.1021/ac011294p. [DOI] [PubMed] [Google Scholar]
- [27].Maehashi K, Katsura T, Kerman K, Takamura Y, Matsumoto K, Tamiya E. Label-free protein biosensor based on aptamer-modified carbon nanotube field-effect transistors. Anal Chem. 2007;79:782–7. doi: 10.1021/ac060830g. [DOI] [PubMed] [Google Scholar]
- [28].Liu F, Dubey M, Takahashi H, Castner DG, Grainger DW. Immobilized antibody orientation analysis using secondary ion mass spectrometry and fluorescence imaging of affinity-generated patterns. Anal Chem. 2010;82:2947–58. doi: 10.1021/ac902964q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Liu Y, Yan J, Howland MC, Kwa T, Revzin A. Micropatterned aptasensors for continuous monitoring of cytokine release from human leukocytes. Anal Chem. 2011;83:8286–92. doi: 10.1021/ac202117g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Xiao Y, Lai RY, Plaxco KW. Preparation of electrode-immobilized, redoxmodified oligonucleotides for electrochemical DNA and aptamer-based sensing. Nat Protoc. 2007;2:2875–80. doi: 10.1038/nprot.2007.413. [DOI] [PubMed] [Google Scholar]
- [31].Zhu H, Macal M, George MD, Dandekar S, Revzin A. A miniature cytometry platform for capture and characterization of T-lymphocytes from human blood. Anal Chim Acta. 2008;608:186–96. doi: 10.1016/j.aca.2007.12.021. [DOI] [PubMed] [Google Scholar]
- [32].Zhu H, Stybayeva G, Macal M, Ramanculov E, George MD, Dandekar S, et al. A microdevice for multiplexed detection of T-cell-secreted cytokines. Lab Chip. 2008;8:2197–205. doi: 10.1039/b810244a. [DOI] [PubMed] [Google Scholar]
- [33].Liu Y, Tuleouva N, Ramanculov E, Revzin A. Aptamer-based electrochemical biosensor for interferon gamma detection. Anal Chem. 2010;82:8131–6. doi: 10.1021/ac101409t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yan X, Gao X, Zhang Z. Isolation and characterization of 2′-amino-modified RNA aptamers for human TNFalpha. Genomics Proteomics Bioinform. 2004;2:32–42. doi: 10.1016/S1672-0229(04)02005-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Rowe AA, Miller EA, Plaxco KW. Reagentless measurement of aminoglycoside antibiotics in blood serum via an electrochemical, ribonucleic acid aptamer-based biosensor. Anal Chem. 2010;82:7090–5. doi: 10.1021/ac101491d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Shimayama T, Nishikawa F, Nishikawa S, Taira K. Nuclease-resistant chimeric ribozymes containing deoxyribonucleotides and phosphorothioate linkages. Nucleic Acids Res. 1993;21:2605–11. doi: 10.1093/nar/21.11.2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wilson C, Keefe AD. Building oligonucleotide therapeutics using non-natural chemistries. Curr Opin Chem Biol. 2006;10:607–14. doi: 10.1016/j.cbpa.2006.10.001. [DOI] [PubMed] [Google Scholar]
- [38].Sin A, Murthy SK, Revzin A, Tompkins RG, Toner M. Enrichment using antibody-coated microfluidic chambers in shear flow: model mixture of human lymphocytes. Biotech Bioeng. 2005;91:816–26. doi: 10.1002/bit.20556. [DOI] [PubMed] [Google Scholar]
- [39].Cheng XH, Irimia D, Dixon M, Sekine K, Demirci U, Zamir L, et al. A microfluidic device for practical label-free CD4+T cell counting of HIV-infected subjects. Lab Chip. 2007;7:170–8. doi: 10.1039/b612966h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Stybayeva G, Mudanyali O, Seo S, Silangcruz J, Macal M, Ramanculov E, et al. Lensfree holographic imaging of antibody microarrays for high-throughput detection of leukocyte numbers and function. Anal Chem. 2010;82:3736–44. doi: 10.1021/ac100142a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].See RH, Kum WWS, Chang AH, Goh SH, Chow AW. Induction of tumor-necrosis-factor and interleukin-1 by purified staphylococcal toxic shock syndrome toxin-1 requires the presence of both monocytes and lymphocytes t. Infect Immun. 1992;60:2612–8. doi: 10.1128/iai.60.7.2612-2618.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.