

Abstract
Rationale: Autoimmunity has long been associated with pulmonary hypertension. Bronchus-associated lymphoid tissue plays important roles in antigen sampling and self-tolerance during infection and inflammation.
Objectives: We reasoned that activated bronchus-associated lymphoid tissue would be evident in rats with pulmonary hypertension, and that loss of self-tolerance would result in production of pathologic autoantibodies that drive vascular remodeling.
Methods: We used animal models, histology, and gene expression assays to evaluate the role of bronchus-associated lymphoid tissue in pulmonary hypertension.
Measurements and Main Results: Bronchus-associated lymphoid tissue was more numerous, larger, and more active in pulmonary hypertension compared with control animals. We found dendritic cells in and around lymphoid tissue, which were composed of CD3+ T cells over a core of CD45RA+ B cells. Antirat IgG and plasma from rats with pulmonary hypertension decorated B cells in lymphoid tissue, resistance vessels, and adventitia of large vessels. Lymphoid tissue in diseased rats was vascularized by aquaporin-1+ high endothelial venules and vascular cell adhesion molecule–positive vessels. Autoantibodies are produced in bronchus-associated lymphoid tissue and, when bound to pulmonary adventitial fibroblasts, change their phenotype to one that may promote inflammation. Passive transfer of autoantibodies into rats caused pulmonary vascular remodeling and pulmonary hypertension. Diminution of lymphoid tissue reversed pulmonary hypertension, whereas immunologic blockade of CCR7 worsened pulmonary hypertension and hastened its onset.
Conclusions: Bronchus-associated lymphoid tissue expands in pulmonary hypertension and is autoimmunologically active. Loss of self-tolerance contributes to pulmonary vascular remodeling and pulmonary hypertension. Lymphoid tissue–directed therapies may be beneficial in treating pulmonary hypertension.
Keywords: hypertension, pulmonary, lymphoid tissue, inflammation, autoimmunity
At a Glance Commentary
Scientific Knowledge on the Subject
Autoimmunity has long been associated with pulmonary hypertension, but little is known about the mechanisms that may drive autoimmune-related pulmonary vascular remodeling.
What This Study Adds to the Field
Bronchus-associated lymphoid tissue–directed therapies may be beneficial in treating pulmonary hypertension.
Pulmonary hypertension (PH) is characterized by progressive increases in pulmonary artery pressure and pulmonary vascular resistance resulting in right ventricular failure (1). The primary histopathologic finding in PH is vascular remodeling, in which the pulmonary vasculature becomes stiff, occluded, and fibrotic (2). Vascular remodeling seems to require lung infiltration of inflammatory cells, and continual influx likely sustains a cycle of perpetual inflammation and remodeling by poorly defined mechanisms (3).
The bronchovascular space is a structure that marries sterile (vasculature) and nonsterile (airway) components. Bronchus-associated lymphoid tissues (BALTs) are tertiary lymphoid organs that provide an elegant immune surveillance apparatus ensuring airway and vessel homeostatic patency. They are histologically similar to lymph nodes containing afferent lymphatic connection, B cells, T cells, antigen-presenting cells, stroma, and a vascular supply (4). The chief antigen-presenting cells in BALT are dendritic cells (DC) expressing OX-62+ (also known as CD103) and/or CCR7+ that organize around airways in interconnected networks (5). DCs scavenge antigens, migrate to CCL19 and CCL21-rich BALT by expression of the cognate receptor CCR7, and orchestrate immune responses and nonresponses (anergy) by T and B cells (5). CCR7−/− mice spontaneously develop PH and display BALT rich in B and T cells, likely because CCR7+ DC are absent to suppress lymphocytes by anergy (6). Recently, it was demonstrated that pulmonary lymphoid neogenesis accompanies idiopathic PH (7). In that study, it was suggested that pulmonary tertiary lymphoid organs could be directing local immune phenomena.
Associations between PH and autoimmunity have long been recognized (8, 9). Antibodies against endothelial cells (10), fibroblasts (11), phospholipids (12), and nuclear antigens (13) have been described. Target antigens have been identified (11), as have contractile effects of IgG binding to vascular cells (14). These findings collectively suggest that local autoantibody production in PH contributes to PH pathology.
We hypothesized that the hypoxia and monocrotaline (MCT) induced models of PH cause lung injury sufficient to stimulate rat BALT, resulting in breakage of self-tolerance. We imagined that subsequent autoantigenic labeling of vascular cells with autoantibody may result from, and perpetuate, the inflammation and vascular remodeling typical of PH. We found that, in PH compared with control animals, BALT was expanded, demonstrated an activated follicular organization, and was extensively vascularized. In MCT rats and less so in hypoxic rats, we measured high titers of plasma IgG that labeled lung vascular proteins in a variety of experimental settings. To determine the pathobiologic contributions of BALTs and autoantibodies in PH, we used pharmacologic, immunologic, and passive transfer strategies in distinct PH models and genetic backgrounds. We found that BALT and autoantibodies are sufficient to cause vascular remodeling and PH.
Methods
Additional details of methodology are provided in the online supplement.
Models of PH
The Institutional Animal Care and Use Committee of the institution approved this study (#32910[02]1E to K.R.S., #91010[03]1E to M.E.Y.). The characterization of the hypoxic mouse, cow, MCT, and chronic hypoxia rat models has been previously summarized (15). For most rat studies, we used male Wistar rats with a single subcutaneous injection of MCT at 60 mg/kg at 6–8 weeks of age. Lung parenchyma from animals was obtained as previously described (15). Hemodynamics were assessed with a Millar catheter placed in the main pulmonary artery by way of the right ventricle; correct placement of the catheter was confirmed by observing a significant rise in diastolic pressure as the catheter moved out of the ventricle. Systemic blood pressure was monitored with another pressure catheter inserted in the femoral artery. Cardiac performance was assessed with a pressure-volume system (Scisense Inc, London, ON, Canada). For analyses, animals were anesthetized with 2% isofluorane, and their body temperature was maintained at 37°C. Total pulmonary vascular resistance index was calculated as mean pulmonary artery pressure/cardiac index, where cardiac index equals cardiac output divided by body weight. Hemodynamic data from animals in the study are provided in the figures. In some control animals and end-stage animals with PH, echocardiographic analyses were performed using a Vevo770 (VisualSonics, Toronto, ON, Canada). Pulse-wave Doppler of pulmonary outflow was recorded in the parasternal short-axis view at the level of the aortic valve. Total pulmonary vascular resistance index was calculated as mean pulmonary artery pressure/cardiac index, where cardiac index equals cardiac output divided by body weight, as described previously.
Passive Autoantibody Transfer
IgGs were purified from 4-week MCT rat plasmas. Alternatively, unfractionated plasma was used in separate experiments. Autoantibodies were injected at 1 mg/ml by tail vein injection twice per week for 3 weeks. Before any injections, preimmune plasma was centrifuged from 1 ml peripheral blood drawn from the tail vein that was collected once per week for 2 weeks.
Statistical Analyses
For data from all studies, GraphPad Prism software (GraphPad, La Jolla, CA) was used to generate graphs and analyze data. Analysis of variance with Bonferroni post-test (P < 0.05) was used to determine statistical differences between groups. Student t test was used for comparison between two groups.
Results
BALT Numbers Increase in PH
BALT has been shown to be involved in maintenance of self-tolerance and prevention of autoimmunity (4). In the rat, BALTs are sparsely present compared with rats with lung inflammation and/or infection (16). We reasoned that differences between rat BALTs from control animals and from PH would offer clues as to the development of BALT in humans. To address this, we quantified the number and size of BALTs from control animals and from two (rat) models of PH. After 4 weeks of either hypoxia or 4 weeks post-MCT injection, BALTs were significantly more abundant and larger compared with control animals (Figure 1A). Similar results were found regardless of rat strain (Wistar, Wistar Kyoto, Sprague-Dawley, and Lewis strains, data not shown), and in hypoxic mice and two bovine models of PH (Figure 1B; see Figure E1 in the online supplement). Notably, the increased number of BALTs and their histologic appearance closely mirror BALT in human idiopathic pulmonary arterial hypertension (7). Thus, across species and rat strains, BALTs are associated with PH, consistent with human idiopathic pulmonary arterial hypertension.
Figure 1.

Bronchus-associated lymphoid tissues (BALT) are more conspicuous in pulmonary hypertension (PH). BALTs are increased in number (A, top left, and hematoxylin and eosin sections, top right) in rats with PH compared with control animals. (B) BALTs are readily apparent in humans and in bovine (representative images from cows, rats, and mice with PH). Representative images and measurements in A taken from lung sections from n = 6 animals per group, with two to three sections per rat from three independent experiments. Bars = 20 μm. Representative image using human section (B, left) taken from analysis of n = 3 patient samples, three sections per sample. *P less than 0.05 compared with control, Student t test. IPAH = idiopathic pulmonary arterial hypertension; MCT = monocrotaline.
BALTs in PH Lung Are Organized and Are Immune-activated
We investigated the presence of immune cells and expression of immune-modulatory proteins. PH BALTs contained abundance of CD3+ T cells, CD45RA+ B cells, and DCs expressing either CCR7, CD11c, and/or OX-62 and in a more focal manner compared with control animals (Figure 2, representative images). In PH rats, DCs were present as a “network” underpinning the adlumenal surface of the associated airway while more robustly expressing the DC maturation marker CD83 (Figure 2). DCs in control rats sparingly populated the bronchovascular milieu, lacked appreciable organization, and expressed less CD83 (Figure 2). Lymphocytes organized into clusters of B cells and T cells in PH BALTs but less so in control BALTs (Figure 2). CD21+ follicular DCs were rare and interspersed among the CD11b+ cells (Figure 2). In MCT but not hypoxic rats, a pronounced increase in proliferating cells was evident as Ki-67 positivity (Figure 2). Antibodies to either CD138 (plasma cells) or activation-induced cytidine deaminase (AID; an enzyme required for antibody class-switching) did not reliably produce staining above background (not shown). Collectively, these results show that, in rats with MCT PH compared with control animals, BALT is increasingly populated with T cells, B cells, and DCs. Furthermore, the patterns of cell clustering and segregation are similar to immune-activated lymphoid tissues in disease contexts.
Figure 2.

Bronchus-associated lymphoid tissues (BALTs) in rats with pulmonary hypertension (PH) are highly organized and more vascularized compared with control animals. In PH versus control animals, CD3+ T cells and CD45RA+ B cells adopt distinct segregational localizations in PH BALT, and are encircled by OX-62+ dendritic cells, and CD11b+, CD11c+, and CD83+ cells (left column of images and top right images). VCAM+ structures are more evident in PH versus control animals, whereas aquaporin (AQP)-1–positive structures seemed to be proportional to BALT size in both PH and control animals (right column of images, middle). CD45RA+ cells did not colocalize with ki-67 immunoreactivity in either control animals or PH, even when control BALT sizes were large (lowest control left image, right column). Note robust ki-67–positive cells (red, lowest right image) in adventitial of bronchovascular space. Representative images and measurements taken from lung sections from n = 6 animals per group, with two to three sections per rat from three independent experiments. All images, original magnification: ×100; bars = 20 μm where applicable. CTL = control; MCT = monocrotaline; VCAM = vascular cell adhesion molecule.
BALTs in PH Lung Are Well Vascularized and Selectively Adhesive
Next, we determined whether BALTs in PH were associated with increased vascularity as seen in secondary immune organs (17). PH BALTs were vascularized comparably with control BALTs when adjusted for increased size. PH lymphatic vessels expressing aquaporin 1 (Figure 2) were evident, as were vascular cell adhesion molecule–positive (Figure 2) high endothelial venules. Aquaporin 1+ and vascular cell adhesion molecule were expressed by high endothelial venule endothelium and by cells lining sinuses consistent with previous localizations (18). Antibodies against peripheral node addressin (PNAd) did not distinguish BALT structures or cellular constituents above background in rat lungs (not shown). To functionally test adhesion differences consistent with the expression differences we found, we incubated purified rat spleen L-selectin+ T cells labeled with fluorescent dye on rat lung sections. We observed increased binding of L-selectin+ T cells to PH BALTs compared with control BALTs (Figures 3A–3C). This binding was blocked by preincubation of lung tissue with PNAd-blocking antibody (Figure 3D).
Figure 3.

Pulmonary hypertension (PH) lung bronchus-associated lymphoid tissues (BALTs) are bound by T cells by peripheral node addressin (PNAd). BALTs in experimental PH bind dye-labeled immunomagnetic bead sorted spleen and thymic rat CD3+ T cells (B and C), but control lung sections (A) do not. The T-cell adhesion may be largely attributed to binding to PNAd in BALTs (white arrows) because preincubation of lung sections with PNAd antibody attenuated T-cell adhesion to the BALT but not the lung parenchyma (D, yellow arrows). Representative images from tissue-cell incubations taken from lung sections from n = 6 animals per group, with two to three sections per rat from three independent experiments. Bars = 20 μm. All binding experiments were performed in triplicate. A = airway; MCT = monocrotaline.
Plasma from PH Rats Contains Antifibroblast Antibodies Produced in BALTs
Tertiary lymphoid tissues are associated with maintenance of self-tolerance and implicated in autoimmune disorders (19). The presence of autoantibodies in humans with PH is well documented, and target antigens have been identified. We investigated whether PH rat plasma contained autoantibodies as in humans with PH, and looked for evidence of autoantibody production occurring in BALT. To do this, we applied plasma from MCT rats (12 of 15) to lung sections from control animals followed by fluorescent-labeled secondary antibody against rat IgG. We found intense adventitial staining (Figure 4B) and small vessel staining (Figure 4B, insert) that was absent if we applied control (Figure 4A, nonhypertensive) rat plasma. Similarly, control rat plasmas applied to lung sections from PH rats, either hypoxic lung (Figure 4C) or MCT (Figure 4D), failed to generate a staining signal. In our 4-week hypoxia experiments with mice, we did not find substantial evidence of autoantibody production, similar to what we found in hypoxic rats (see Figure E1). Plasmas from a few hypoxic rats (n = 3 of 15) weakly stained control rat lung tissue (not shown). Protein G–mediated IgG removal or serial dilution of the PH plasmas eliminated autoantibody staining (not shown), excluding the possibility of background contributions from the secondary antibody or nonspecific binding of plasma proteins. To identify antigen specificity, we incubated Alexa-594 conjugated vimentin (Figure 4E), phosphatidylinositol 3 kinase (Figure 4E), or Hsp27 (not shown) with rat lung sections, because these were identified as fibroblast autoantigens in human PH (11). Pulmonary artery adventitia and lung parenchyma in PH rat sections, but not control animals (not shown), were labeled with these fluorantigens, suggesting the presence of autoantibodies for those antigens. Binding could be competed away with unlabeled antigens (not shown). Double staining for CD45RA and antirat IgG colabeled a distinct population of cells in BALTs from MCT rats but not control animals (Figure 4F, arrow). These results suggest that in situ autoantibody production is ongoing in MCT rat BALTs and plasma cells. We quantified plasma IgG as PH developed in the MCT rats (Figure 4G) and found that MCT rats were increased over control animals at 7 days (1,120 ± 24 μg/ml, tapering at Week 2 [2,400 ± 74 μg/ml] and Week 3 [1,280 ± 68 μg/ml]), but then dramatically increasing at 4 weeks (4,160 ± 74 μg/ml). To determine the extent of tissue antigenicity, we electrophoresed control rat tissue lysates and immunoblotted with PH rat plasma as primary sera and antirat IgG as secondary (Figure 4H). Blotting with pooled (n = 3) control rat plasma produced a single band in all lanes at the size expected for IgG. In contrast, blotting with pooled (n = 3 per time point) MCT plasmas produced additional bands in lung, thyroid, heart, and skeletal muscle lysate lanes beginning at Week 1 post-MCT, and continuingly evident in 2- and 3-week MCT plasmas. Plasma from 4-week MCT rats produced banding in a range of sizes in all tissue lysates. This finding is consistent with reports of interepitope and intraepitope autoimmune spreading (20, 21).
Figure 4.

Rats with pulmonary hypertension (PH) circulate autoantibodies. (A) Control (CTL) lung sections incubated with plasma from normal (no PH) rats is not bound by sequentially applied Alexa 488–conjugated antirat IgG. In contrast, control lung sections incubated with plasma from rats with monocrotaline (MCT) induced PH can be bound by antirat IgG (B, green signal indicated by yellow arrows) indicating that the PH plasma contains IgG that bind self-antigens (autoantibodies). The inset in B shows resistance sized vessels (50–500 μm internal diameter) are also labeled with autoantibodies in PH plasma. (C and D) Lung sections from rats with hypoxic PH or MCT-PH incubated with control plasmas lack autoantibody binding. MCT-PH lung sections incubated with fluor-labeled phosphatidyl inositol 3 kinase (Pi3K) or vimentin (E, red signal, yellow arrows) indicate the presence of autoantibodies in situ. (F) Lung sections from MCT rats, but not control rats, demonstrate the presence of rat IgGs (green) localized to regions with CD45RA+ B cells (red). (G) Plasma IgG titers increase in MCT-PH rats compared with control rats, peaking at Week 4 and correlating with increasing mean pulmonary artery (PA) pressure. (H) Immunoblots using rat plasma as the primary antibody reveal autoimmune epitope spreading. Compared with the lack of autoantibodies in control rat plasma, MCT-PH plasma contains autoantibodies that increasingly recognize a wider array of tissue lysates, peaking at 4 weeks post-MCT. Representative images taken from lung sections from n = 6 animals per group, with two to three sections per rat from three independent experiments. Bars = 20 μm. ELISAs were performed in three separate experiments in triplicate on rat plasmas from n = 3 rats per group. Immunoblots shown are representative of tissue lysates run in three separate experiments. Blotting used pools (n = 3) of indicated rat plasmas, performed independently using three separate pools.
We wanted to visualize bona fide autoantibody binding of cellular targets in vitro. When culturing rat pulmonary artery adventitial cells, we found that MCT plasmas and a few hypoxic rat plasmas, but not control animals, contained antibodies predominantly to fibroblasts, and to a lesser extent α smooth muscle actin expressing pulmonary artery cells and pulmonary artery endothelial cells (Figure 5B). Staining patterns varied between individual rat plasmas, ranging from intense nuclear, to cytoplasmic, to perinuclear, or filamentous. Preincubation of PH plasma with protein G eliminated staining of vascular cells (not shown). Identical results were obtained using human pulmonary artery fibroblasts incubated with human PH plasmas (Figure 5A), mirroring previously published results with human aortic vascular smooth muscle cells (14).
Figure 5.

Human and monocrotaline (MCT) rat autoantibodies label pulmonary artery fibroblasts. (A) Compared with control (CTL) human plasma, plasma from patients with pulmonary hypertension (PH) (verified in clinical laboratory workup to have autoantibodies) label human lung fibroblasts in nuclear, punctate, and filamentous patterns. (B) Similarly, rat plasmas label rat lung fibroblasts in nuclear, punctate, and filamentous patterns. IgG from PH rats, but not human IgG or preimmune plasma IgG, stimulate PA fibs toward a phenotype that is proinflammatory (C, increased IL-6 and CCL2 secretion) and proadhesive (D, increased expression of intercellular adhesion molecule [ICAM]). Cell cultures and ELISAs were performed in three separate experiments in triplicate with rat plasmas from the indicated rats per group, compared with n = 3 control media experiments. Quantitative polymerase chain reaction was performed in triplicate using mRNA isolated from fibroblasts used in three independent experiments. All images, original magnification: ×100; bars = 20 μm where applicable. *P less than 0.05 compared with control, Student t test.
PH Plasma Antifibroblast Antibodies Stimulate Pulmonary Adventitial Fibroblasts to Produce Proadhesive and Proinflammatory Cytokines and Chemokines
Binding of autoantibodies induces proinflammatory phenotypes in target cells, including fibroblasts (22). Because we found evidence of autoantibodies against pulmonary vascular cells primarily in the MCT PH rat plasmas, we tested whether binding of autoantibodies to pulmonary adventitial fibroblasts would modulate their function. In both human cell/human plasma and rat cell/rat plasma contexts, we found that PH plasmas (MCT for rats), but not controls, stimulated pulmonary adventitial fibroblasts to secrete IL-1β and IL-6 (Figure 5C), and to express intercellular adhesion molecule-1 (Figure 5D).
Strategies Aimed at Diminution of BALT Biology Prevent and Reverse PH
To test for functional roles for BALT and autoantibody production in PH, we first reviewed the literature and considered four potential control nodes: (1) monocrotalinogenic lung cell apoptosis, (2) CCR7-mediated cell homing, (3) lymphatic vascularization, and (4) BALT formation and maintenance. Accordingly, we chose salubrinal, a cell stress protectant, to modulate the unfolded protein response toward a survival phenotype, away from a proapoptotic phenotype (control node 1). This was based on our recent observations that salubrinal protects against MCT-induced pneumotoxicity and pulmonary vascular remodeling (23). Salubrinal, when coadministered with MCT and then twice per week thereafter for 4 weeks, significantly reduced BALT size and number and largely prevented autoantibody formation in rats (Figure 6A). Conversely, salubrinal administered with the same regimen but initiated 2 weeks after MCT did not prevent MCT-induced increases in BALT size, number, or autoantibody development. Similar results were obtained with the vascular endothelial growth factor receptor 3 inhibitor MAZ-51 (control node 3, lymphatics), which attenuated all endpoints by prevention but less so by reversal regimen (Figure 6B). Antagonism of CCR7 by CCR7-Ig potentiated MCT-PH, BALT, and plasma IgG, with lung remodeling highly similar to the CCR7−/− mouse phenotype (Figure 6C, control node 2). CCR7 antagonism in MCT rats was associated with increased AID, decreased CCR7, CCL19, CCL21, lymphotoxin β receptor (LTBR), and CXCL13 in BALT. Finally, we applied LTBR blockade (LTBR-Ig, control node 4) and found prevention and reversal of MCT-induced PH and vascular remodeling. BALTs were almost completely absent in LTBR-Ig–treated MCT rats, and we could not detect plasma autoantibodies (Figure 6D). Laser capture microdissection and quantitative polymerase chain reaction of BALT showed dramatically decreased expression of lymphotoxin α receptor, CXCL13, and AID (tightly linked with class switching recombination and somatic hypermutation events occurring in B cell selection [24]) compared with control and MCT rats (Figure 6E). These genes were chosen for analysis based on studies of activated lymphoid tissue in disease associated with immune dysfunction and autoimmunity (4, 5, 24). Figure 7 summarizes results from pharmacologic and immunologic interventions in MCT rats, in either prevention or reversal mode.
Figure 6.

Strategies aimed at diminution of bronchus-associated lymphoid tissues (BALTs) biology prevent and reverse pulmonary hypertension (PH). (A) Preventing lung cell apoptosis (and presumably self-antigen burden) by salubrinal (Sal), an unfolded protein response modulator, prevents and reverses pulmonary vascular remodeling and PH in a manner associated with decreased BALT size and number. (B) Pharmacologic VEGFR3 antagonism prevents and reverses monocrotaline (MCT)-PH and is associated with decreased macrophage influx, decreased blood and lymphatic vascularization, and smaller BALT size and numbers. (C) CCR7 controls migration of lung CCR7+ dendritic cells (DC) and CCR7+ T cells into BALT; pharmacologic CCR7 antagonism worsens MCT-PH and is associated with larger BALT size and numbers. (D) Lymphotoxin β receptor (LTBR) control animals BALT formation and maintenance; pharmacologic LTBR antagonism prevents and reverses MCT-PH and is associated with decreased blood and lymphatic vascularization, and smaller BALT size and numbers. In A–D, pressure measurement, BALT counts, and representative lung section images were taken from n = 6 animals per group, with two to three sections per rat from three independent experiments. All images, original magnification: ×100. *P less than 0.05 compared with control, Student t test. (E) Representative laser capture microdissection of BALT from rat lung section. Black arrows show the microdissected BALT region. The table lists the mRNAs isolated from the rat groups and their respective mean fold changes in expression relative to the control lung BALT. n = 2–3 BALT per section, 3–5 sections per rat, n = 6 animals per group. For each fold change equal to or greater than ± 1.2 fold, P less than 0.05 compared with control using Student t test. CTL = control; Maz = Maz51 VEGFR3 inhibitor.
Figure 7.

Summary of in vivo data from experiments aimed at bronchus-associated lymphoid tissues (BALT) biology. LTBR = lymphotoxin β receptor; PA = pulmonary artery.
Passive Transfer of Autoantibodies Causes Pulmonary Vascular Remodeling and Hypertension
To confront the question of whether autoimmune-related pathologies directly contribute to development of PH, we turned to a “gain of function” strategy. We injected autoantibody-containing plasmas from MCT rats into naive rats. Compared with injection of preimmune plasmas or adjuvant (saline) alone (Figure 8A), or human IgG (Figure 8B), injection of either autoantibody-positive plasmas (Figure 8C) or purified IgG from MCT rats (Figure 8D) induced BALT, and bronchovascular and resistance vessel remodeling (Figures 8C and 8D, far left and middle images) that led to moderately severe PH with right and left ventricular hypertrophy and fibrosis (Figures 8C and 8D, far right images, blue collagen deposition). We again found high titers of plasma IgG in autoantibody-injected PH rats (Figure 8E, upper) that, when isolated, labeled lung vascular cells in culture and in tissue sections (Figure 8, lower).
Figure 8.
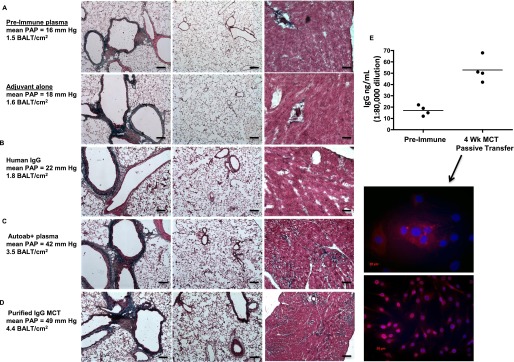
Pulmonary hypertension (PH) can be caused by autoantibody passive transfer from monocrotaline (MCT) rats into naive rats. (A) Naive rats injected with either preimmune sera, adjuvant (saline) alone, or adjuvant mixed with control human IgG (B) do not develop pulmonary vascular remodeling or PH. Lung (left and middle images) and heart (right images) histology is normal, as is pulmonary artery pressure (PAP) in these rats. (C) Lung remodeling and heart fibrosis with elevated PAP in rats injected with plasma (C) or purified IgG (D) from MCT rats. Note the more intense blue staining in heart sections from rats receiving autoantibody transfer (far right images, C and D). (E) High titers of IgG in rats 4 weeks after cell immunizations or passive antibody transfers have ceased. Note low titers in control rat groups. IgG from rats, 4 weeks after apoptotic lung cell immunizations or autoantibodies have ceased, label nuclei and perinuclear compartments of cultured pulmonary artery fibroblasts. Representative images and measurements taken from lung sections from n = 6 animals per group, with two to three sections per rat from three independent experiments. Cell cultures and ELISAs were performed in three separate experiments in triplicate with rat plasmas from the indicated rats per group, compared with n = 3 control media experiments. All images, original magnification: ×100; bars = 20 μm where applicable. BALT = bronchus-associated lymphoid tissues.
Discussion
BALT in PH displays unique cellular consistency, vascular architecture, and genomic programming permissive for production of autoantibodies. BALTs increase in size and number, are vascularized to facilitate T-cell binding, and are a source for local production of activating antifibroblast antibodies. Intracellular proteins vimentin and Hsp27 are potential autoantigens in rats with MCT-induced PH. Autoantibodies induce phenotypic change in pulmonary adventitial fibroblasts consistent with a proinflammatory phenotype. The distinct gene expression programs of BALT from control versus PH rats mirror other studies of activated lymphoid tissue capable of producing autoantibodies. We established that the signaling networks of CCR7, VEGFR3, LTBR, and the unfolded protein response all impact the biology of BALT, autoantibodies, and PH by distinct yet converging mechanisms. In so far as (1) finding plasma IgGs that bound lung vascular autoantigens, (2) isolating autoantibodies, (3) causing disease by introduction of either isolated autoantibodies or immunizations that induce them, and (4) reisolating autoantibodies, we conclude that autoantibodies contribute to the pathobiology of PH. Thus, lung vascular cells are important sources of autoantigens in PH, and BALTs may be principal sites of autoantibody production. This work provides mechanistic evidence that locally produced autoantibodies against lung vascular antigens can cause pulmonary vascular remodeling and PH.
PH remains a disease without a cure. The specific disease mechanisms remain elusive, despite the recent emphasis to describe the contributions of inflammation to vascular remodeling. Evidence of autoimmunity in some forms of PH has been established for more than 40 years. Our data address fundamental questions related to the natural history of autoimmune phenomena associated with PH.
In humans, ectopic lymphoid tissues form in response to local infection or sterile inflammation. Recirculating lymphocytes enter lymphoid tissues by high endothelial venules and leave by lymphatics. BALTs provide an environment to optimize encounters between DCs, macrophages, and other antigen-presenting cells with T and B cells. This facilitates anergy, maintenance of self-tolerance, and mobilization of appropriate adaptive immune responses. BALT develops after sustained signaling of IL-6 in the lung (25), in the absence of CCR7 (6), and with the requirement of IL-17 (26). Here, the BALT in MCT rats are bona fide lymphoid follicles and not lymphocytic aggregates by virtue of lymphocyte segregation, the presence of high endothelial venule, the presence of follicular DCs, gene expression repertoire, and antibody class switching. These characteristics were absent from lymphoid follicles in control rat lungs, which were also smaller and less numerous. We speculate that the duration and extent of lung injury are critical factors that influence BALT development and loss of tolerance. MCT is a broad pneumotoxin that causes lung cell apoptosis, airway cytokine flux, airway constriction, and decreased respiratory compliance. We recently found that MCT causes a robust activation of the unfolded protein response and CCAAT/enhancer-binding protein homologous protein (CHOP)-mediated apoptosis (23) in bronchial epithelium within 48 hours of MCT administration. Salubrinal-mediated unfolded protein response modulation reduced the number and extent of apoptotic cells. We speculate that the mechanism by which reduced apoptosis correlates with reduced BALT numbers and size is related to a decrease in self-antigen processing. In hypoxia, the pattern of apoptosis induced is cell type–specific in acute hypoxia (27) versus chronic hypoxia (15). Furthermore, the extent of apoptosis is much less obvious compared with MCT. Therefore, we speculate that autoimmunity occurs as a secondary event during the course of PH and perhaps in proportion to severity. Accordingly, PH may develop in the absence of autoantibody production, likely caused by variable exposure to self-antigen epitopes. Although beyond the scope of our efforts here, it would be interesting to examine autoimmunity in the vascular endothelial growth factor receptor-2 blockade (sugen 5,416) plus hypoxia model. For a severe PH model, we chose the MCT rat over the sugen-hypoxia rat model because the bronchovascular remodeling (the niche where BALT arises) seems to be more intense in the MCT model. Because the sugen-hypoxia model seems to be apoptosis-dependent, we anticipate that BALT will be more numerous and larger compared with control animals. The question of whether autoantibodies are generated in sugen-hypoxic rats needs to be investigated.
In PH rats, we found more BALT that was larger, more organized, and vascularized. This is likely a response to increased sampling load of self-antigens following lung injury and thus favors more interactions between DC and lymphocytes, particularly in the context of anergy. Our data implicate CCR7 as a critical chemokine required for DC egress from the lung parenchyma to lymphoid tissues, similar to its roles in gut inflammation (28). Although we found localized gene expression and cytokines supportive of lymphoid neogenesis, the mechanics of the process are not addressed here. Blockade of LTBR impacts several aspects of BALT neogenesis and maintenance (29). In our studies, BALT was required for autoantibody generation and PH induced by MCT. It could be argued that, because we intervened with LTBR-Ig at 14 days post-MCT, the disease process was not fully established. However, we found autoantibodies within the first week post-MCT and moderate PH is present at 2 weeks (∼35 mm Hg).
After DCs clear apoptotic lung cells, they must interact with immune cells using costimulatory molecules displayed on the reciprocal cell surfaces. PH in athymic rats is prevented by immune reconstitution with FoxP3+ Tregs (30). Our data synergize with those findings in that CCR7 blockade inhibits the ability of DCs to coordinate an appropriate Treg response. An alternative explanation is that subpopulations of Tregs exist (possibly even FoxP3 negative or low) that are CCR7+. If these cells are prevented from migrating properly to lymphoid tissues, they cannot engage in anergistic interactions with DC. Tregs and TH17 cells may be instrumental in modulating the activity of self-reactive B cells. B cells can be activated by both innate (Toll-like receptors) and acquired immune mechanisms, and can reciprocally influence T cells and DC. In this regard, it is interesting that we observed increased binding of T cells to BALT and vascular structures in the lung, which we could block by antibodies to PNAd (Figure 3). Thus, BALT in the setting of PH seems to have differential expression (Figure 2) of proteins that play critical roles in immune cell function. Along these lines, myeloid-derived suppressor cells may also play a role in the autoimmune pathogenesis of PH. We have observed increased numbers of circulating myeloid-derived suppressor cells in patients with PH (31), and similar phenotypic cells (DC-SIGN or CD11b+/MHCII−) localize to remodeled pulmonary vasculature in human (32) and animal models (15) of PH. Establishing the functionally cooperative roles of subsets of regulatory myeloid and lymphocytic cells in the development of PH and BALT is possible using the models and approaches we present here.
We induced pulmonary vascular remodeling, PH, and cardiac fibrosis in naive rats by injecting them with MCT-derived plasma autoantibodies. One of the most striking differences was the extent of the fibrosis in both ventricles of the heart of the autoantibody-recipient rats, which we attribute to epitope spreading. An alternative explanation is that rats 4 weeks post-MCT are undergoing multiorgan failure and produce autoantibodies accordingly. We found no obvious histopathology in organs other than lung and heart (spleen, kidney, liver, skeletal muscle, thyroid) harvested from rats that were given autoantibodies. However, given the clear evidence in Figure 4H that these organs contain autoantigenic targets, further investigation is warranted. Future studies will address the mechanism and extent of the autoantibody binding in lung, the kinetics of the putative pathology that ensues, and interepitope autoimmune spreading. It will be important to determine whether the autoantibody production remains a local phenomenon (BALT), or is a systemic process (lymph nodes). In addition, future studies will test whether vascular cell senescence caused by autoantibody binding contributes to the phenotypic remodeling in the pulmonary vasculature.
Autoimmune phenomena in PH have been described for 40 years. This work builds on the observations of dozens of laboratories over that time. Recent findings in inflammation and autoimmune biology in PH are reshaping current research perspectives in the field. Here we report that autoantibodies in PH can be pathologic, and strategies aimed at their diminution can be therapeutically efficacious.
Acknowledgments
Acknowledgment
The authors thank Dr. Jeff Browning at Biogen Idec for kindly providing LTBR-Ig and MOPC-21 reagents, technical support, and scientific advisement. They thank Julie Harral for her expertise in rat hemodynamic measurements. They also thank Nicole Spoelstra at the University of Colorado Denver Laser Capture Microdissection Core.
Footnotes
Supported by a Children's Hospital Research Institute Research Scholar Award (M.E.Y.) and National Institutes of Health Grants HL-14985 and HL-81506 (K.R.S.).
Author Contributions: M.E.Y. conceived of the study, conducted experiments, interpreted data, and wrote the paper. K.L.C. and P.J.C. conducted experiments, interpreted data, and edited the manuscript. D.D.I. and K.R.S. edited the manuscript.
Originally Published in Press as DOI: 10.1164/rccm.201302-0403OC on October 4, 2013
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, Elliott CG, Gaine SP, Gladwin MT, Jing ZC, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S43–S54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 2.Tuder RM, Abman SH, Braun T, Capron F, Stevens T, Thistlethwaite PA, Haworth SG. Development and pathology of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S3–S9. doi: 10.1016/j.jacc.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 3.Hassoun PM, Mouthon L, Barberà JA, Eddahibi S, Flores SC, Grimminger F, Jones PL, Maitland ML, Michelakis ED, Morrell NW, et al. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol. 2009;54:S10–19. doi: 10.1016/j.jacc.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 4.Randall TD. Bronchus-associated lymphoid tissue (BALT) structure and function. Adv Immunol. 2010;107:187–241. doi: 10.1016/B978-0-12-381300-8.00007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foo SY, Phipps S. Regulation of inducible BALT formation and contribution to immunity and pathology. Mucosal Immunol. 2010;3:537–544. doi: 10.1038/mi.2010.52. [DOI] [PubMed] [Google Scholar]
- 6.Larsen KO, Yndestad A, Sjaastad I, Løberg EM, Goverud IL, Halvorsen B, Jia J, Andreassen AK, Husberg C, Jonasson S, et al. Lack of CCR7 induces pulmonary hypertension involving perivascular leukocyte infiltration and inflammation. Am J Physiol Lung Cell Mol Physiol. 2011;301:L50–L59. doi: 10.1152/ajplung.00048.2010. [DOI] [PubMed] [Google Scholar]
- 7.Perros F, Dorfmüller P, Montani D, Hammad H, Waelput W, Girerd B, Raymond N, Mercier O, Mussot S, Cohen-Kaminsky S, et al. Pulmonary lymphoid neogenesis in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;185:311–321. doi: 10.1164/rccm.201105-0927OC. [DOI] [PubMed] [Google Scholar]
- 8.Badesch DB, Wynne KM, Bonvallet S, Voelkel NF, Ridgway C, Groves BM. Hypothyroidism and primary pulmonary hypertension: an autoimmune pathogenetic link? Ann Intern Med. 1993;119:44–46. doi: 10.7326/0003-4819-119-1-199307010-00008. [DOI] [PubMed] [Google Scholar]
- 9.Nicolls MR, Taraseviciene-Stewart L, Rai PR, Badesch DB, Voelkel NF. Autoimmunity and pulmonary hypertension: a perspective. Eur Respir J. 2005;26:1110–1118. doi: 10.1183/09031936.05.00045705. [DOI] [PubMed] [Google Scholar]
- 10.Tamby MC, Chanseaud Y, Humbert M, Fermanian J, Guilpain P, Garcia-de-la-Peña-Lefebvre P, Brunet S, Servettaz A, Weill B, Simonneau G, et al. Anti-endothelial cell antibodies in idiopathic and systemic sclerosis associated pulmonary arterial hypertension. Thorax. 2005;60:765–772. doi: 10.1136/thx.2004.029082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Terrier B, Tamby MC, Camoin L, Guilpain P, Broussard C, Bussone G, Yaïci A, Hotellier F, Simonneau G, Guillevin L, et al. Identification of target antigens of antifibroblast antibodies in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;177:1128–1134. doi: 10.1164/rccm.200707-1015OC. [DOI] [PubMed] [Google Scholar]
- 12.Karmochkine M, Cacoub P, Dorent R, Laroche P, Nataf P, Piette JC, Boffa MC, Gandjbakhch I. High prevalence of antiphospholipid antibodies in precapillary pulmonary hypertension. J Rheumatol. 1996;23:286–290. [PubMed] [Google Scholar]
- 13.Kherbeck N, Tamby MC, Bussone G, Dib H, Perros F, Humbert M, Mouthon L. The role of inflammation and autoimmunity in the pathophysiology of pulmonary arterial hypertension. Clin Rev Allergy Immunol. 2013;44:31–38. doi: 10.1007/s12016-011-8265-z. [DOI] [PubMed] [Google Scholar]
- 14.Bussone G, Tamby MC, Calzas C, Kherbeck N, Sahbatou Y, Sanson C, Ghazal K, Dib H, Weksler BB, Broussard C, et al. IgG from patients with pulmonary arterial hypertension and/or systemic sclerosis binds to vascular smooth muscle cells and induces cell contraction. Annals Rheum Dis 2012;71:596–605. [DOI] [PubMed]
- 15.Stenmark KR, Meyrick B, Galiè N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1013–1032. doi: 10.1152/ajplung.00217.2009. [DOI] [PubMed] [Google Scholar]
- 16.Bánfi A, Tiszlavicz L, Székely E, Peták F, Tóth-Szüki V, Baráti L, Bari F, Novák Z. Development of bronchus-associated lymphoid tissue hyperplasia following lipopolysaccharide-induced lung inflammation in rats. Exp Lung Res. 2009;35:186–197. doi: 10.1080/01902140802495862. [DOI] [PubMed] [Google Scholar]
- 17.Kawamata N, Xu B, Nishijima H, Aoyama K, Kusumoto M, Takeuchi T, Tei C, Michie SA, Matsuyama T. Expression of endothelia and lymphocyte adhesion molecules in bronchus-associated lymphoid tissue (BALT) in adult human lung. Respir Res. 2009;10:97. doi: 10.1186/1465-9921-10-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ohtani O, Ohtani Y. Structure and function of rat lymph nodes. Arch Histol Cytol. 2008;71:69–76. doi: 10.1679/aohc.71.69. [DOI] [PubMed] [Google Scholar]
- 19.Rahman ZS. Impaired clearance of apoptotic cells in germinal centers: implications for loss of B cell tolerance and induction of autoimmunity. Immunol Res. 2011;51:125–133. doi: 10.1007/s12026-011-8248-4. [DOI] [PubMed] [Google Scholar]
- 20.Lv H, Havari E, Pinto S, Gottumukkala RV, Cornivelli L, Raddassi K, Matsui T, Rosenzweig A, Bronson RT, Smith R, et al. Impaired thymic tolerance to α-myosin directs autoimmunity to the heart in mice and humans. J Clin Invest. 2011;121:1561–1573. doi: 10.1172/JCI44583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hintermann E, Holdener M, Bayer M, Loges S, Pfeilschifter JM, Granier C, Manns MP, Christen U. Epitope spreading of the anti-CYP2D6 antibody response in patients with autoimmune hepatitis and in the CYP2D6 mouse model. J Autoimmun. 2011;37:242–253. doi: 10.1016/j.jaut.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 22.Fineschi S, Goffin L, Rezzonico R, Cozzi F, Dayer JM, Meroni PL, Chizzolini C.Antifibroblast antibodies in systemic sclerosis induce fibroblasts to produce profibrotic chemokines, with partial exploitation of toll-like receptor 4. Arth Rheum 2008;58:3913–3923. [DOI] [PubMed]
- 23.Yeager ME, Belchenko DD, Nguyen CM, Colvin KL, Ivy DD, Stenmark KR. Endothelin-1, the unfolded protein response, and persistent inflammation: role of pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol. 2012;46:14–22. doi: 10.1165/rcmb.2010-0506OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Staszewski O, Baker RE, Ucher AJ, Martier R, Stavnezer J, Guikema JE. Activation-induced cytidine deaminase induces reproducible DNA breaks at many non-Ig loci in activated B cells. Mol Cell. 2011;41:232–242. doi: 10.1016/j.molcel.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goya S, Matsuoka H, Mori M, Morishita H, Kida H, Kobashi Y, Kato T, Taguchi Y, Osaki T, Tachibana I, et al. Sustained interleukin-6 signalling leads to the development of lymphoid organ-like structures in the lung. J Pathol. 2003;200:82–87. doi: 10.1002/path.1321. [DOI] [PubMed] [Google Scholar]
- 26.Rangel-Moreno J, Carragher DM, de la Luz Garcia-Hernandez M, Hwang JY, Kusser K, Hartson L, Kolls JK, Khader SA, Randall TD.The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat Immunol2011;12:639–646. [DOI] [PMC free article] [PubMed]
- 27.Z'graggen BR, Tornic J, Müller-Edenborn B, Reyes L, Booy C, Beck Schimmer B.Acute lung injury: apoptosis in effector and target cells of the upper and lower airway compartment. Clin Exper Immunol 2010;161:324–331. [DOI] [PMC free article] [PubMed]
- 28.McNamee EN, Masterson JC, Jedlicka P, Collins CB, Williams IR, Rivera-Nieves J. Ectopic lymphoid tissue alters the chemokine gradient, increases lymphocyte retention and exacerbates murine ileitis. Gut. 2013;62:53–62. doi: 10.1136/gutjnl-2011-301272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Browning JL. Inhibition of the lymphotoxin pathway as a therapy for autoimmune disease. Immunol Rev. 2008;223:202–220. doi: 10.1111/j.1600-065X.2008.00633.x. [DOI] [PubMed] [Google Scholar]
- 30.Tamosiuniene R, Tian W, Dhillon G, Wang L, Sung YK, Gera L, Patterson AJ, Agrawal R, Rabinovitch M, Ambler K, et al. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ Res. 2011;109:867–879. doi: 10.1161/CIRCRESAHA.110.236927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yeager ME, Nguyen CM, Belchenko DD, Colvin KL, Takatsuki S, Ivy DD, Stenmark KR.Circulating myeloid-derived suppressor cells are increased and activated in pulmonary hypertension. Chest 2012;141:944–952. [DOI] [PMC free article] [PubMed]
- 32.Perros F, Dorfmüller P, Souza R, Durand-Gasselin I, Mussot S, Mazmanian M, Hervé P, Emilie D, Simonneau G, Humbert M. Dendritic cell recruitment in lesions of human and experimental pulmonary hypertension. Eur Respir J. 2007;29:462–468. doi: 10.1183/09031936.00094706. [DOI] [PubMed] [Google Scholar]