

Abstract
Rationale: Rhinovirus infection is followed by significantly increased frequencies of positive, potentially pathogenic sputum cultures in chronic obstructive pulmonary disease (COPD). However, it remains unclear whether these represent de novo infections or an increased load of organisms from the complex microbial communities (microbiome) in the lower airways.
Objectives: To investigate the effect of rhinovirus infection on the airway bacterial microbiome.
Methods: Subjects with COPD (n = 14) and healthy control subjects with normal lung function (n = 17) were infected with rhinovirus. Induced sputum was collected at baseline before rhinovirus inoculation and again on Days 5, 15, and 42 after rhinovirus infection and DNA was extracted. The V3–V5 region of the bacterial 16S ribosomal RNA gene was amplified and pyrosequenced, resulting in 370,849 high-quality reads from 112 of the possible 124 time points.
Measurements and Main Results: At 15 days after rhinovirus infection, there was a sixfold increase in 16S copy number (P = 0.007) and a 16% rise in numbers of proteobacterial sequences, most notably in potentially pathogenic Haemophilus influenzae (P = 2.7 × 10-20), from a preexisting community. These changes occurred only in the sputum microbiome of subjects with COPD and were still evident 42 days after infection. This was in contrast to the temporal stability demonstrated in the microbiome of healthy smokers and nonsmokers.
Conclusions: After rhinovirus infection, there is a rise in bacterial burden and a significant outgrowth of Haemophilus influenzae from the existing microbiota of subjects with COPD. This is not observed in healthy individuals. Our findings suggest that rhinovirus infection in COPD alters the respiratory microbiome and may precipitate secondary bacterial infections.
Keywords: rhinovirus, chronic obstructive pulmonary disease, bacteria, microbiome
At a Glance Commentary
Scientific Knowledge on the Subject
After rhinovirus exacerbations of chronic obstructive pulmonary disease (COPD), sputum cultures are often positive for potential bacterial pathogens. It is unclear whether these represent true de novo infections or outgrowth from preexisting microbial communities in the lower airways.
What This Study Adds to the Field
We report that in subjects with COPD after rhinovirus infection, there is a rise in bacterial burden and an outgrowth from bacteria present at baseline. This does not occur in healthy individuals. This suggests that rhinovirus may play a critical role in precipitating secondary bacterial infections. Preventing or actively managing viral infections in COPD could mitigate the frequent secondary bacterial complications of viral exacerbations.
Chronic obstructive pulmonary disease (COPD) is a growing global health epidemic, predicted to be the fourth leading cause of mortality worldwide by 2030 (1). Despite the chronic nature of COPD, acute exacerbations are the major cause of mortality, accounting for almost 70% of health care costs and accelerating the progressive decline in lung function (2). The great majority of exacerbations are caused by respiratory infections with bacteria and viruses, each of which has been detected in about 50% of cases, with coinfection common (3). The concurrent presence of bacteria and viruses during exacerbations of COPD has been shown to be associated with a greater decline in lung function and prolonged hospital stay (3, 4).
Current understanding of the interactions between viruses and bacteria in exacerbations of obstructive airway disease is based predominantly upon classical microbial culture techniques. These have suggested that the lower airways are sterile in healthy individuals, and that bacteria are detectable in approximately one-third of stable subjects with moderate to severe COPD (5, 6). However, culture-based studies identify only a fraction of the bacteria present in a sample and suffer from well-documented limitations (7, 8). Molecular culture-independent techniques have identified complex microbial communities in the lower airways, with a distinct microbiota reported for a number of conditions and including COPD (9–12). These techniques rely on phylogenetic relationships between the sequences of highly conserved genes, such as the 16S ribosomal RNA (rRNA) gene. They can build a picture of the complete microbial community in an environment (the microbiome) and offer a more comprehensive analysis than classical culture-based techniques. To date, there have been no sequence-based studies looking at the effect of viral infection on the respiratory microbiome.
The most common viruses detected during COPD exacerbations are rhinoviruses (13). We have developed a human model of COPD exacerbation that uses experimental rhinovirus infection and induces the clinical, physiological, and inflammatory features typical of COPD exacerbations (14). In 60% of patients with COPD, rhinovirus infection is followed by positive sputum bacterial cultures (15), but it remains unclear whether these represent de novo infections or an increase in load of preexisting organisms from the lower airways. The aim of this study, therefore, was to investigate the effect of rhinovirus infection on the respiratory microbiome in COPD. Combining the in vivo model with molecular culture-independent methodologies, we have examined the bacterial communities in sputum before, during, and after rhinovirus infection to establish what changes, if any, occurred to the microbiome. Some of the results of these studies have been previously reported in the form of an abstract (16).
Methods
Subjects and Sampling
Approval for the study was obtained from the local research ethics committee (study nos. 00/BA/459E and 07/H0712/138) and informed consent was obtained from all subjects. Fourteen subjects with mild COPD (Global Initiative for Chronic Obstructive Lung Disease [GOLD] stage 2) and 17 control subjects (10 nonsmokers and 7 smokers) without any obstructive airway disease were included in this study. Subjects had no history of asthma or atopy, no other significant systemic or respiratory conditions, and no history of respiratory tract infection, exacerbation, or antibiotic use in the 3 months before the study. Subjects with COPD had a postbronchodilator FEV1 between 50 and 80% of predicted and an FEV1/FVC ratio less than 70%. The subjects were inoculated intranasally with low-dose rhinovirus-16 (10 TCID50), using an atomizer as previously described (15). Induced sputum was collected at baseline before RV inoculation (Day 0) and again on Days 5, 15, and 42 after rhinovirus infection (17).
DNA Extractions
Genomic DNA was extracted from induced sputum according to a modified protocol provided with the QIAamp DNA mini kit (Qiagen, Manchester, UK) (18). Full details are provided in the online supplement.
454 Pyrosequencing and Sequence Analysis
The V3–V5 region of the bacterial 16S rRNA gene was amplified with the forward primer 357F (19) and the modified reverse primer 926R (20) (barcoded to tag each polymerase chain reaction [PCR] product [21]). Quadruplicate 25-μl PCRs were set up, amplified, purified, and prepared for sequencing as previously described (18). Full details are provided in the online supplement.
Single-direction pyrosequencing using the GS Junior Titanium emPCR kit (Lib-L) was performed, using the 454 Life Sciences GS Junior (Roche Diagnostics, Oakland, CA). The barcoded pyrosequence reads were processed using QIIME (22). Initial denoising was performed to remove sequencing errors (23), and then Chimera Slayer was used to remove PCR-generated artifacts (24). Sequence reads were removed if they contained ambiguous bases or mismatches in the primer sequences, homopolymer runs, or a mean window quality score of less than 25. Sequences were clustered into operational taxonomic units (OTUs) at 97% identity (25), aligned to full-length 16S rRNA sequences (26), and assigned a taxonomic identity with the Ribosomal Database Project (RDP) Classifier (27). Any sequences that were present only once (singletons) or any OTUs present in only one sample were removed.
Representative sequences (most common) from each OTU of interest were aligned with the online SINA aligner (http://www.arb-silva.de/aligner/) and then imported into the ARB phylogenetic package (http://www.arb-home.de/) running on Bio-Linux 6.0 (http://nebc.nerc.ac.uk/tools/bio-linux/bio-linux-6.0). Alignments were merged with the reference Silva alignment (SSURef_108) and the 375-bp region between the Escherichia coli reference positions, 533 and 908, were used to generate phylogenetic trees. Phylogenetic trees of the OTU sequences and nearest neighbors were constructed with ARB’s neighbor-joining package with 1,000 bootstrap replicates and rooted with outgroups from related bacterial families.
16S Quantitative PCR
Triplicate 10-μl quantitative PCRs (qPCRs) were set up containing 1 μl of a 10-fold dilution of template DNA, 0.2 μl of forward primer 357F (10 μM), 0.2 μl of reverse primer 926R (10 μM), 5 μl of KAPA SYBR FAST Universal 2× qPCR master mix (Kapa Biosystems, Woburn, MA), and 3.6 μl of water. For data acquisition, the following cycling sequence was used: 1 cycle of 95°C for 3 minutes; 40 cycles of 94°C for 20 seconds; 1 cycle of 50°C for 30 seconds; and 1 cycle of 72°C for 30 seconds. After the PCR, a dissociation curve (melting curve) was constructed in the range of 65–95°C. Each run contained nontemplate controls and a 10-fold dilution series of the Pseudomonas aeruginosa PA01 16S gene cloned into a plasmid of known size. The standard curve samples were used to extrapolate the total 16S rRNA copy number from cycle threshold values for the sputum specimens. All data were analyzed with Corbett rotor-gene 6.1 software (Qiagen, Venlo, the Netherlands).
Statistical Analysis
Metastats was used to perform nonparametric t-test comparisons of microbial communities between groups (28). Significant changes in OTU abundance within phyla were assessed using two-tailed Fisher’s exact tests with the P values corrected by multiple hypothesis testing using the false discovery rate. Alpha-diversity indices were calculated in QIIME using the Shannon index (29), the Chao1 index (30), and the equitability index (31). Beta-diversity was calculated using weighted and unweighted UniFrac (32) and the Bray–Curtis measure of dissimilarity in QIIME (33). Other statistical analysis was performed with GraphPad Prism for Windows (GraphPad Software, San Diego, CA).
Results
Subjects, Sampling, and Sequencing
Successful RV inoculation was confirmed in all 14 subjects with COPD and all 17 control subjects by PCR for rhinovirus in nasal lavage fluid (15). Virus load peaked on Day 5 in sputum and remained significantly elevated over baseline up to Day 15 in both cohorts. Although on Day 5 sputum virus load was 1–3 logs higher in the COPD group compared with control subjects, this was not statistically significant (14). All of the subjects (see Table 1 for demographics) completed the study, and none required treatment with corticosteroids or antibiotics.
TABLE 1.
CLINICAL CHARACTERISTICS OF STUDY SUBJECTS
| COPD (n = 14) | Control Subjects (n = 17) | P Value (COPD vs. Control Subjects) | |
|---|---|---|---|
| Age, yr |
57 ± 8.9 |
56 ± 9.2 |
NS |
| Sex (% male) |
69 |
64 |
NS |
| Smoker (ex or current) |
100% |
47% |
<0.01 |
| FEV1 PP |
69 ± 7 |
104 ± 11 |
<0.001 |
| FEV1/FVC | 58 ± 8.3 | 79 ± 3.8 | <0.001 |
Definition of abbreviations: COPD = chronic obstructive pulmonary disease; NS = not significant; PP = percent predicted.
Data are presented as percentage value or mean ± SD as appropriate.
Genomic DNA was successfully extracted from 116 of the possible 124 time points (5 control and 3 COPD time points yielded insufficient samples). A further four samples yielded inadequate bacterial DNA to sequence (three control subjects and one subject with COPD). After denoising, chimera checking, and singleton removal, a total of 370,849 high-quality 16S rRNA reads remained. The distribution of reads was between 927 and 14,851 per sample. To control for bias of per-sample read coverage, sequences were rarefied (randomly resampled) to the same minimum of 927 for all subjects. The bacterial sequences were then clustered by sequence similarity into OTUs, which approximate to classical bacterial taxonomy. After rarefaction to 927 reads, 105,165 sequences remained, representing 456 OTUs in 112 samples. This final curated data set was used in all following analyses. The full data set can be downloaded from http://lungen.bioinformatics.ic.ac.uk/data/microbiome_rv_copd.
Bacterial Quantification
Quantification of the bacterial 16S rRNA gene in the baseline sputum samples demonstrated high copy numbers (mean, 1.660 × 109 copies/ml of sputum) with no significant difference between the two cohorts (P = 0.08) (Figure 1A). Fifteen days after rhinovirus infection, there was a sixfold increase in 16S rRNA copy number from baseline in the COPD cohort (P = 0.007) (Figure 1B). In the control cohort, however, there was only a smaller and nonsignificant 2.5-fold increase in 16S rRNA copy number from the baseline (P = 0.07) (Figure 1C). On Day 15 after rhinovirus infection, the elevated bacterial copy number of 16S rRNA correlated with the concentrations of sputum inflammatory cells (P = 0.0001), neutrophils (P = 0.001), and neutrophil elastase levels (P = 0.045) (Spearman’s rank correlation) (Figure 1D).
Figure 1.

16S ribosomal RNA (rRNA) gene copy number at baseline and after rhinovirus (RV) infection and its correlation with inflammatory markers. (A) There is no significant difference between the bacterial load before RV infection in the chronic obstructive pulmonary disease (COPD) and control cohorts (P = 0.08). (B and C) After RV infection, there is an increase in bacterial load on Day 15 compared with baseline in (B) the COPD cohort (P = 0.0068) but not in (C) the control cohort (P = 0.072). (D) On Day 15, the bacterial copy number of 16S rRNA correlates with the concentrations of sputum inflammatory cells (P = 0.0001), neutrophils (P = 0.001), and sputum neutrophil elastase levels (P = 0.045) (Spearman’s rank correlation).
Baseline Sputum Microbiome in Health and Disease
We began by comparing the baseline microbiota of the subjects with COPD with the healthy control subjects before rhinovirus infection. At the phylum level, the microbiota of the subjects with COPD was dominated by Firmicutes (65%) with large numbers of Bacteroidetes (17%) and Proteobacteria (5%) also present. Streptococcus was the most common genus (42.5% of total reads) followed by Veillonella (15.2%) and Prevotella (15.0%). In the control subjects, the same phyla predominated, with Firmicutes making up 52% of the total reads, Bacteroidetes 18%, and Actinobacteria and Proteobacteria accounting for 12 and 11% respectively. Again, Streptococcus was the most common genus (31.5% of total reads) followed by Prevotella (14.6%) and then Veillonella (10.3%) (Figure 2).
Figure 2.

A phylogenetic tree and heat map of bacterial 16S ribosomal RNA (rRNA) sequences derived from sputum at baseline. This depicts the top 100 operational taxonomic units (OTUs) organized phylogenetically by tree with abundance indicated by the color (darker blue, more abundant). The samples are grouped into chronic obstructive pulmonary disease (COPD), smokers, and nonsmoking control subjects (NS). Taxonomy assignments at the phylum level are shown in the inner column and are color-coded.
Using principal coordinate analysis (PCoA) of the UniFrac and Bray–Curtis distance matrices, there were no distinct clustering patterns between the two groups at baseline. There were no differences in the richness, diversity, or evenness of the microbial populations between subjects with COPD and control subjects at baseline. Nevertheless, we identified significant differences between the abundances of specific phyla in the patients with COPD and control subjects at baseline. In particular, a higher number of proteobacterial sequences were observed in the control subjects (P = 2.2 × 10–16, odds ratio [OR] = 0.35) with a subsequent reduction in the numbers of Firmicutes (P = 2.2 × 10–16, OR = 1.70). At the OTU level, this was driven by the presence of significantly fewer Veillonellaceae in the control group (P = 3.9 × 10–14, OR = 1.31).
Changes in the Microbiome after RV Infection
Having identified differences in the microbiome at baseline, we went on to examine the changes that occurred after rhinovirus infection.
To assess differences in overall bacterial community composition, we employed the Bray–Curtis measure of dissimilarity, where 0 means the two groups have the same composition (i.e., share all species), and 1 means the two sites do not share any species. We observed in the COPD group that the bacterial communities on Day 15 were significantly more dissimilar from the baseline community than at other time points (Figure 3). These differences were also demonstrated with the alternative UniFrac metric. In the control group at the phylum level, no significant change from baseline was seen (Figure 4A). This is reflected in the clustering of samples from each time point together on PCoA using both weighted and unweighted UniFrac, as well as on Bray–Curtis distance matrices (data not shown).
Figure 3.

Mean (±SEM) Bray–Curtis dissimilarity between communities and their respective baseline. A Bray–Curtis measure of dissimilarity of 0 means the two groups have the same composition (i.e., share all species), and 1 means the two sites do not share any species. There is a significant difference between the bacterial communities on Day 15 in the subjects with chronic obstructive pulmonary disease (COPD) compared with the baseline community. *P < 0.001 (two-tailed t test).
Figure 4.

Distribution of bacterial phyla at each time point after rhinovirus (RV) inoculation. (A) In the control subjects, no significant difference was observed between the percentage compositions at the phylum level between any time points. (B) In subjects with chronic obstructive pulmonary disease (COPD), significant increases in Proteobacteria (dark blue) were observed on Day 15 after RV inoculation compared with baseline (P = 2.2 × 10–16).
In the subjects with COPD, there was a 16% rise in the number of proteobacterial sequences on Day 15 after rhinovirus infection (P = 2.2 × 10–16, OR = 0.18). This was associated with a 12% and 5% decline in the Firmicutes and Bacteroidetes phyla, respectively (Figure 4B). Despite these changes, there were no significant differences in the richness, diversity, or evenness of the microbial populations between time points (see Figure E1 in the online supplement). The response to rhinovirus in the COPD cohort was not uniform, and there were a number of subjects in whom there was a large change in the relative abundance of the main phyla (Figure 5). The observed decline in the Firmicutes and Bacteroidetes phyla was due to reduction in the numbers of Streptococcaceae, Veillonellaceae, and Prevotellaceae. Within the Proteobacteria phylum, there was a 21% increase in the average relative abundance of a Haemophilus species (P = 2.7 × 10–20, OR = 0.4) as well as a 9.5% rise in the average relative abundance of Neisseriaceae between baseline and Day 15 (P = 0.0008, OR = 0.7) (Figure E2). Incorporating the representative sequences of both of these OTUs into phylogenetic trees constructed from SILVA reference sequences (18) enabled us to confidently identify these OTUs as Haemophilus influenzae (Figure 6) and Neisseria subflava or N. flavescens (Figure E3).
Figure 5.

The distribution of the top four phyla grouped at each time point. (A) In the control subjects, no significant change from baseline was observed in any of the phyla after rhinovirus (RV) infection. (B) The response to RV in the chronic obstructive pulmonary disease (COPD) cohort was not uniform, but there was a significant increase from baseline in Proteobacteria on Day 15 after RV inoculation (P = 2.2 × 10–16).
Figure 6.
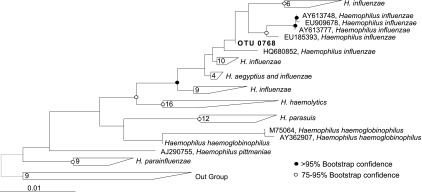
Phylogenetic identification of Haemophilus sp. operational taxonomic unit (OTU). Phylogenetic analysis of the representative sequence of OTU0768 (boldface type) shows there is strong clustering of this bacterium within the Haemophilus genus. Bootstrapping analysis provides a method to judge the strength of confidence for nodes on phylogenetic trees, and a value greater than 95% seen here supports confident assignment of this OTU as Haemophilus influenzae. The tree was rooted with a near neighbor outgroup constructed with sequences from Morganella morganii, Proteus mirabilis, and Providencia stuartii.
Importantly, both of these OTUs were present at baseline in lower numbers in patients with COPD, and at 42 days after initial rhinovirus infection, Haemophilus influenzae remained elevated compared with baseline.
Culture versus Culture-Independent Analysis
Induced sputum from all of the subjects at each time point underwent standard clinical microbial culture. None of the baseline sputum cultures yielded growth of any potentially pathogenic bacteria. After rhinovirus infection, sputum cultures for two of the subjects with COPD identified potential bacterial pathogens, and both of these were also identified via 16S RNA gene sequencing.
The sputum culture of one subject with COPD demonstrated Streptococcus pneumoniae on Day 5. The sequencing data identified the presence of a Streptococcus species, which made up 18% of the total reads present at this time point. This OTU was present in this subject at baseline in similar numbers, and on Day 15, when it accounted for 37% of the reads. Despite this, cultures at both time points were negative.
The sputum cultures of the second subject with COPD were positive for Haemophilus influenzae on Day 15. Although Haemophilus influenzae was identified by 16S rRNA gene sequencing, it accounted for less than 10% of the total reads, whereas a Streptococcus species, undetected by culture, was the most abundant OTU in that sample, accounting for 51% of the reads.
Interestingly, two subjects with COPD in whom bacterial cultures were negative for potential pathogens had a microbiome almost entirely composed of one OTU: a Neisseria species (84%) and Haemophilus influenzae (94%), respectively.
Discussion
For the first time, we have demonstrated the effect of rhinovirus infection on the respiratory microbiome. In this study, we have shown that after rhinovirus infection there was a significant rise in numbers of proteobacterial sequences, in particular the potentially pathogenic Haemophilus influenza, which had been present at baseline in lower sequence numbers, and these remained elevated at 42 days. The changes occurred only in the sputum microbiome of subjects with COPD, in contrast to the temporal stability demonstrated in the microbiome of healthy smokers and nonsmokers. Although we have previously demonstrated bacterial infections after rhinovirus infection in subjects with COPD, using sputum culture, we were not able to distinguish new infections from an increased load of organisms previously present in the airway (15). These findings suggest outgrowth occurred from an existing bacterial community present at baseline rather than acquisition of new bacterial species.
The baseline bacterial communities of both the control subjects and subjects with COPD contained Streptococcus, Prevotella, Fusobacterium, Haemophilus, and Pseudomonas, which have previously been reported to be present in the airways of healthy subjects, patients with asthma, and patients with COPD (34). Although the upper respiratory tract microbiome differs between smokers and nonsmokers, no differences have been detected in the lower respiratory microbial communities when looking at bronchoalveolar lavage (BAL) and lung tissue samples (11, 12). Looking at the healthy control subjects, we saw no significant differences in the sputum microbiome between smokers and nonsmokers. There are also a number of similarities between the baseline COPD microbiome presented here and previously published results. Sze and colleagues also found lower numbers of proteobacterial sequences in subjects with COPD compared with both smokers and nonsmokers (12), and the distribution of the genera in the BAL of the two subjects with mild COPD described by Erb-Downward and colleagues (11) is similar to that demonstrated in the sputum of our subjects with mild COPD. Despite differing sampling modalities and processing techniques, the similarities between the baseline COPD microbiome presented here and previously published results (11, 34) suggest that microbial airway communities can be robustly accessed by sputum, BAL, and bronchial brushings.
We report a 16S RNA gene bacterial copy number in sputum similar to that observed in subjects with cystic fibrosis (107 to 1010 16S rRNA copies/ml sputum) (35). In studies involving BAL (11) and lung tissue (12), no difference between the bacterial load in COPD and healthy subjects has been observed. In agreement with these findings, we also report no significant difference between the bacterial load in sputum between the subjects with COPD and healthy control subjects at baseline. After rhinovirus infection, however, there was an increase in 16S bacterial copy number in the COPD cohort, which again was not observed in the control subjects. The elevated 16S rRNA gene copy number 15 days after RV infection correlates with the total number of inflammatory cells, neutrophil count, and neutrophil elastase levels in sputum, suggesting the higher bacterial load could be driving a neutrophilic inflammatory response.
There are a number of abnormalities in the COPD pulmonary immune system, including impaired mucociliary clearance (36), increased epithelial permeability (37), dysfunctional phagocytosis (38), and impaired interferon production (39). These defects may all lead to ineffective bacterial clearance and allow overgrowth of pathogenic bacteria within a community. Our specific discovery of an increase in the numbers of potentially pathogenic Haemophilus influenzae in subjects with COPD is consistent with the increased adhesion of H. influenzae in nasal epithelial cells seen in response to rhinovirus infection (40). Impaired clearance of nontypeable Haemophilus influenzae has been demonstrated in a mouse model of emphysema (41), suggesting our observed differences could be accounted for by increased adhesion of H. influenzae and impaired pulmonary clearance.
Although prior studies of naturally occurring exacerbations have reported new strain acquisition or a strain change as an important trigger of exacerbations, they have not employed culture-independent techniques nor have they had the resolution that we were able to achieve with pyrosequencing (42). In addition, the studies have a number of weaknesses, including only a single sampling time point during exacerbations, varying times from onset to presentation, and the effects of clinically necessary treatment. By employing this experimental rhinovirus model, not only were we able to prospectively sample to establish a baseline microbiota before infection, but we could also sample at fixed multiple time points in relation to infection onset, could withhold treatment, and could include subjects without COPD as a control group.
Sputum has been used widely in studies of cystic fibrosis and bronchiectasis microbiota, but it is used infrequently in studies of other respiratory conditions. The use of induced sputum here allowed us to sample subjects serially over a prolonged period to monitor the change in the lung microbiota, something that would not have been possible with more invasive techniques. We were therefore able to show that the post–rhinovirus infection alterations in the microbiota are more prolonged, lasting at least 6 weeks postinfection (Figure 4B).
One potential limitation of our study was that we did not sample the oral microbial communities for comparison with the lung microbiota and consequently cannot comment on how they are related, or if they are related at all. There remains some controversy as to the exact relationship between the two environments and whether the lung microbiome is truly independent. The two sites are anatomically contiguous, and a large number of oral bacteria are seen among the lung microbiota, which undoubtedly represents microaspiration of oral organisms (43–45). Studies characterizing the lung microbiota using lung biopsy samples have demonstrated similar bacterial communities without traversing the upper airways and thereby eliminating potential carryover of upper respiratory tract organisms during sampling (11). The use of induced sputum in this study also avoided invasive sampling and associated potential contamination of the lower airway samples.
Another limitation of using an experimental model is that we have restricted rhinovirus infection to a small group of patients with milder COPD, with an FEV1 greater than 50% predicted. The response to rhinovirus was not uniform across the COPD cohort, with some subjects experiencing much larger changes in microbial community composition in response to rhinovirus than others. There was no difference in the rhinovirus sputum load, inflammatory mediators, or symptom scores in these subjects, so the reason for their response is unclear. Bacterial colonization and impaired immunity are even more prevalent in patients with more severe COPD and lower FEV1, and it is therefore likely that the effects of rhinovirus infection would be even greater in patients with more severe COPD.
In summary, we demonstrate that after RV infection there is a rise in bacterial burden and a significant outgrowth of Haemophilus influenzae from the existing microbiota of subjects with COPD. This is not observed in healthy smokers or nonsmokers. This suggests that rhinovirus infection in COPD results in alterations in the respiratory microbiome and may precipitate secondary bacterial infections. These findings now need to be explored further in patients with more severe disease as well as in naturally occurring viral exacerbations of COPD.
Acknowledgments
Acknowledgment
The authors gratefully acknowledge the individuals who participated in the studies that contributed to this work. This work is dedicated to the memory of Joseph Footitt.
Footnotes
Supported by the Asmarley Charitable Trust, Medical Research Council Program Grant G0600879, Wellcome Trust Grant 083567/Z/07/Z for the Centre for Respiratory Infection, Wellcome Trust Grant 096964/Z/11/Z, the Imperial College and the National Institute for Health Research (NIHR) Biomedical Research Centre funding scheme, the NIHR Clinical Lecturer funding scheme, an unrestricted grant from GlaxoSmithKline, and a grant from Pfizer UK.
Author Contributions: P.L.M. conceived of the project, designed and performed experiments, analyzed the data, and wrote the manuscript; P.M. conceived of the project, designed experiments, collected samples, and wrote the manuscript; S.A.G.W.-O., D.H., and M.J.C. performed statistical analyses, interpreted the results, and helped write the manuscript; J.F. conceived of the project, designed experiments, and collected samples; M.-B.T.-T., S.E., and O.M.K. enrolled patients and performed the research; W.O.C.C., S.L.J., and M.F.M. conceived of the project, designed experiments, coordinated collaborations, and wrote the manuscript. J.F. died before the publication of this article. The corresponding author, M.F.M., has therefore supplied the information regarding J.F.’s contribution to the manuscript and his competing interests, and it is correct to the best of her knowledge.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Originally Published in Press as DOI: 10.1164/rccm.201302-0341OC on August 30, 2013
This article has an online supplement, which is available from this issue’s table of contents at www.atsjournals.org
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Barnes PJ. Chronic obstructive pulmonary disease: a growing but neglected global epidemic. PLoS Med. 2007;4:e112. doi: 10.1371/journal.pmed.0040112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Donaldson GC. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax. 2002;57:847–852. doi: 10.1136/thorax.57.10.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Papi A, Bellettato CM, Braccioni F, Romagnoli M, Casolari P, Caramori G, Fabbri LM, Johnston SL. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med. 2006;173:1114–1121. doi: 10.1164/rccm.200506-859OC. [DOI] [PubMed] [Google Scholar]
- 4.Seemungal T, Sykes A ICEAD Contributors. Recent advances in exacerbations of COPD. Thorax. 2008;63:850–852. doi: 10.1136/thx.2008.099127. [DOI] [PubMed] [Google Scholar]
- 5.Sethi S, Maloney J, Grove L, Wrona C, Berenson CS. Airway inflammation and bronchial bacterial colonization in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173:991–998. doi: 10.1164/rccm.200509-1525OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosell A, Monsó E, Soler N, Torres F, Angrill J, Riise G, Zalacaín R, Morera J, Torres A. Microbiologic determinants of exacerbation in chronic obstructive pulmonary disease. Arch Intern Med. 2005;165:891–897. doi: 10.1001/archinte.165.8.891. [DOI] [PubMed] [Google Scholar]
- 7.Suau A, Bonnet R, Sutren M, Godon JJ, Gibson GR, Collins MD, Doré J. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl Environ Microbiol. 1999;65:4799–4807. doi: 10.1128/aem.65.11.4799-4807.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Staley JT, Konopka A. Measurement of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Annu Rev Microbiol. 1985;39:321–346. doi: 10.1146/annurev.mi.39.100185.001541. [DOI] [PubMed] [Google Scholar]
- 9.Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, Davies J, Ervine A, Poulter L, Pachter L, et al. Disordered microbial communities in asthmatic airways. PLoS One. 2010;5:e8578. doi: 10.1371/journal.pone.0008578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang YJ, Kim E, Cox MJ, Brodie EL, Brown R, Wiener-Kronish JP, Lynch SV. A persistent and diverse airway microbiota present during chronic obstructive pulmonary disease exacerbations. OMICS. 2010;14:9–59. doi: 10.1089/omi.2009.0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, Schmidt LA, Young VB, Toews GB, Curtis JL, Sundaram B, et al. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS One. 2011;6:e16384. doi: 10.1371/journal.pone.0016384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sze MA, Dimitriu PA, Hayashi S, Elliott WM, McDonough JE, Gosselink JV, Cooper J, Sin DD, Mohn WW, Hogg JC. The lung tissue microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;185:1073–1080. doi: 10.1164/rccm.201111-2075OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mohan A, Chandra S, Agarwal D, Guleria R, Broor S, Gaur B, Pandey RM. Prevalence of viral infection detected by PCR and RT-PCR in patients with acute exacerbation of COPD: a systematic review. Respirology. 2010;15:536–542. doi: 10.1111/j.1440-1843.2010.01722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mallia P, Message SD, Gielen V, Contoli M, Gray K, Kebadze T, Aniscenko J, Laza-Stanca V, Edwards MR, Slater L, et al. Experimental rhinovirus infection as a human model of chronic obstructive pulmonary disease exacerbation. Am J Respir Crit Care. 2010;183:1–44. doi: 10.1164/rccm.201006-0833OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mallia P, Footitt J, Sotero R, Jepson A, Contoli M, Trujillo-Torralbo M-B, Kebadze T, Aniscenko J, Oleszkiewicz G, Gray K, et al. Rhinovirus infection induces degradation of antimicrobial peptides and secondary bacterial infection in COPD. Am J Respir Crit Care Med. 2012;186:1117–1124. doi: 10.1164/rccm.201205-0806OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Molyneaux PL, Mallia P, Duff RM, Moffatt MF, Cookson WO, Johnston SL. Investigating the role of rhinovirus infection in precipitating bacterial infections in COPD using culture independent molecular microbiology [abstract] Am J Respir Crit Care Med. 2011;183:A3121. [Google Scholar]
- 17.Mallia P, Message SD, Kebadze T, Parker HL, Kon OM, Johnston SL. An experimental model of rhinovirus induced chronic obstructive pulmonary disease exacerbations: a pilot study. Respir Res. 2006;7:116. doi: 10.1186/1465-9921-7-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cardenas PA, Cooper PJ, Cox MJ, Chico M, Arias C, Moffatt MF, Cookson WO. Upper airways microbiota in antibiotic-naïve wheezing and healthy infants from the tropics of rural Ecuador. PLoS One. 2012;7:e46803. doi: 10.1371/journal.pone.0046803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muyzer G, De Waal EC, Uitterlinden AG. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction–amplified genes coding for 16S rRNA. Appl Environ Microbiol. 1993;59:695–700. doi: 10.1128/aem.59.3.695-700.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muyzer G, Teske A, Wirsen CO, Jannasch HW. Phylogenetic relationships of Thiomicrospira species and their identification in deep-sea hydrothermal vent samples by denaturing gradient gel electrophoresis of 16S rDNA fragments. Arch Microbiol. 1995;164:165–172. doi: 10.1007/BF02529967. [DOI] [PubMed] [Google Scholar]
- 21.Fierer N, Hamady M, Lauber CL, Knight R. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc Natl Acad Sci USA. 2008;105:17994–17999. doi: 10.1073/pnas.0807920105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quince C, Lanzen A, Curtis TP, Davenport RJ, Hall N, Head IM, Read LF, Sloan WT. Accurate determination of microbial diversity from 454 pyrosequencing data. Nat Methods. 2009;6:639–641. doi: 10.1038/nmeth.1361. [DOI] [PubMed] [Google Scholar]
- 24.Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK, Sodergren E, et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011;21:494–504. doi: 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 26.Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010;26:266–267. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.White JR, Nagarajan N, Pop M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLOS Comput Biol. 2009;5:e1000352. doi: 10.1371/journal.pcbi.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shannon CE. A mathematical theory of communication. Bell Syst Tech J. 1948;27:379–423. [Google Scholar]
- 30.Chao A. Nonparametric estimation of the number of classes in a population. Scand J Stat. 1984;11:265–270. [Google Scholar]
- 31.Magurran AE. Princeton, NJ: Princeton University Press; 1988. Ecological diversity and its measurement. [Google Scholar]
- 32.Lozupone C, Hamady M, Knight R. UniFrac—an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics. 2006;7:371. doi: 10.1186/1471-2105-7-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bray JR, Curtis JT. An ordination of the upland forest communities of southern Wisconsin. Ecol Monogr. 1957;27:325–349. [Google Scholar]
- 34.Han MK, Huang YJ, Lipuma JJ, Boushey HA, Boucher RC, Cookson WO, Curtis JL, Erb-Downward J, Lynch SV, Sethi S, et al. Significance of the microbiome in obstructive lung disease. Thorax. 2012;67:456–463. doi: 10.1136/thoraxjnl-2011-201183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao J, Schloss PD, Kalikin LM, Carmody LA, Foster BK, Petrosino JF, Cavalcoli JD, VanDevanter DR, Murray S, Li JZ, et al. Decade-long bacterial community dynamics in cystic fibrosis airways. Proc Natl Acad Sci USA. 2012;109:5809–5814. doi: 10.1073/pnas.1120577109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Verra F, Escudier E, Lebargy F, Bernaudin JF, De Crémoux H, Bignon J, Verra F, Escudier E, Lebargy F, Bernaudin JF, et al. Ciliary abnormalities in bronchial epithelium of smokers, ex-smokers, and nonsmokers. Am J Respir Crit Care Med. 1995;151:630–634. doi: 10.1164/ajrccm/151.3_Pt_1.630. [DOI] [PubMed] [Google Scholar]
- 37.Olivera DS, Boggs SE, Beenhouwer C, Aden J, Knall C. Cellular mechanisms of mainstream cigarette smoke–induced lung epithelial tight junction permeability changes in vitro. Inhal Toxicol. 2007;19:13–22. doi: 10.1080/08958370600985768. [DOI] [PubMed] [Google Scholar]
- 38.Donnelly LE, Barnes PJ. Defective phagocytosis in airways disease. Chest. 2012;141:1055–1062. doi: 10.1378/chest.11-2348. [DOI] [PubMed] [Google Scholar]
- 39.Beasley V, Joshi PV, Singanayagam A, Molyneaux PL, Johnston SL, Mallia P. Lung microbiology and exacerbations in COPD. Int J Chron Obstruct Pulmon Dis. 2012;7:555–569. doi: 10.2147/COPD.S28286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang JH, Kwon HJ, Jang YJ. Rhinovirus enhances various bacterial adhesions to nasal epithelial cells simultaneously. Laryngoscope. 2009;119:1406–1411. doi: 10.1002/lary.20498. [DOI] [PubMed] [Google Scholar]
- 41.Tandon MK, Phillips M, Waterer G, Dunkley M, Comans P, Clancy R. Oral immunotherapy with inactivated nontypeable Haemophilus influenzae reduces severity of acute exacerbations in severe COPD. Chest. 2010;137:805–811. doi: 10.1378/chest.09-1382. [DOI] [PubMed] [Google Scholar]
- 42.Sethi S, Evans N, Grant BJB, Murphy TF. New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. N Engl J Med. 2002;347:465–471. doi: 10.1056/NEJMoa012561. [DOI] [PubMed] [Google Scholar]
- 43.Morris A, Beck JM, Schloss PD, Campbell TB, Crothers K, Curtis JL, Flores SC, Fontenot AP, Ghedin E, Huang L, et al. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med. 2013;187:1067–1075. doi: 10.1164/rccm.201210-1913OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, Bushman FD, Collman RG. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. 2011;184:957–963. doi: 10.1164/rccm.201104-0655OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Charlson ES, Diamond JM, Bittinger K, Fitzgerald AS, Yadav A, Haas AR, Bushman FD, Collman RG. Lung-enriched organisms and aberrant bacterial and fungal respiratory microbiota after lung transplant. Am J Respir Crit Care Med. 2012;186:536–545. doi: 10.1164/rccm.201204-0693OC. [DOI] [PMC free article] [PubMed] [Google Scholar]