

Abstract
Aims
The optic atrophy 1 (OPA1) protein is an essential protein involved in the fusion of the mitochondrial inner membrane. Despite its high level of expression, the role of OPA1 in the heart is largely unknown. We investigated the role of this protein in Opa1+/− mice, having a 50% reduction in OPA1 protein expression in cardiac tissue.
Methods and Results
In mutant mice, cardiac function assessed by echocardiography was not significantly different from that of the Opa1+/+. Electron and fluorescence microscopy revealed altered morphology of the Opa1+/− mitochondrial network; unexpectedly, mitochondria were larger with the presence of clusters of fused mitochondria and altered cristae. In permeabilized mutant ventricular fibers, mitochondrial functional properties were maintained, but direct energy channeling between mitochondria and myofilaments was weakened. Importantly, the mitochondrial permeability transition pore (PTP) opening in isolated permeabilized cardiomyocytes and in isolated mitochondria was significantly less sensitive to mitochondrial calcium accumulation. Finally, 6 weeks after transversal aortic constriction (TAC), Opa1+/− hearts demonstrated hypertrophy almost two-fold higher (p<0.01) than in wild type mice with altered ejection fraction (decrease of 43% versus 22% in Opa1+/+ mice, p<0.05).
Conclusions
These results suggest that, in adult cardiomyocytes, OPA1 plays an important role in mitochondrial morphology and PTP functioning. These properties may be critical for cardiac function under conditions of chronic pressure overload.
Keywords: Adaptation, Biological; Animals; Down-Regulation; GTP Phosphohydrolases; metabolism; Mice; Mice, Knockout; Mitochondria; genetics; metabolism; ultrastructure; Mitochondrial Membrane Transport Proteins; genetics; metabolism; Mitochondrial Membranes; metabolism; Mitochondrial Proteins; genetics; physiology; Myocytes, Cardiac; cytology; metabolism; Optic Atrophy, Autosomal Dominant; genetics; metabolism; physiopathology; Permeability; Pressure
Keywords: cardiac energy metabolism, mitochondria, mitochondria dynamics, permeability transition pore, hypertrophy
Introduction
Changes in mitochondrial morphology appear essential for the function or dysfunction of most tissues. Mitochondrial fusion and fission are highly regulated and tightly balanced processes. In mammalian cells, mitochondrial fission is regulated by dynamin-related protein 1 (DRP1) and its receptor FIS1 (Fission mediator protein 1) (for review, see1). In turn, mitochondrial fusion is mediated by two mitofusins (MFN1 and MFN2),2 which are involved in outer membrane fusion, and OPA1 (Optic atrophic type 1 protein), which is closely linked to the inner membrane.3 Mutations of genes encoding these proteins lead to severe diseases, especially neurological disorders, such as Charcot-Marie-Tooth disease type 2A for Mfn2 or autosomal dominant inherited optic atrophy (DOA) for Opa1 mutations.4–7
OPA1 is a ubiquitously expressed protein4,8 and its level is particularly upregulated in the heart. Because OPA1 is involved in the regulation of the dynamics of mitochondria that occupy about 30% of the cardiac cell volume, OPA1 may play a role in cardiac energy metabolism. We have previously shown that the energetic transfer between mitochondria and energy-consumers performed by direct adenylic nucleotide channeling (DANC)9 is closely related to the cell architecture and mitochondrial network.10,11 Finally, because decreased mitochondrial content and mitochondrial fragmentation are key features of cardiac diseases,12,13 alterations of mitochondrial morphology could be of major pathophysiological relevance.14 Indeed, a heterozygous mutation of Opa1 in Drosophila15 is associated with decreased heart rate and cardiac arrhythmia, suggesting that a loss of OPA1 could induce cardiac function abnormalities. Heart tube–specific knockdown of Opa1 also induces heart tube dilation with profound contractile impairment.16 In heart failure, a decrease in OPA1 protein content associated with mitochondrial fragmentation17 has been shown but the exact role of OPA1 depletion in the adult mammalian heart has not yet been assessed.
Recently, a mouse model carrying a splice mutation in the Opa1 gene leading to haploinsufficiency was described.18,19 While homozygous mutants die in utero, heterozygous mutants are viable but exhibit an age-dependent loss of retinal ganglion cells (characteristic of DOA) with a disorganization of mitochondrial cristae. This mouse model thus appears to be of great interest in studying the role of OPA1 in mitochondrial morphology, organization and function in the heart.
Methods
Supplementary Methods are available at Cardiovascular Research online.
Animals
The B6;C3-Opa1329-355del mouse line (herein referred to as Opa1+/−) has been described in details before.18–20 Basal cardiac phenotype was determined in six-month-old male mice while ten-week-old mice were submitted to transverse aortic constriction (TAC) to estimate the ability of mutated mouse hearts to adapt to pathological stress. Anesthesia was induced by intraperitoneal injection of Ketamine (50mg.kg−1) and Xylazine (8mg.kg−1). Aortic stenosis was induced by placing a silk suture around the aorta after thoracic incision. Age-matched controls underwent the same procedure without placement of suture. Mice were studied six weeks after surgery. All mice were anesthetized by intraperitoneal injection of pentobarbital (200mg.kg−1). Depth of anesthesia was checked by toe pinch before the start of surgery. After thoracotomy, the animals’ hearts were excised and rinsed in ice-cold calcium-free Krebs solution equilibrated with 95% O2 – 5% CO2.
All experiments were performed in conformity with the European Community guiding principles in the care and use of animals (Directive 2010/63/EU of the European Parliament). Authorizations to conduct animal experiments were obtained from the French Ministère de l’Agriculture, de la Pêche et de l’Alimentation (no. 92-284, June 27, 2007).
Biochemical analysis
Frozen tissues were used to measure activities of total CK, citrate synthase and mitochondrial complex I by standard spectrophotometric assays as previously described.11
Echocardiography
Transthoracic echocardiography was performed using a 12 MHz transducer (Vivid 7, General Electric Healthcare) under 2.5% isoflurane gas anesthesia. Two-dimensional-guided (2D) M-mode echocardiography was used to determine wall thickness and left ventricular chamber volume at systole and diastole and contractile parameters, such as fractional shortening (FS) or ejection fraction (EF).
Cardiomyocyte isolation
Retrograde heart perfusion according to the Langendorff method was performed to isolate cardiomyocytes as previously shown.21 The cells were plated onto dishes coated with laminin and were kept at room temperature for 2 hours.
Morphological description of mitochondrial network by fluorescence microscopy
Freshly isolated cardiomyocytes were incubated for 45 min at room temperature with 500nM Mito-Tracker Orange dye to label mitochondria and with 1μM calcein to estimate global cell volume (Invitrogen). 3D reconstruction was performed using IMARIS software (Bitplane Company, Zurich, Switzerland).
Electron microscopy
Samples of left ventricular papillary muscle from six-month-old mice were prepared for electron microscopy as previously described.10
Analysis of in situ mitochondrial function
Mitochondrial respiration was studied in situ in saponin-permeabilized fibers, as previously described.22 Fibers were exposed to increasing [ADP], and the ADP-stimulated respiration above basal oxygen consumption (Vo) was plotted to determine the apparent Km for ADP and maximal respiration rate (Vmax). The acceptor control ratio (ACR), an index of oxidation-phosphorylation coupling, was calculated as Vmax/V0 in the presence of glutamate/malate as substrates.
Estimation of energy transfer to sarcoplasmic reticulum and myofilaments
For measurements of SR and myofibrillar function, papillary muscle fibers were dissected and permeabilized for 30 minutes in saponin (50μg/ml), as previously described.9 The contribution of the energetic transfer system, the creatine kinase system (CK) and the DANC, to the provision of ATP for sarcoplasmic reticulum ATPase (SERCA) function was estimated by measuring SR calcium content in saponin-permeabilized fibers after loading under various energetic conditions, as previously described.10,11 The pMgATP/rigor tension relation was used for studying the ability of the different energetic systems to supply ATP for myosin ATPase.23
Mitochondrial permeability transition pore (PTP) experiments
For PTP experiments, fresh cardiomyocytes were co-loaded in culture medium with 5μM Rhod-2 and 1μM calcein (Invitrogen) for 45 min at room temperature and washed with culture medium solution.
In additional experiments, mitochondria from Opa1+/+ and Opa1+/− were isolated according to24. Swelling and depolarization of mitochondria were monitored after Ca2+ addition as described in25.
Quantitative RT-PCR
RNA was extract with Trizol reagent (Invitrogen), and transcribed into cDNA with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, France). Real-time PCR was performed using TaqMan Low Density Array (TLDA) technology (see supplemental data).
Statistical Analysis
Values are expressed as means±sem. The statistical significance of the difference between the Opa1+/+ and Opa1+/− groups was estimated by Student’s t-test, and when necessary by two-way ANOVA using Newman-Keuls post-hoc test. Values of p<0.05 were considered significant.
Results
Anatomical characteristics and heart function
The heterozygous mutation in Opa1 resulted in an approximately 50% reduction of OPA1 protein (Figure 1A) in line with previous results.18 Six-month-old Opa1+/− mice displayed normal body weight and growth (Figure 1B). There was no difference in heart weight between Opa1+/+ and Opa1+/− mice (Figure 1C). Heart rate, left ventricular (LV) end-diastolic and end-systolic volumes as well as ejection fraction assessed by echocardiography were normal in Opa1+/− (Figure 1, D–F). Similarly, contractile reserve, revealed by infusion of dobutamine (a β-adrenergic receptor agonist), was not compromised in Opa1+/− mice (Figure 1G). Thus, in adult Opa1+/− mice, cardiac function was normal and able to respond normally to an acute stimulation.
Figure 1.
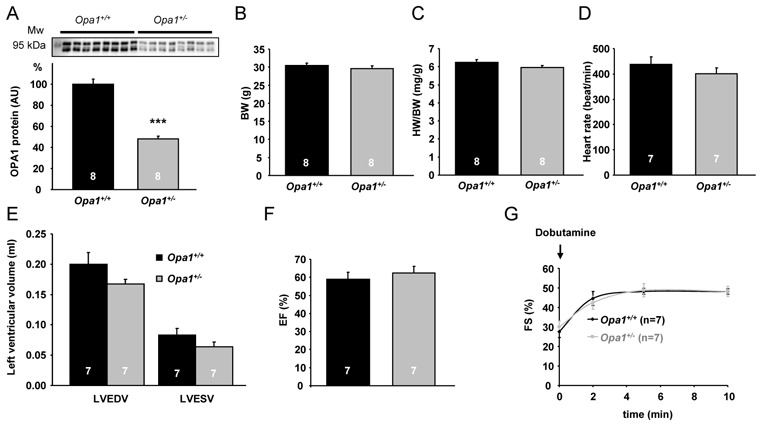
Anatomical characteristics and cardiac function of 6-month-old Opa1+/+ and Opa1+/− mice. (A) Western blot analysis of OPA1 protein. Upper panel: a representative original recording. Lower panel: mean values of OPA1 protein. ***p<0.001 versus Opa1+/+. (B) Body weight (BW). (C) Heart weight to body weight (HW/BW) ratio. (D) Heart rate. (E) LV end-diastolic volume (LVEDV) and LV end-systolic volume (LVESV). (F) LV ejection fraction (EF). (G) LV fractional shortening (FS) after dobutamine stimulation.
Cardiomyocyte ultrastructure and mitochondrial network organization
Compared with Opa1+/+, the mitochondrial network in Opa1+/− cardiomyocytes appeared to be more heterogeneous, with larger mitochondria and/or an increase in the number of mitochondrial clusters and an increase of volume dispersion (Figure 2A). IMARIS analysis revealed that Opa1+/− cardiomyocyte and global mitochondrial volumes were identical to control (Figure 2B and C). However, the distribution of mitochondrial volumes was shifted, with an increase in the percentage of large mitochondria (p<0.01) and a decrease in the proportion of small mitochondria in Opa1+/− cardiomyocytes (p<0.05) (Figure 2E). This is consistent with a decreased number of mitochondria per cell (Figure 2D). These results show that partial OPA1 deficiency affects mitochondrial network morphology of adult cardiomyocytes by modifying the distribution and morphology of individual mitochondria rather than the global mitochondrial volume.
Figure 2.
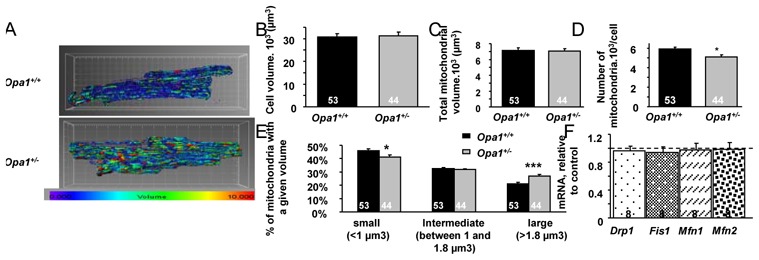
Morphological description of the mitochondrial network. (A) Representative pictures obtained with Mito-Tracker orange and IMARIS software. Each mitochondrion is identified by a color in accordance with its volume (the smallest mitochondria are blue and the largest are red). (B) Cell volume measured by calcein loading and IMARIS analysis. (C) Total mitochondrial volume quantified by IMARIS. (D) Number of mitochondria per cell. (E) Percentage of mitochondrial size categories. (F) Level of proteins involved in mitochondrial dynamics (Drp1, Mfn1, Mfn2) in Opa1+/+ and Opa1+/− cardiac tissue estimated by Western Blot analysis (normalized to citrate synthase level). *p<0.05; ***p<0.001 versus Opa1+/+.
To describe more precisely the structure of Opa1+/− mitochondria, we performed detailed electron microscopic analysis of cardiac tissue. Compared to Opa1+/+ hearts where mitochondria form regular rows that extended along myofibrils (Figure 3, A and B, upper panel, and A, lower panel), the arrangement of mitochondria in Opa1+/− cardiomyocytes was mostly irregular (Figure 3C, upper panel), with clusters of mitochondria (Figure 3, C and D, upper panel) and high variability in size and shape (Figure 3B, upper panel), which was clearly visible in transverse sections (Figure 3D, upper panel). Mitochondrial cristae of Opa1+/− formed semi-circular areas alternating with areas of disintegrated cristae within the same mitochondrion and unusual areas displaying slight spatial deformation of cristae inside mitochondria (Figure 3B, lower panel). Other mitochondria displayed fragmented cristae (Figure 3C, lower panel) together with foci in the matrix that were devoid of cristae and the formation of free spaces between the outer and inner mitochondrial membranes (Figure 3C, lower panel). Typically, the presence of large mitochondria was accompanied by the presence of dark material that coincided with the disappearance of the outer and inner mitochondrial membranes. These defects may represent the sites where abnormal non-completed mitochondrial fusion took place due to the deficiency in OPA1 (Figure 3D, lower panel).
Figure 3.
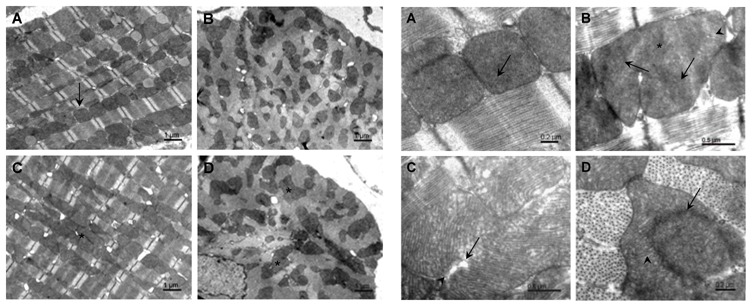
Electron micrographs of mitochondria in cardiomyocytes from Opa1+/+ and Opa1+/− mice. Upper Panel: Longitudinal (A) and transverse (B) sections of Opa1+/+ cardiomyocytes show individual mitochondria (arrow). Longitudinal (C) and transverse (D) sections of Opa1+/− cardiomyocytes demonstrate mitochondrial clusters (asterisks).
Lower Panel: (A) Opa1+/+ cardiomyocytes. Cristae are homogeneously distributed in the mitochondria (arrow). B–D Opa1+/− cardiomyocytes showing enlarged mitochondria and incompletely fused cristae. (B.) Cristae spreading in different directions (arrows); area of disintegrated cristae (asterisks); area of deformation of cristae (arrowhead). (C) Fragmented cristae (arrow); separation between inner and outer mitochondrial membranes (arrowhead). (D) Regions of incomplete mitochondrial fusion (arrow). Note that the peripheral part of the mitochondrion contains normal cristae (arrowhead).
We confirmed by EM analysis an increase of the mean surface of individual mitochondria (Figure 1S in supplemental data) showing that the observed larger mitochondria observed in fluorescence were not clusters of small mitochondria, and that OPA1 partial deletion in the heart results in large mitochondria with incomplete fusion of inner membranes.
We also determined whether partial OPA1 deletion was accompanied by compensatory responses of other proteins involved in mitochondrial dynamics. Gene expression of Drp1, Fis1, Mfn1 and Mfn2 was similar between control and mutant mice (Figure 2S), showing no modification at the transcription level. Western Blot experiments also revealed no compensation at the protein level (Figure 2F).
Oxidative capacity and energy transfer between mitochondria and myofilaments
We investigated whether the decrease of OPA1 expression could altered the intrinsic mitochondrial function. Basal and maximal respirations (when complex I, complex I and II, or complex II alone were activated) were not significantly different between the two groups (Figure 4A). Consequently, the coupling between oxidation and phosphorylation did not show any notable difference (Figure 4B). The sensitivity of mitochondrial respiration to ADP was estimated with and without creatine to study the coupling of oxidative phosphorylation with mitochondrial creatine kinase (mi-CK). The Km for ADP without (KmADP) and with creatine (KmADP+Cr) was significantly lower in mutant mice (Figure 4C). However, the mi-CK functional efficacy assessed by KmADP/KmADP+Cr was not significantly different between the two groups (2.6±0.3 versus 2.4±0.6). Finally, when respiration was measured with octanoyl-carnitine as substrate, Opa1+/− cardiac mitochondria were less able to oxidize lipids than control mice (Figure 4D).
Figure 4.
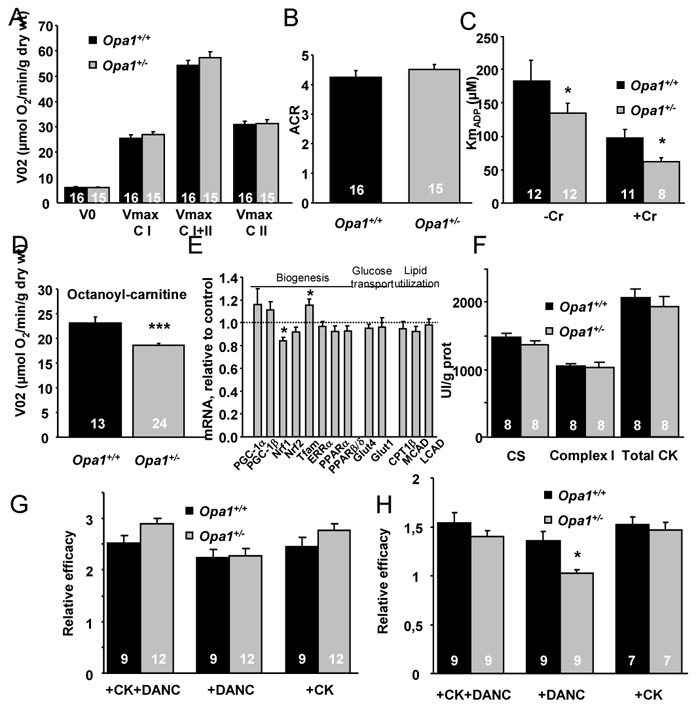
Cardiac energy metabolism of Opa1+/+ and Opa1+/− mice. (A) Oxygen consumption rate of skinned ventricular fibers in the absence (Vo) or presence of 2mM ADP (Vmax) with 10mM glutamate and 4mM malate (complex I, CI), 15mM succinate (both CI and complex II CI+II), and after complex I inhibition with 2mM amytal (CII). (B) Acceptor control ratio. (C) Apparent Km of oxygen consumption for ADP with (+Cr) or without (−Cr) 12 mM creatine. (D) Oxygen consumption rate with 2 mM ADP and 4 mM malate and 0.1mM octanoyl-carnitine. (E) mRNA levels of proteins involved in mitochondrial biogenesis, glucose transport, and lipid utilization. (F) Citrate synthase (CS), complex I and total creatine kinase (CK) activities. (G–H) Efficacy of CK and mitochondria (DANC) to support SERCA (G) or myosin ATPase (H) in permeabilized fibers. *p<0.05 versus OPA1+/+ ***p<0.001 versus OPA1+/+
We also determined whether factors involved in mitochondrial biogenesis (PGC-1α and β, NRF, TFAM, ERRα, PPARα), glucose transport (GLUT1 and 4), and β-oxidation (CPT1, MCAD, LCAD) were modified. Except for a small decrease in NRF1 and a small increase in TFAM, none of the mRNA levels of these genes was modified (Figure 4E). Similarly, activity of citrate synthase (CS), Complex I and total CK were not significantly different in Opa1+/− mice compared with Opa1+/+ mice (Figure 4F). Finally, level of oxidative stress was similar in Opa1+/− and Opa1+/+ mice as estimated by Oxyblot kit and by measuring aconitase/fumarase activities, Hsp60 protein level, SOD and GPX1 mRNA levels. (Figure 3S). The same result was obtained for markers of autophagy which were not modified (Figure 4S).
All these data demonstrate that downregulation of OPA1 does not alter respiratory capacity of mitochondria, oxidative stress status or mitochondrial mass but may display metabolic reprogramming at the level of substrate utilization.
Because energetic transfers, especially direct adenylic nucleotide channeling (DANC) between energy-producers (mitochondria) and energy-consumers (SERCA and ATPase of myofilaments), depend on the specific arrangement of the organelles in the cell, we measured energy transfer to SERCA in saponin-permeabilized ventricular fibers. The efficacy of each system (DANC or CK) was estimated relative to the efficacy of exogenous ATP only. The estimated maximal SR loading capacity under optimal energetic conditions in the presence of exogenous ATP, PCr and working mitochondria was similar in both groups of mice. CK efficacy as well as DANC efficacy were also similar in Opa1+/− and Opa1+/+ mice, suggesting that the adenine nucleotide channeling between mitochondria and SERCA was not altered by OPA1 deficiency (Figure 4G). EM images obtained in transversal sections confirmed that there was also no apparent change in SR-mitochondrial contacts (Figure 5S).
The ability of these different energetic systems to support myosin-ATPase activity was then assessed by measuring the rigor tension developed by permeabilized fibers when the MgATP concentration was progressively decreased. The ability of DANC plus CK or CK alone to provide high ATP/ADP ratio in the myofibrillar compartment was similar for the two groups (Figure 4H). However, DANC in Opa1+/− mice was significantly less efficient than in Opa1+/+ mice: in Opa1+/+ fibers, working mitochondria shifted pMgATP50 for rigor by 1.36±0.09 units, whereas, in Opa1+/− fibers, this parameter was as low as 1.03±0.04 (p<0.01). This showed that direct adenine nucleotide channeling from mitochondria to myosin ATPase was altered in hearts of Opa1+/− mice.
Mitochondrial Ca2+ retention capacity and permeability transition pore properties
The impact of partial Opa1 deficiency on the mitochondrial calcium retention capacity (CRC) and PTP state was assessed in situ in permeabilized cardiomyocytes by fluorescence techniques. Following 2μM calcium addition, intramitochondrial [Ca2+] progressively increased, as indicated by the rise in Rhod-2 fluorescence, until PTP opening induced calcium loss from the mitochondrial matrix (Figure 5A, upper panel). Similarly, PTP opening provoked the release of intramitochondrial calcein, which induced a decrease in calcein fluorescence (Figure 5A, lower panel). The addition of cyclosporin A (CsA) greatly reduced the leakage of calcein and Rhod-2 fluorescence, confirming that the observed phenomenon was indeed PTP opening. In comparison with Opa1+/+ cardiomyocytes, Opa1+/− cells loaded more calcium and PTP opening was delayed (Figure 5A and B). Statistical analysis demonstrated a significant increase in mitochondrial calcium retention capacity in Opa1+/− cardiomyocytes and, consequently, a shift in the onset of Ca2+ leakage from mitochondria (Figure 5B, upper panel), suggesting a change in PTP opening probability. This finding was confirmed by the analysis of calcein traces, which showed a delay in calcein leak and PTP opening, as well as a decrease in the rate of calcein leak in Opa1+/− cardiomyocytes (Figure 5B, lower panel). These results demonstrated a link between mutation in Opa1 and changes in mitochondrial PTP function and mitochondrial calcium retention capacity.
Figure 5.
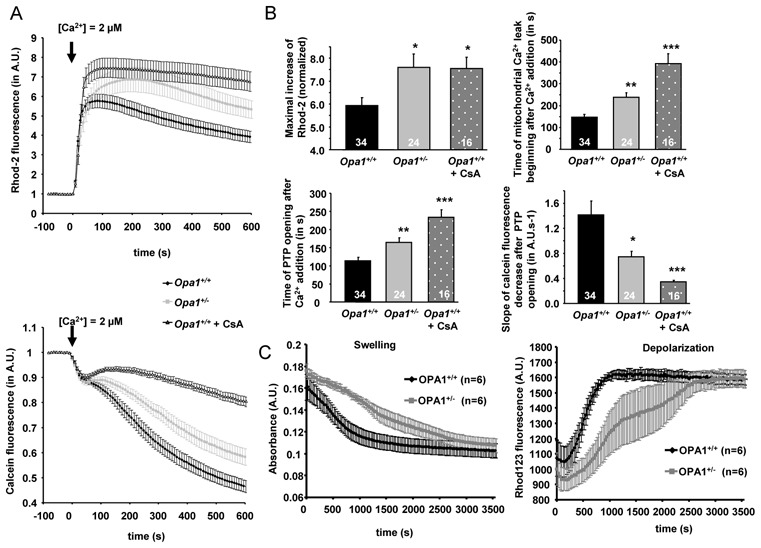
Kinetics of mitochondrial calcium loading and PTP opening in permeabilized cardiomyocytes and isolated mitochondria of Opa1+/+ and Opa1+/− mice. (A) Upper panel: time course of mitochondrial calcium accumulation following Ca2+ addition (indicated by arrow) measured by Rhod-2 fluorescence. Lower panel: time course of mitochondrial calcein fluorescence following Ca2+ addition (indicated by arrow). (B) Mean values of mitochondrial calcium loading capacity and PTP opening properties. Upper panel, maximal increase of Rhod-2 fluorescence (left panel) and time to the beginning of mitochondrial Ca2+ leak after Ca2+ addition (right panel). Lower panel, time to PTP opening after Ca2+ addition (left panel), and slope of calcein fluorescence decrease after PTP opening (right panel). *p<0.05; **p<0.01; ***p<0.001 versus Opa1+/+. (C) Time course of swelling (left) and depolarization (right) of isolated mitochondria after addition of 25 μM Ca2+.
We confirmed these results using isolated mitochondria (Figure 5C). Swelling and depolarization of mitochondria were monitored after addition of 25μM Ca2+. We observed no difference in resting membrane potential, as assessed by Rhod123 fluorescence. Furthermore, after Ca2+ addition, as in isolated permeabilized cardiomyocytes, a delay in the opening of mPTP on Opa1+/− mice was observed. Indeed, t1/2 for depolarization and swelling curves were 8.7±0.7 min and 10.4±0.8 min respectively in Opa1+/+, but increased to 22.0±4.7 min and 24.0±4.4 min in Opa1+/− mice (p<0.05). Thus, the modifications of PTP function were intrinsically linked to the lack of OPA1 in mitochondria.
Myocardial hypertrophy and myocardial dysfunction induced by pressure overload
To estimate the role of OPA1 in cardiac response to mechanical stress and to unmask possible loss in functional reserve, we performed echocardiographic analysis 6 weeks after transaortic constriction (TAC). In both sham groups (Opa1+/+-sham and Opa1+/−-sham), heart weight/body weight ratio (Figure 6A) and parameters of the cardiac function (Figure 6, C–E) were similar. On the other hand, hearts from Opa1+/−-TAC mice showed an almost two-fold greater hypertrophy (+72%) than did Opa1+/+-TAC (+40%) (Figure 6A). The high level of hypertrophy was associated with significant alterations of cardiac function. In contrast to Opa1+/+ hearts where TAC did not induce significant changes in the contractile indices (except in left-ventricular end-systolic volume), in the Opa1+/− group, TAC led to significant ventricular dilation, as shown by the increase in left ventricle end-diastolic volume and ventricular dysfunction, evidenced by a decrease in end-systolic volume, and a 43% reduction in left ventricle fractional shortening (Figure 6, C–E). Cardiac output index was also calculated, but no significant difference between groups was observed (Opa1+/+-sham: 2.18±0.23; Opa1+/+-TAC: 2.12±0.18; Opa1+/−-sham: 1.99±0.13; Opa1+/−-TAC: 1.87±0.17 ml/min/g). Indices of heart failure, such as pulmonary edema, tended to appear in Opa1+/− mice (Figure 6B). As expected, markers of mitochondrial content or biogenesis (Table 2S) were downregulated, and markers of stress or fibrosis (Figure 6S) were upregulated by TAC, but no marked difference was observed between Opa1+/− and Opa1+/+ mice. Taken together, this suggests that the heart of Opa1+/− mice have an increased sensitivity to mechanical stress.
Figure 6.
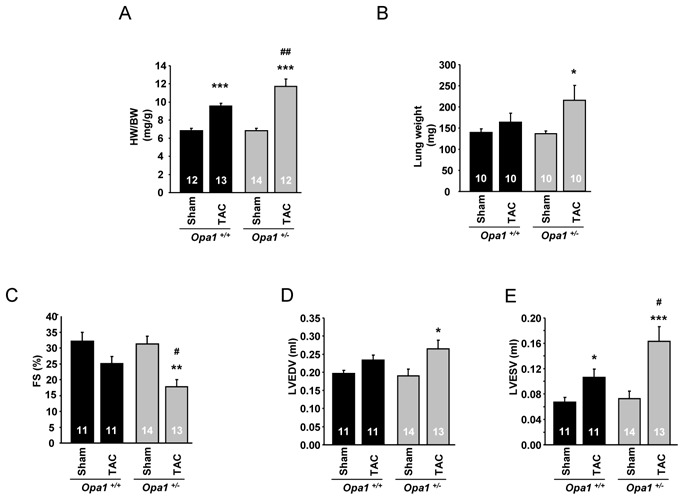
Anatomical characteristics and echocardiographic data of Opa1+/− mice following transverse aortic constriction (TAC). (A) Heart weight to body weight ratio. (B) Lung weight. (C) Fractional shortening. (D) Left ventricle end-diastolic diameter. (E) Left ventricle end-systolic volume. *p<0.05; **p<0.01; ***p<0.001 versus sham. #p<0.05; ##p<0.01 versus Opa1+/+-TAC.
Discussion
The role of OPA1 in the heart has been poorly explored, despite the high expression level of OPA1 and other dynamin-related proteins in this organ.15–17,26–29 Adult cardiomyocytes are highly oxidative cells and possess a large amount of mitochondria. However, a fast dynamic of these organelles was not clearly demonstrated.27 Obviously, this does not mean that dynamin-related proteins are silent in the adult heart and/or have no important physiological roles in this organ. We thus investigated the consequences of the decreased OPA1 expression on the intracellular energetic pathways and mouse heart function. The present results show that, 1) OPA1 depletion induced mitochondrial network remodeling, while 2) oxidative capacity and respiratory chain function were not altered, but 3) direct channeling of ATP and ADP between mitochondria and myosin-ATPases was reduced in mutant mice, and 4) mitochondria of Opa1+/− mice accumulated more calcium and presented a delay in calcium-induced PTP opening, and finally 5) despite a normal cardiac function at basal state, the mutant mice were more sensitive to prolonged hemodynamic stress.
OPA1 downregulation induced clear changes in mitochondrial morphology and cytoarchitecture. Indeed, in accordance with roles already attributed to OPA1,30 we observed alterations of mitochondrial cristae. However, unexpectedly, we observed heterogeneity in mitochondrial size with the appearance of larger mitochondria in Opa1+/− cardiomyocytes. Although modifications of the mitochondrial network were expected, these results are surprising because several previous studies had shown that the downregulation of proteins involved in fusion induces fragmentation of the mitochondria.31–33 However, our results are consistent with a recent study, showing that while Mfn2 KO mice exhibit mitochondrial fragmentation in neonatal cardiomyocytes, the opposite result, i.e., larger mitochondria, was observed in adult myocardium.29 Thus, our results can be explained by the specific architectural constraints of the adult cardiomyocyte that limit mitochondrial movements in contrast to neonatal cardiac cells. Indeed, the adult cardiac cell is a paradigm of highly compartmentalized cells possessing a sophisticated subcellular architecture in which repeated arrays of sarcomeres, T-tubules, sarcoplasmic reticulum (SR) and mitochondria interact. Therefore, it appears that a deficit in fusion protein may result in incomplete fusion in a complex cell type such as the adult cardiomyocyte, and in fragmentation in a simpler cell model. It can be proposed that, in a constrained environment, a deficit in OPA1 by impairing fusion of inner mitochondrial membranes, would compromise further fission mechanisms.
A primary consequence of mitochondrial morphology alterations was a decrease in the Km for ADP in Opa1+/− mice, which could result from disturbances in the cell architecture,34 leading to weaker interactions between mitochondria and the surrounding organelles, such that mitochondria are more open to the cytosol.10 A second consequence is an impairment of energy transfer between mitochondria and myofilaments.9,11 Indeed, our results show, for the first time, that a mere perturbation of the mitochondrial morphology may impair adenine nucleotide compartmentation, thereby confirming a link between cellular architecture and energy fluxes.10,11 In the heart, the local energy-providing systems (DANC and CK) are redundant and therefore alterations in DANC have no major impact on basal cardiac function.9 However, under conditions of chronic stress, deficient energy transfer may have a deleterious impact on cardiac function.10,23
In the heart of Opa1+/− mice, mitochondrial oxidative capacities and respiratory chain complexes were not changed. Similarly, in leukocytes from ADOA patients, which exhibit only a partial loss of OPA1 expression, no alteration in mitochondrial oxidative capacity was observed.35 However, almost complete loss of OPA1 resulted in severely altered mitochondrial respiration.36 The only change in metabolic phenotype found in the present study was an alteration of free fatty-acid utilization by mitochondria. The mechanism responsible for this alteration is not clear. β-oxidation does not seem to be involved because MCAD or LCAD RNAs were not modified. One can hypothesize that lack of OPA1 could disturb the organization of mitochondrial inner membranes and thus affect the transport of free fatty acids in the matrix. Alternatively, mitochondrial Ca2+ flux modifications, due to altered PTP properties (see below), can also affect metabolic substrate utilization.37
The most unexpected effect of OPA1 deficiency is related to PTP function and mitochondrial calcium retention capacity. Using Rhod-2 and calcein, we observed higher CRC and a delayed PTP opening under calcium stimulation. Interestingly, Mfn2 KO mice presented the same characteristics29 suggesting a link between PTP function and dynamin-related proteins. Various hypotheses can be proposed to explain how OPA1 can influence PTP function and enhance the capacity for Ca2+ retention. First, because mitochondria from Opa1+/− mice are larger, they could, in principle, load more calcium,28,38,39 but recent results suggest that a direct relationship between mitochondrial volume and Ca2+ uptake capacity is unlikely.29 Second, because OPA1 is located in the inner mitochondrial membrane and participates in the organization of mitochondrial cristae,30 loss of this protein could disorganize the environment of intermembrane space and disturb the downstream signaling events.3,40 In this regard, the PTP, which needs proximity of the mitochondrial internal and external membranes to form, could be sensitive to OPA1 deletion and cristae disorganization. Finally, one can not exclude that due to its location, OPA1 could be a structural part of the supramolecular complex that forms the PTP.
A higher CRC and delayed PTP opening have been described as a cardioprotective mechanism against ischemia/reperfusion injury.28,29 However, we hypothesize that, in our model, these mitochondrial characteristics can be detrimental to the heart. Indeed, the TAC-induced hemodynamic stress resulted in greater cardiac hypertrophy and chamber dilation, and lower fractional shortening in Opa1+/− mice. In both strains at this stage, however, cardiac output index was still maintained due to the chamber enlargement. One possible explanation for the increased remodeling in Opa1+/− mice is the lower sensitivity of PTP to calcium accumulation in the mitochondrial matrix, which could induce mitochondrial calcium overload. Indeed, it has suggested that higher intramitochondrial calcium content would reduce the metabolic reserve capacity of the heart.37 Another possibility could be the partial loss of DANC efficacy found in our study. Finally, it is reasonable to suggest that the impairment of the mitochondrial dynamics does not allow an appropriate and homogeneous adaptation of the mitochondrial network to the increase in the energetic demand of stress, thereby exacerbating cardiac remodeling.
OPA1 has been proposed to be involved in the organization of cristae, and its deletion could modify the propensity of mitochondria to release cytochrome c. However, the correlation between the PTP, the morphology of cristae and the release of cytochrome c is not straightforward.41 Further experiments are needed to address this important issue.
In conclusion, we have established that the decreased expression of the OPA1 protein in a mouse model mimicking human ADOA disease has consequences for the morphology of mitochondria and for energetic transfers between organelles, as well as for mitochondrial CRC and PTP properties. These alterations could explain the sensitization of the heart to hemodynamic stress and show that OPA1 plays an important role in cardiac physiology.
Acknowledgments
Funding
This work was partly supported by a grant VEGA 2/0174/09. This work was supported by grants from the European Union contract LSHM-CT-2005-018833/EUGeneHeart and a grant from University of Paris Sud (Attractivité). R V-C, C B and F J are scientists at Centre National de la Recherche Scientifique.
We thank Dr. R. Fischmeister for continuous support. We thank Eric Jacquet and Romain Barbet for TDLA analysis (IMAGIF qPCR-Platform, UPR-2301 CNRS Gif/Yvette). We also thank Valérie Domergue-Dupont and the animal core facility of IFR141 for efficient handling and preparation of the animals. We are grateful to Bernd Wissinger (Genetics Laboratory, Institute for Ophthalmic Research, Centre for Ophthalmology, University of Tübingen, Germany) for sharing the Opa1+/− mouse line.
Footnotes
Conflict of interest
None
References
- 1.Liesa M, Palacin M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev. 2009;89:799–845. doi: 10.1152/physrev.00030.2008. [DOI] [PubMed] [Google Scholar]
- 2.Legros F, Lombes A, Frachon P, Rojo M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol Biol Cell. 2002;13:4343–4354. doi: 10.1091/mbc.E02-06-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olichon A, Emorine LJ, Descoins E, Pelloquin L, Brichese L, Gas N, et al. The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett. 2002;523:171–176. doi: 10.1016/s0014-5793(02)02985-x. [DOI] [PubMed] [Google Scholar]
- 4.Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26:211–215. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- 5.Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- 6.Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- 7.Votruba M, Moore AT, Bhattacharya SS. Clinical features, molecular genetics, and pathophysiology of dominant optic atrophy. J Med Genet. 1998;35:793–800. doi: 10.1136/jmg.35.10.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delettre C, Griffoin JM, Kaplan J, Dollfus H, Lorenz B, Faivre L, et al. Mutation spectrum and splicing variants in the OPA1 gene. Hum Genet. 2001;109:584–591. doi: 10.1007/s00439-001-0633-y. [DOI] [PubMed] [Google Scholar]
- 9.Kaasik A, Veksler V, Boehm E, Novotova M, Minajeva A, Ventura-Clapier R. Energetic crosstalk between organelles: architectural integration of energy production and utilization. Circ Res. 2001;89:153–159. doi: 10.1161/hh1401.093440. [DOI] [PubMed] [Google Scholar]
- 10.Wilding JR, Joubert F, de Araujo C, Fortin D, Novotova M, Veksler V, et al. Altered energy transfer from mitochondria to sarcoplasmic reticulum after cytoarchitectural perturbations in mice hearts. J Physiol. 2006;575:191–200. doi: 10.1113/jphysiol.2006.114116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piquereau J, Novotova M, Fortin D, Garnier A, Ventura-Clapier R, Veksler V, et al. Postnatal development of mouse heart: formation of energetic microdomains. J Physiol. 2010;588:2443–2454. doi: 10.1113/jphysiol.2010.189670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ventura-Clapier R, Garnier A, Veksler V. Energy metabolism in heart failure. J-Physiol. 2004;555:1–13. doi: 10.1113/jphysiol.2003.055095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ventura-Clapier R, Garnier A, Veksler V, Joubert F. Bioenergetics of the failing heart. Biochim Biophys Acta. 2011;1813:1360–1372. doi: 10.1016/j.bbamcr.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 14.Ong SB, Hausenloy DJ. Mitochondrial morphology and cardiovascular disease. Cardiovasc Res. 2010;88:16–29. doi: 10.1093/cvr/cvq237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shahrestani P, Leung HT, Le PK, Pak WL, Tse S, Ocorr K, et al. Heterozygous mutation of Drosophila Opa1 causes the development of multiple organ abnormalities in an age-dependent and organ-specific manner. PLoS One. 2009;4:e6867. doi: 10.1371/journal.pone.0006867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dorn GW, 2nd, Clark CF, Eschenbacher WH, Kang MY, Engelhard JT, Warner SJ, et al. MARF and Opa1 control mitochondrial and cardiac function in Drosophila. Circ Res. 2011;108:12–17. doi: 10.1161/CIRCRESAHA.110.236745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen L, Gong Q, Stice JP, Knowlton AA. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc Res. 2009;84:91–99. doi: 10.1093/cvr/cvp181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alavi MV, Bette S, Schimpf S, Schuettauf F, Schraermeyer U, Wehrl HF, et al. A splice site mutation in the murine Opa1 gene features pathology of autosomal dominant optic atrophy. Brain. 2007;130:1029–1042. doi: 10.1093/brain/awm005. [DOI] [PubMed] [Google Scholar]
- 19.Alavi MV, Fuhrmann N, Nguyen HP, Yu-Wai-Man P, Heiduschka P, Chinnery PF, et al. Subtle neurological and metabolic abnormalities in an Opa1 mouse model of autosomal dominant optic atrophy. Exp Neurol. 2009;220:404–409. doi: 10.1016/j.expneurol.2009.09.026. [DOI] [PubMed] [Google Scholar]
- 20.Heiduschka P, Schnichels S, Fuhrmann N, Hofmeister S, Schraermeyer U, Wissinger B, et al. Electrophysiological and histologic assessment of retinal ganglion cell fate in a mouse model for OPA1-associated autosomal dominant optic atrophy. Invest Ophthalmol Vis Sci. 2010;51:1424–1431. doi: 10.1167/iovs.09-3606. [DOI] [PubMed] [Google Scholar]
- 21.Verde I, Vandecasteele G, Lezoualc’h F, Fischmeister R. Characterization of the cyclic nucleotide phosphodiesterase subtypes involved in the regulation of the L-type Ca2+ current in rat ventricular myocytes. Br J Pharmacol. 1999;127:65–74. doi: 10.1038/sj.bjp.0702506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuznetsov AV, Veksler V, Gellerich FN, Saks V, Margreiter R, Kunz WS. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat Protoc. 2008;3:965–976. doi: 10.1038/nprot.2008.61. [DOI] [PubMed] [Google Scholar]
- 23.Joubert F, Wilding JR, Fortin D, Domergue-Dupont V, Novotova M, Ventura-Clapier R, et al. Local energetic regulation of sarcoplasmic and myosin ATPase is differently impaired in rats with heart failure. J Physiol. 2008;586:5181–5192. doi: 10.1113/jphysiol.2008.157677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem. 1977;252:8731–8739. [PubMed] [Google Scholar]
- 25.Belzacq-Casagrande AS, Martel C, Pertuiset C, Borgne-Sanchez A, Jacotot E, Brenner C. Pharmacological screening and enzymatic assays for apoptosis. Front Biosci. 2009;14:3550–3562. doi: 10.2741/3470. [DOI] [PubMed] [Google Scholar]
- 26.Fang L, Moore XL, Gao XM, Dart AM, Lim YL, Du XJ. Down-regulation of mitofusin-2 expression in cardiac hypertrophy in vitro and in vivo. Life Sci. 2007;80:2154–2160. doi: 10.1016/j.lfs.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 27.Beraud N, Pelloux S, Usson Y, Kuznetsov AV, Ronot X, Tourneur Y, et al. Mitochondrial dynamics in heart cells: very low amplitude high frequency fluctuations in adult cardiomyocytes and flow motion in non beating Hl-1 cells. J Bioenerg Biomembr. 2009;41:195–214. doi: 10.1007/s10863-009-9214-x. [DOI] [PubMed] [Google Scholar]
- 28.Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–2022. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]
- 29.Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O’Shea KM, et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol. 2011;31:1309–1328. doi: 10.1128/MCB.00911-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 31.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci U S A. 2004;101:15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, et al. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. 2003;278:7743–7746. doi: 10.1074/jbc.C200677200. [DOI] [PubMed] [Google Scholar]
- 34.Saks V, Kuznetsov A, Andrienko T, Usson Y, Appaix F, Guerrero K, et al. Heterogeneity of ADP Diffusion and Regulation of Respiration in Cardiac Cells. Biophys J. 2003;84:3436–3456. doi: 10.1016/S0006-3495(03)70065-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mayorov VI, Lowrey AJ, Biousse V, Newman NJ, Cline SD, Brown MD. Mitochondrial oxidative phosphorylation in autosomal dominant optic atrophy. BMC Biochem. 2008;9:22. doi: 10.1186/1471-2091-9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280:26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- 37.Elrod JW, Wong R, Mishra S, Vagnozzi RJ, Sakthievel B, Goonasekera SA, et al. Cyclophilin D controls mitochondrial pore-dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J Clin Invest. 2010;120:3680–3687. doi: 10.1172/JCI43171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nogueira V, Devin A, Walter L, Rigoulet M, Leverve X, Fontaine E. Effects of decreasing mitochondrial volume on the regulation of the permeability transition pore. J Bioenerg Biomembr. 2005;37:25–33. doi: 10.1007/s10863-005-4120-3. [DOI] [PubMed] [Google Scholar]
- 39.Iijima T, Tanaka K, Matsubara S, Kawakami H, Mishima T, Suga K, et al. Calcium loading capacity and morphological changes in mitochondria in an ischemic preconditioned model. Neurosci Lett. 2008;448:268–272. doi: 10.1016/j.neulet.2008.10.056. [DOI] [PubMed] [Google Scholar]
- 40.Wasilewski M, Scorrano L. The changing shape of mitochondrial apoptosis. Trends Endocrinol Metab. 2009;20:287–294. doi: 10.1016/j.tem.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 41.Sun MG, Williams J, Munoz-Pinedo C, Perkins GA, Brown JM, Ellisman MH, et al. Correlated three-dimensional light and electron microscopy reveals transformation of mitochondria during apoptosis. Nat Cell Biol. 2007;9:1057–1065. doi: 10.1038/ncb1630. [DOI] [PubMed] [Google Scholar]