

Abstract
Design of inhibitors for N-myristoyltransferase (NMT), an enzyme responsible for protein trafficking in P. falciparum, the most lethal species of parasites that cause malaria, is described. Chemistry-driven optimization of compound 1 from a focused NMT inhibitor library led to the identification of two early lead compounds 4 and 25, which showed good enzyme and cellular potency and excellent selectivity over human NMT. These molecules provide a valuable starting point for further development.
Keywords: Anti-Malarial Target, Inhibitor Design, Crystal Structure, Selectivity, Mutagenesis
INTRODUCTION
Malaria, a disease caused by protozoan parasites of the genus Plasmodium, is a demanding health problem. The most lethal form, caused by P. falciparum, was responsible for nearly 655,000 deaths in 2010, mainly of children below the age of five.1 Although recent vaccine trials are giving promising results,2 chemotherapy remains the mainstay of anti-malarial treatment. Since chloroquine has lost its efficacy in most endemic areas due to the rapid development of drug resistance,3 artemisinin-based combination therapies serve as the current gold standard.4 Recently, signs of emerging resistance to artemisinins5 have led to renewed efforts to develop novel anti-malarial agents, with purely synthetic tri/tetraoxane and spiroindolone among the most promising candidates.6-10
The enzyme N-myristoyltransferase (NMT) represents a promising drug target,11 since it has been shown to be essential in many organisms.12-15 NMT, a monomeric enzyme ubiquitous in eukaryotes, catalyzes an irreversible co-translational or post-translational transfer of myristate (C14:0 fatty acid) from myristoyl-coenzyme A (myr-CoA) to the N-terminal glycine of a subset of eukaryotic proteins.16, 17 Although genetic knockdown of NMT in P. falciparum has yet to be achieved presumably due to its essentiality, myristoylated proteins such as glideosme-associated protein-45, ADP-ribosylation factor-1 and calcium-dependent protein kinase-1 have been characterized and shown to be critical for parasite viability.18-21 Furthermore, a novel inducible knockout of NMT in the closely related mammalian pathogen P. berghei has demonstrated an acute dependence on NMT activity in the parasite life cycle.22 It seems likely that inhibition of myristoylation also has an equal potential to disrupt key biological pathways in P. falciparum.
N-myristoylation by NMT follows an ordered Bi-Bi mechanism, in which the binding of myr-CoA to NMT occurs prior to the protein substrate.23 Binding of myr-CoA facilitates the opening of a second pocket, to which the protein substrate binds. Transfer of myristate to the protein substrate takes place via a nucleophilic addition-elimination reaction and this is followed by ordered release of CoA and the myristoylated protein. In contrast to the myr-CoA pocket which is highly conserved across species, the peptide binding cavity is more divergent. As a result, inhibitors of fungal NMTs have been discovered by several research groups that are competitive with the protein substrate and show excellent selectivity relative to human NMT.24-29
A “piggy-back” approach30 was used to identify compound 1 (Figure 1) as a hit by screening a focused library of reported C. albicans NMT (CaNMT)24-29 and T. brucei NMT (TbNMT) inhibitors.31, 32 Compound 1, initially developed by Roche in an antifungal campaign,29 showed moderate inhibition against PfNMT and promising selectivity over human NMT1 (HsNMT1).
Figure 1.
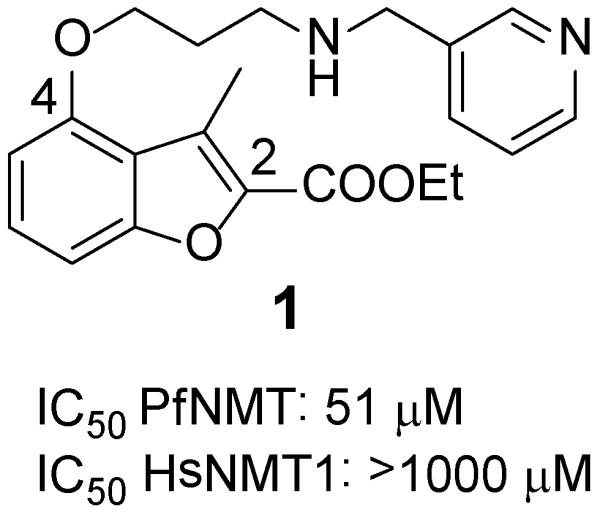
Structure and biological activity of compound 1
Herein we report the design of potent and selective P. falciparum NMT (PfNMT) inhibitors based on the structure of compound 1. Crystal structures of NMT inhibitors enabled experimentally derived structure-activity relationships (SAR) to be interpreted and mutagenesis studies provided a rational basis for human enzyme selectivity.
RESULTS AND DISCUSSION
Investigation of the C-4 side chain
In the absence of structural information for Plasmodium NMT at that time, a chemistry-driven approach was adopted. Based on the structure of compound 1, C-4 and C-2 side chains were identified as promising sites for modification (Figure 2). Exploration of 24 variations in the C-4 side chain revealed SAR, which are similar to those Roche found in their CaNMT project:24 (i) trimethylene is an optimal chain length; (ii) a secondary amine X is preferred (Tables S1-S3, Supporting Information). Conformational restriction of the trimethylene side chain by replacement with 4-piperidinol, resulted in the discovery of compound 2 (Figure 2). 2 showed 3-fold activity improvement over 1 while maintaining good human selectivity. In addition, 2 is more drug-like than 1 in terms of lower molecular weight, fewer rotatable bonds and is also more synthetically tractable. Therefore, 4-piperidinol was considered to be an optimal C-4 side chain. More importantly, this compound also displayed moderate anti-parasitic activity with an EC50 value of 15 μM.
Figure 2.

Modification of C-4 side chain of compound 1
Investigation of the C-2 side chain
The study of the C-2 side chain began by varying the R2 group of the ester and replacement of the latter by an amide (Scheme 1). An N-Boc piperidinol side chain was incorporated into the readily synthesized benzofuran scaffold33 via a Mitsunobu reaction, followed by hydrolysis of the ethyl ester to form the key intermediate I-2. The ester and amide with different R2 groups were synthesized under standard coupling conditions. Larger R2 groups, especially those including an aromatic ring (4, 5 vs. 2, 6 and 12, 13 vs. 9, 10), give rise to better enzyme inhibition in both series. Compound 4 represents a significant improvement in potency and has a sub-μM IC50 value of 0.27 μM. Introduction of a methylene moiety between R2 and the ester group was a key factor in this enhanced potency (4 vs. 3), suggesting the importance of flexibility in the linker group Y. Furthermore, spacers longer than methylene significantly reduced the affinity (7, 8 vs. 4), indicating that this spacing is optimal. Surprisingly, a huge loss of activity was found when the Y linker was altered from a methylene ester to a methylene amide (4 vs. 12); however, the deleterious effect of the ester to amide swap is largely mitigated when the phenyl R2 group is replaced by a napthyl group (5 vs. 13), prompting further investigation of the linker group Y to understand its role in binding.
Scheme 1.

a.
a Reagents: (a) DIAD, PPh3, N-Boc-4-OH piperidine, THF, r.t., 4 hr, 95%; (b) 5% TFA in DCM, r.t., 2 hr, quantitative; (c) LiOH, MeOH/H2O, 50 °C, 4 hr, 90%; (d) EDCI, HOBt, DIPEA, R2OH, CH3CN, r.t., 4 hr, 70-90%; (e) PyBOP, DIPEA, R2NH2, DMF/DCM, r.t., 3 hr, 50-70%.
Investigation of linker group Y
Compounds containing rigid Y linkers with similar length (14-17) were initially synthesized (Scheme 2). These inhibitors (3, 11, 14 to 17, Table 2) were found to be only moderately active compared to their methylene ester analogues, such as 4, reinforcing the earlier conclusion that the flexibility of the Y linker group is crucial. Compounds with various types of flexible Y linkers (18-25, Table 2) were then prepared (Scheme 3). It is clear that a flexible linker alone, such as ethylene 18, does not confer inhibitory activity. Introduction of a heteroatom in the ethylene linker improved enzyme affinity (19, 20 vs. 18). Compounds 22-25 were designed to mimic the original methylene ester, with a view to finding a more suitable replacement (Table 2). Both the carbonyl and the ester oxygen atoms in the methylene ester were found to contribute to potency since replacement of either atom by methylene led to a 40-fold activity drop (21, 22 vs. 4). Removing a rotatable bond of the ethylene gave a 3-fold improvement in affinity (23 vs. 22), which appears to contradict the earlier interpretation that flexibility in the Y linker is important. It is possible that this particular rigid linker directs the pendant phenyl group into a specific hydrophobic cavity. The dramatic loss of activity in 24 is unexpected and cannot be explained by the shorter length of the linker Y compared to the methylene ester because phenyl ketone 14, which has an even shorter linker, retains some inhibitory activity with an IC50 of 22 μM. Therefore, it is likely that the methylene sulfone is incompatible with the binding site of the enzyme. 1,2,4-oxadiazole 25, an ester bioisostere, exhibited promising potency and selectivity although it was still less active than its ester counterpart. Given that none of the other Y linkers synthesized showed comparable inhibition, the methylene ester was selected as an optimal linker group (Figure 3). Next, a standard set of substituents (methyl, chloro and methoxy) were employed to find suitable substituents on the pendant phenyl ring in the methylene ester series. However, of the seven synthesized compounds, only the meta-methoxy substituent in 26 gave comparable potency to the parent compound 4 (Table S4, Supporting Information), indicating a tight R2 group binding pocket in the enzyme.
Scheme 2.

a
aReagents: (a) PhCOCH2Br, K2CO3, CH3CN, 80°C, 24 hr, 40%; (b) DIAD, PPh3, N-Boc-4-OH piperidine, THF, r.t., 4 hr, 90%; (c) 5% TFA in DCM, r.t., quantitative; (d) LiAlH4, THF, 0 °C to r.t., 2 hr, 90%; (e) MnO2, DCM, r.t., 24 hr, 77%; (f) benzyltriphenyl phosphonium bromide, NaOH, THF/H2O, r.t, 1 hr, 70% (including cis- and trans-isomers); (g) Pd/C, 1,4-cyclohexadiene, EtOH, 80 °C, 4 hr, 40%; (f) i. R2(NH2)C=N-OH, EDCI, HOBt, DIPEA, CH3CN, r.t., 4 hr; ii. 4Å molecular sieve, dry toluene, 110 °C, 12 hr, 70% over two steps.
Table 2.
Activities of inhibitors containing different Y linkers against PfNMT and P. falciparuma
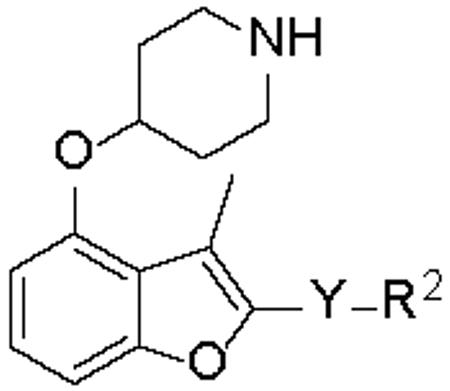
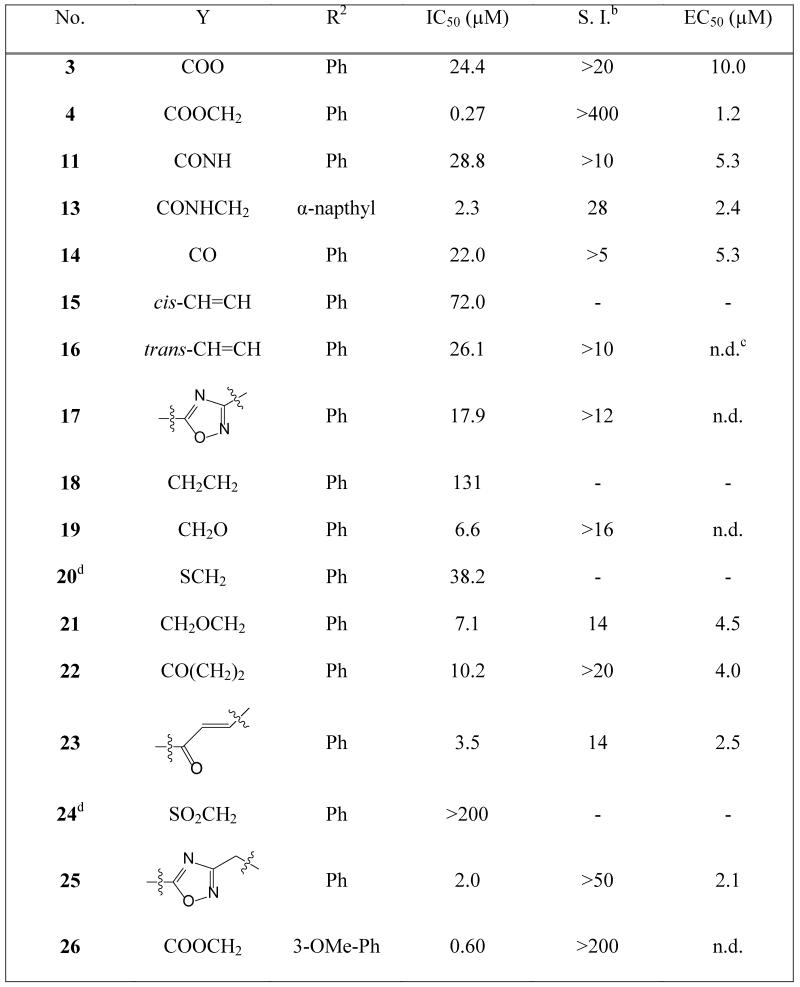
|
The IC50(PfNMT) and EC50(P. falciparum, 3D7 line) values were averaged from two independent dose-response curves, the standard deviation was within 30% of the resulting values;
S.I. = Selectivity Index, calculated as IC50(HsNMT1) / IC50(PfNMT);
n.d. = not determined;
there is no methyl group at the C-3 position of the benzofuran scaffold.
Scheme 3.

a
a Reagents: (a) DIAD, PPh3, N-Tr-4-OH piperidine, THF, r.t., 4 hr, 65%; (b) LiAlH4, THF, 0 °C to r.t., 2 hr, 90%; (c) DIAD, PPh3, phenol, THF, r.t., 4 hr, 5%; (d) 0.1% TFA, 0.2% H2O in DCM, r.t., 90%; (e) NaH, BnBr, DMF, r.t., 15%; (f) CH3COCH2Cl, K2CO3, CH3CN, 80°C, 24 hr, 20%; (g) DIAD, PPh3, N-Boc-4-OH piperidine, THF, r.t., 4 hr, 90%; (h) benzaldehyde, NaOH, EtOH/H2O, 3 hr, 45%; (i) 5% TFA in DCM, r.t., quantitative; (j) Pd/C, 1,4-cyclohexadiene, EtOH, 80 °C, 4 hr, 40%; (k) ethyl carbazate, EtOH/H2O, 80°C, 24 hr, 24%; (l) SOCl2, DMF, −20°C to r.t., 3 hr, 54%; (m) BnCl, K2CO3, dry acetone, 60°C, 6 hr, 24%; (n) m-CPBA, DCM, r.t., 1 hr, 50%.
Figure 3.

Investigation of C-2 side chain
These compounds in Table 2 were also applied to synchronized P. falciparum trophozoites (3D7 line). Consistent with the premise that NMT is a good target in Plasmodium, it was found that these inhibitors also showed promising anti-plasmodial effects in vitro, with the most potent PfNMT inhibitor 4 giving the highest anti-parasitic activity. These results imply that optimized PfNMT inhibitors have excellent potential to be developed into anti-malarial therapeutics.
Structure Studies
Although attempts to crystallize PfNMT have been unsuccessful, we have been able to determine co-crystal structures of P. vivax NMT (PvNMT) with a non-hydrolysable myr-CoA analogue (NHM)34 and a series of benzofuran inhibitors. PvNMT shares 81% sequence identity with PfNMT, with only 2 out of 23 residues situated within 5Å of the ligand differing between the two enzymes (Y212 and Y334 in PvNMT are each replaced by Phe in PfNMT, Figure S1, Supporting Information). Selected compounds from Table 2 were assayed against PvNMT and similar levels of potency (<3-fold difference) to PfNMT were observed. Therefore, we consider that structures of PvNMT complexes can be used to rationalize the experimental SAR for the PfNMT inhibitors.
Inhibitor 26 occupies what is expected to be the peptide binding pocket of PvNMT (Figure S2, Supporting information). Its key interactions with the enzyme are illustrated in Figure 4. The secondary amino group of the piperidine establishes an ion-pair interaction with the C-terminal carboxylate (L410) and a cation-dipole interaction with the phenolic hydroxyl of Y107. The ion-pair mimics the interaction of the α-amino groups of substrate peptides with the buried α-carboxylate of NMT (as observed in structures of S. cerevisiae NMT, ScNMT);35 moreover, similar interactions are formed by inhibitors of CaNMT29 and TbNMT.31 The carbonyl oxygen together with the oxygen atom of the benzofuran ring in 26 participate in water-mediated hydrogen bonds with the hydroxyl of Y334, which may account for the superiority of the methylene ester over its less polar alkyl or ether/thioether equivalents (18-21 vs. 4). The methylene spacer in 26 allows the pendant aromatic group to be directed into a tight pocket, in agreement with the experimental SAR, where the phenyl group makes π-π interactions with F105 and forms further apolar interactions with residues V96, E97, D98, F103 and R104 that contribute to a loop that closes over the peptide binding pocket. Shorter (3 vs. 4) or longer (7, 8 vs. 4) spacers than the methylene ester result in 10- to 100-fold lower inhibition, presumably because they prevent or compromise the binding of the attached phenyl group to this pocket. Meanwhile the meta-methoxy oxygen in 26 forms a hydrogen bond with the hydroxyl of S319.
Figure 4.

Crystal structure of 26 (1.50 Å resolution, PDB code: 4B14) co-crystallized with P. vivax NMT. Distances are given in Å. Atoms are colored: C yellow (enzyme) and green (inhibitor 26), N blue, O red, H2O red sphere.
Inhibitors 22, 25 and 26 occupy almost identical binding positions in the enzyme (Figure 5a). Interestingly, binding of these ligands rigidify the side chain of H213, which shows two distinct conformations in the unligated structure, but adopts a single conformation that forms water-mediated hydrogen bonds with Y linkers in these compounds (Figure 5a). This indicates an important role for H213 in binding, which is also documented in the binding of a peptide substrate in ScNMT.36 As seen from the experimental SAR, an amide Y linker disfavored. The reason for this seemingly surprising result is clarified by examination of the bound structures of these two compounds. It is found that the carbonyl group in 13 adopts the opposite orientation to that of its ester equivalent in 26 (Figure 5b). We believe that this reflects different conformations in the free ligands, presumably due to a steric clash between methyl in the benzofuran scaffold and the hydrogen atom in the amide (Figure 6). Furthermore, the side chain of H213 is observed to move away from the ligand upon the binding of 13 (Figure 5b). This amide carbonyl is hence too far away from either Y334 or H213 to form an interaction and the altered conformation of the spacer might affect the binding geometry of the pendant phenyl ring, which accounts for the large activity difference between amide 12 and ester 4. However, this difference is largely mitigated when the phenyl R2 group is replaced by a napthyl group (13 vs. 4), which makes more extensive π-π interactions with F105.
Figure 5.

Structural overlays of benzofuran NMT inhibitors: (a) 22 (1.84 Å resolution, PDB code: 4B12, C purple), 25 (1.58 Å resolution, PDB code: 4B13, C cyan) and 26 (PDB code: 4B14, C green) share identical binding positions and rigidify the movement of H213 (binary complex, 1.56 Å resolution, PDB code: 4B10, C grey) to a single conformation (ternary complexes, C yellow); (b) opposite orientation adopted by the carbonyl moiety in 13 (1.59 Å resolution, PDB code: 4B11, C magenta) compared to the one in 26 (PDB code: 4B14, C green) push H213 away from the ligand. Atoms are colored: N blue, O red, H2O red sphere.
Figure 6.
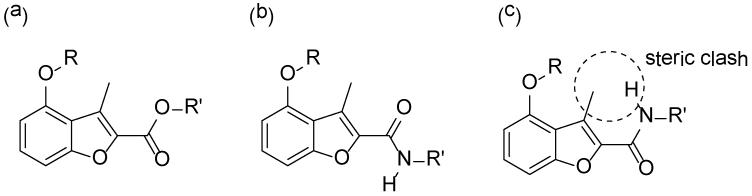
Geometry comparison of ester versus amide inhibitors: (a) Representation of the binding mode of the ester-containing inhibitor 26; (b) Representation of the binding mode of the amide-containing inhibitor 13; (c) Alternative conformation of inhibitor 13 does not adopt the conformation found in (a), due to a steric clash between the amide NH and the methyl group.
Selectivity Studies
High inhibitor selectivity between Plasmodial and human NMTs is desirable because NMT is also expressed in human cells. Indeed, our benzofuran inhibitors displayed good to excellent selectivity over HsNMT1 (Table 2). This is surprising in the light of a recent high throughput screening campaign which suggests that strongly selective PfNMT inhibitors would not be easy to identify.37 The structures were therefore examined with a view to rationalizing the selectivity of the benzofuran compounds.
We compared the structures of PvNMT in its binary complex with NHM and in the ternary complex with NHM and 26. The side chain of Y211 adopts an alternate conformation upon binding 26 so that there are stacking interactions with the benzofuran ring (Figure 7a). Following superimposition of the structures of PvNMT-NHM-26 and HsNMT1-Myr-CoA, it is apparent that the equivalent tyrosine (Y296) in the human enzyme occupies a position that would clash with 26 (Figure 7b). Given the weak inhibition of HsNMT1 by 26, we speculated that a higher energy barrier associated with the conformational change of Y296 in HsNMT1 might be restricting binding. In contrast to 26, 27 (MRT57965),38 a PvNMT inhibitor showing little human selectivity, is accommodated in the Plasmodial enzyme with only minor changes in the conformation of Y211 (Figure 7c). It is predicted that similarly small conformational changes of Y296 would be required to accommodate this inhibitor in HsNMT1 (Figure 7d). We therefore hypothesized that steric hindrance by Y296 in HsNMT1 contributes to human selectivity for these benzofuran inhibitors.
Figure 7.

Investigation of selectivity over HsNMT1: (a) conformational change of Y211 in PvNMT-NHM (PDB code: 4B10) upon the binding of 26 (PDB code: 4B14); (b) HsNMT1-Myr-CoA (PDB code: 3IU1) overlaid with PvNMT-NHM-26 complex (PDB code: 4B14). This indicates a clash between Y296 in HsNMT1 and 26; (c) conformational change of Y211 in PvNMT-NHM (PDB code: 4B10) upon binding 27(PDB code: 4A95); (d) overlay of HsNMT1-Myr-CoA (PDB code: 3IU1) with PvNMT-NHM-27 (PDB code: 4A95). This suggests minimal conformational changes of that particular tyrosine when binding this inhibitor. Atoms are colored: C yellow (enzymes in the ternary complex), C grey (enzymes in the binary complex), C green (inhibitors), N blue, O red, S gold.
To test this hypothesis, site-directed mutagenesis was used to substitute this tyrosine by alanine in both PvNMT and HsNMT1. The inhibitory activities of 25, 26 and 27 against the wild type and mutated NMTs were measured (Table 3) and used to calculate the ratio of the IC50 values for HsNMT1 and PvNMT (the selectivity index, SI) for the pair of wild type enzymes and for the pair of alanine-substituted mutants. The alanine mutations are found to have significantly less selectivity for 26 and 25 than the wild-type enzymes, with decreases in the SI of 20-fold and >12-fold respectively. In contrast, the change in the SI for 27 was only two-fold. Mutation of the human NMT leads to a four- to five-fold increase in affinity for compounds 25 and 26. This is consistent with the suggestion that changing tyrosine to the smaller alanine in HsNMT1 removes the potential clash and lowers the energy barrier required to accommodate these two inhibitors. On the other hand, under the conditions that only small conformational change is required to accommodate ligand binding (25, 26 and 27 in PvNMT and 27 in HsNMT1), such mutation exclusively led to weaker enzyme affinity, implying that this tyrosine facilitates inhibitor binding to NMTs unless the residue clashes with the bound ligand. The data experimentally demonstrate the contribution of this tyrosine (Y211 in PvNMT and Y296 in HsNMT1) to the human selectivity.
Table 3.
The inhibitory activity for mutants of PvNMT and HsNMT1a
| Inhibitor | IC50 (μM) | S.I.b | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Pv (WTc) | Pv (Y211A) |
Hs1 (WT) | Hs1 (Y296A) |
WT | MUd | |
| 26 | 0.6 | 2.7 | 45 | 10 | 75 | 3.7 |
| 25 | 6.2 | 16.6 | >500 | 110 | >80 | 6.6 |
| 27 e | 32.7 | 150 | 115 | 218 | 3.5 | 1.5 |
The IC50 values were averaged from two independent dose-response curves; the standard deviation was within 30% of the resulting values;
S. I.=Selectivity Index, calculated by IC50(HsNMT1) / IC50(PvNMT);
WT = wild type;
MU = mutants;
Wild-type enzyme result for 27 was different from that reported by Goncalves38 due to different assay conditions, including the concentrations of enzyme, myr-CoA and different peptide substrate.
CONCLUSION AND OUTLOOK
Chemistry-driven optimization of a screening hit allowed us to identify PfNMT inhibitor 4 as an early lead candidate with good enzyme and cellular potency and excellent selectivity over human NMT. With the aid of structural information, the superiority of a benzyl ester side chain was rationalized and the interactions involved with the linker were mapped. Furthermore, the structural basis for inhibitory selectivity between HsNMT1 and PfNMT was found to be due to a clash between the inhibitor and residue Y296 in HsNMT1.
Potential biological instability of the ester moiety may limit the scope of inhibitor 4. Oxadiazole 25, an ester bioisostere, showed some advantages given its biological stability and comparable cellular potency and human selectivity to 4; synthesis of oxadiazole analogues shall be the focus of further studies. As suggested by Figure 4, two hotspot residues (F105 and S319 in PvNMT, which are also conserved in PfNMT), are identified to involve a π-π interaction and hydrogen bond with aromatic R2 respectively. However, as the unsubstituted phenyl ring is still the best R2 among PfNMT inhibitors so far, it is unlikely that the hydrogen bond with S319 has been formed in PfNMT. Forming this missing hydrogen bond without affecting human selectivity, possibly by using N-containing heterocycles, has potential to further improve the enzyme potency. Finally, understanding the influence of physiochemical properties (such as pKa and cLogP) on the cellular potency is also important.
EXPERIMENTAL SECTION
CHEMISTRY
General Methods
All solvents and reagents were purchased from commercial sources and used without further purification. The compounds were spotted on silica TLC plates (Merck, Si60, F254), visualized under UV-light at 254 nm or iodine over silica. Purification of the compounds for biological tests was performed on a Gilson semi-preparative reverse phase HPLC system (Anachem Ltd., Luton, UK) equipped with a HICHROM C18 Column (250 × 21.2 mm), #306 pumps and a Gilson 155 UV/Vis detector. UV detection was at 220 nm. The mobile phase consisted of water plus 0.1% TFA (solvent A) and methanol plus 0.1% TFA (solvent B) with a gradient of 30% B for 2 min changing to 98% B over 30 min, maintaining for 2 min, and then down to 2% B over 0.5 min at a flow rate of 12 mL/min. The purities of the reported compounds for biological assay tests were evaluated by analytical LC-MS which was carried out on a Waters 2767 system equipped with a photodiode array and a mass spectrometer using an X-Bridge™ C18 column (5 μM, 4.6 × 100 mM). The mobile phase consisted of water plus 0.1% formic acid (solvent A) and methanol plus 0.1 formic acid (solvent B) with a gradient starting from 2% to 98% B over the first 10 min, maintaining for 2 min, and down to 2% B over 1 min followed by the maintenance period for another 4 min at 2% B at a flow rate of 1.2 mL/min. One-dimensional 1H- and 13C-NMR spectra as well as two dimensional NMR spectra were recorded on Bruker AV at 400, 500 MHz or 100, 125 MHz respectively. Chemical shifts are reported in ppm. Mass spectra were obtained from the Mass Spectrometry Service of Department of Chemistry, Imperial College London. Purity (>95%) and molecular mass of compounds for biological tests were confirmed by LC-MS and high resolution of mass spectrometry.
General Procedure of Mitsunobu Reaction
t-Butyl 4-{[2-(ethoxycarbonyl)-3-methyl-1-benzofuran-4-yl]oxy} piperidine-1-carboxylate (I-1)
To a stirred solution of ethyl 4-hydroxy-3-methylbenzofuran-2-carboxylate33 (2 g, 9.1 mmol), t-Butyl 4-hydrxypiperidine-1-carboxylate (4.56 g, 22.7 mmol) and triphenylphosphine (5.95 g, 22.7 mmol) in anhydrous THF (20 mL) was added DIAD (4.34 mL, 22.7 mmol) dropwise at room temperature. The resulting mixture was stirred at the same temperature for 4 hours, and then concentrated in vacuo. The residue was purified by column chromatography over silica gel, eluting with hexane: ethyl acetate, 5:1 to afford the title compound as colourless oil (3.52 g, 96% yield). 1H-NMR (CDCl3, 400 MHz): δ 7.32 (t, J=8.0 Hz, 1H), 7.13 (d, J=8.0 Hz, 1H), 6.65 (d, J=8.0 Hz, 1H), 4.74-4.69 (m, 1H), 4.46 (q, J=7.2 Hz, 2H), 3.68-3.61 (m, 2H), 3.58-3.52 (m, 2H), 2.78 (s, 3H), 2.04-1.97 (m, 2H), 1.96-1.86 (m, 2H), 1.50 (s, 9H), 1.46 (t, J=7.2 Hz, 3H).
General Procedure for N-Boc Deprotection
Ethyl 3-methyl-4-(piperidin-4-yloxy) benzofuran-2-carboxylate (2)
A mixture of compound I-1 (10 mg, 0.025 mmol) and TFA (50 μL) in DCM (1 mL) was stirred at room temperature for 2 hours. The reaction mixture was evaporated under pressure to dryness, which was further purified by semi-preparative reverse phase HPLC to give the title compound as a white solid (9.8 mg, 94% yield). 1H-NMR (CD3OD, 400 MHz): δ 7.41 (t, J=8.4 Hz, 1H), 7.15 (d, J=8.4 Hz, 1H), 6.88 (d, J=8.0 Hz, 1H), 4.98-4.93 (m, 1H), 4.42 (q, J=7.2 Hz, 2H), 3.50-3.40 (m, 2H), 3.33-3.28 (m, 2H), 2.80 (s, 3H), 2.34-2.24 (m, 2H), 2.22-2.12 (m, 2H), 1.43 (t, J=7.2 Hz, 3H). 13C-NMR (CD3OD, 100 MHz): δ 161.87, 157.38, 154.55, 141.09, 130.09, 127.02, 120.30, 106.50, 106.16, 69.95, 62.13, 42.01, 28.28, 14.63, 11.82. Calculated exact mass for the protonated molecule (C17H22NO4): 304.1549; measured accurate mass (ESI): 304.1561. LC-MS purity: 100%, Rt = 11.39 min.
Prototypical Procedure for Ester Analogues 3-8 and 26
Phenyl 3-methyl-4-(piperidin-4-yloxy)-1-benzofuran-2-carboxylate(3)
A mixture of compound I-2 (44 mg, 0.12 mmol), EDCI (25 mg, 0.13 mmol), hydroxybenzotriazole (22 mg, 0.15 mmol) in anhydrous acetonitrile (3 mL) was stirred at room temperature for 30 minutes, and then treated with phenol (12.1 mg, 0.13 mmol) and DIPEA (41 μL, 0.24 mmol). The resulting mixture was further stirred at room temperature for another 12 hours. After that, the solution was evaporated to dryness in vacuo. The residue was re-dissolved in ethyl acetate (20 mL) and washed with 0.5 M NaOH and brine sequentially. The organic layer was dried over anhydrous sodium sulphate and concentrated in vacuo, to produce the N-Boc precursor without further purification.
The Boc-deprotection in DCM (2 mL) containing 5% TFA was carried out according to the preparation of compound 2, to afford the title compound as a white solid (28.1 mg, 51% overall yield). 1H-NMR (CD3OD, 400 MHz): δ 7.50-7.43 (m, 3H), 7.32 (t, J=7.6 Hz, 1H), 7.26 (d, J=7.6 Hz, 2H), 7.18 (d, J=8.0 Hz, 1H), 6.90 (d, J=8.0 Hz, 1H), 4.94-4.90 (m, 1H), 3.48-3.41 (m, 2H), 3.35-3.29 (m, 2H), 2.85 (s, 3H), 2.35-2.27 (m, 2H), 2.22-2.14 (m, 2H). 13C-NMR (CD3OD, 100 MHz): δ 160.02, 157.66, 154.69, 151.71, 140.26, 130.67, 130.62, 129.16, 127.21, 122.79, 120.29, 106.62, 106.19, 70.09, 41.99, 28.26, 12.05. Calculated exact mass for the protonated molecule (C21H22NO4): 352.1549; measured accurate mass (ESI): 352.1545. LC-MS purity: 100%, Rt = 12.17 min.
Prototypical Procedure for amide analogues 9-13
N-isopropyl-3-methyl-4-(piperidin-4-yloxy)benzofuran-2-carboxamide(9)
To a solution of compound I-2 (30 mg, 0.08 mmol) in DMF/DCM (2 mL, 1:1) was sequentially added DIPEA (15 μL, 0.088 mmol), PyBOP (46 mg, 0.088 mmol). The above mixture was stirred at room temperature for 30 minutes, followed by the addition of isopropylamine (7.6 μL, 0.088 mmol). The resulting mixture was further stirred at room temperature for 3 hours. After that, the reaction mixture was diluted with DCM (15 mL) and partitioned with 0.5M NaOH solution (15 mL), and then the aqueous layer was further extracted by DCM (10 mL). The combined organic layers were washed brine, dried over anhydrous sodium sulphate and concentrated in vacuo. The residue was purified by column chromatography over silica gel, to afford the N-Boc precursor.
The Boc-deprotection in DCM (2 mL) containing 5% TFA was carried out according to the preparation of compound 2, to afford the title compound as a white solid (11.5 mg, 34% overall yield). 1H-NMR (CD3OD, 400 MHz): δ 7.37 (t, J=8.4 Hz, 1H), 7.16 (d, J=8.4 Hz, 1H), 6.87 (d, J=8.4 Hz, 1H), 4.98-4.93 (m, 1H), 4.30-4.96 (m, 1H), 3.46-3.40 (m, 2H), 3.32-3.29 (m, 2H), 2.80 (s, 3H), 2.32-2.25 (m, 2H), 2.21-2.13 (m, 2H), 1.29 (d, J=6.4 Hz, 6H). 13C-NMR (CD3OD, 100 MHz): δ 161.52, 156.45, 154.30, 143.11, 129.19, 123.31, 120.78, 106.62, 106.05, 69.78, 42.32, 41.92, 28.23, 22.58, 11.52. Calculated exact mass for the protonated molecule (C18H25N2O3): 317.1865; measured accurate mass (ESI): 317.1867. LC-MS purity: 100%, Rt = 10.75 min.
Procedure for alkenes 15 and 16 and alkyl 18
4-({3-Methyl-2-[(E)-2-phenylethenyl]-1-benzofuran-4-yl}oxy)piperidine(16)
To a mixture of compound I-4 (50 mg, 0.14 mmol) and benzyltriphenyl phosphonium bromide (65 mg, 0.15 mmol) in DCM/water (2 mL, 1:1), 10M of NaOH solution (50 μL) was added. The resulting mixture was stirred at room temperature for 1 hour. After that, the reaction mixture was diluted with DCM (20 mL) and washed sequentially with water and brine (15 mL each). The organic layer was concentrated to give the mixture of the cis/trans N-Boc precursors.
The Boc-deprotection in DCM (2 mL) containing 5% TFA was carried out according to the preparation of compound 2, to isolate the title compound as 1st major fraction from preparative HPLC (white solid, 4.9 mg, 8% overall yield). 1H-NMR (CD3OD, 400 MHz): δ 7.41 (d, J=8.0 Hz, 2H), 7.34-7.26 (m, 3H), 7.17 (t, J=8.0 Hz, 1H), 6.89 (d, J=8.0 Hz, 1H), 6.78 (d, J=8.0 Hz, 1H), 6.72 (d, J=12.4 Hz, 1H), 6.51 (d, J=12.4 Hz, 1H), 4.89-4.85 (m, 1H), 3.44-3.37 (m, 2H), 3.31-3.26 (m, 2H), 2.54 (s, 3H), 2.32-2.22 (m, 2H), 2.19-2.11 (m, 2H). 13C-NMR (CD3OD, 125 MHz): δ 156.93, 152.96, 150.05, 138.72, 132.03, 130.13, 128.88, 128.52, 127.58, 126.68, 120.99, 116.34, 106.39, 105.56, 69.57, 41.95, 28.32, 11.06. Calculated exact mass for the protonated molecule (C22H24NO2): 334.1807; measured accurate mass (ESI): 334.1813. LC-MS purity: 96% (trans:cis = 3:1), Rt = 13.22 min.
4-({3-methyl-2-[(Z)-2-phenylethenyl]-1-benzofuran-4-yl}oxy)piperidine(15)
The title compound was isolated as 2nd major fraction from the above preparative HPLC (off-white solid, 14.6 mg, 23% overall yield). 1H-NMR (CD3OD, 400 MHz): δ 7.58 (d, J=7.2 Hz, 2H), 7.38 (t, J=8.0 Hz, 2H), 7.27 (t, J=7.2 Hz, 1H), 7.23-7.18 (m, 3H), 7.07 (d, J=8.0 Hz, 1H), 6.78 (d, J=8.0 Hz, 1H), 4.90-4.82 (m, 1H), 3.46-3.40 (m, 2H), 3.32-3.27 (m, 2H), 2.53 (s, 3H), 2.35-2.22 (m, 2H), 2.21-2.12 (m, 2H). 13C-NMR (CD3OD, 100 MHz): δ 157.20, 153.02, 150.95, 138.43, 129.81, 129.62, 128.88, 127.58, 126.80, 121.67, 115.10, 114.66, 106.55, 105.43, 69.62, 41.94, 28.34, 10.52. Calculated exact mass for the protonated molecule (C22H24NO2): 334.1807; measured accurate mass (ESI): 334.1799. LC-MS purity: 97%, Rt = 13.64 min.
4-{[3-Methyl-2-(2-phenylethyl)-1-benzofuran-4-yl]oxy}piperidine(18)
A mixture of compound 15 (13 mg, 0.039 mmol), palladium on carbon (10% w/w, 5 mg, 0.008 mmol) and 1,4-cyclohexadiene (70 μL) in ethanol (1 mL) was stirred at 80 °C for 4 hours. The reaction mixture was then concentrated in vacuo and the residue was purified by semi-preparative reverse phase HPLC to give the title compound as colourless oil (6.6 mg, 38% yield). 1H-NMR (CD3OD, 500 MHz): δ 7.23 (t, J=8.0 Hz, 2H), 7.18-7.11 (m, 4H), 7.03 (d, J=8.0 Hz, 1H), 6.74 (d, J=8.0 Hz, 1H), 4.85-4.81 (m, 1H), 3.40-3.35 (m, 2H), 3.30-3.25 (m, 2H), 3.00 (brs, 4H), 2.25-2.19 (m, 2H), 2.15-2.09 (m, 2H), 2.11 (s, 3H). 13C-NMR (CD3OD, 125 MHz): δ 157.06, 153.11, 152.38, 142.37, 129.55, 129.31, 127.08, 125.06, 121.20, 111.15, 106.34, 105.55, 69.41, 41.90, 35.44, 29.07, 28.31, 10.17. Calculated exact mass for the protonated molecule (C22H26NO2): 336.1964; measured accurate mass (ESI): 336.1964. LC-MS purity: 100%, Rt = 13.30 min.
Procedure for ethers 19 and 21
4-{[3-Methyl-2-(phenoxymethyl)-1-benzofuran-4-yl]oxy} piperidine (19)
To a stirred solution of compound I-5 (30 mg, 0.06 mmol), phenol (14.1 mg, 0.15 mmol) and triphenylphosphine (39 mg, 0.15 mmol) in anhydrous THF (2 mL) was added DIAD (29 μL, 0.15 mmol) at room temperature. The resulting mixture was stirred at the same temperature for 4 hours, and then concentrated in vacuo. The residue was purified by column chromatography over silica gel, to afford the N-trityl precursor.
The trityl-deprotection was carried out in DCM (1 mL), containing 0.1% of TFA and 0.2% of water, at room temperature for 12 hours. After that, the reaction mixture was concentrated in vacuo and the residue was purified by semi-preparative reverse phase HPLC, with no TFA in the mobile phases, to afford the title compound as colorless oil (0.4 mg, 2% overall yield). 1H-NMR (CD3OD, 400 MHz): δ 7.32-7.28 (m, 2H), 7.24 (t, J=8.0 Hz, 1H), 7.09 (d, J=8.0 Hz, 1H), 7.04 (d, J=8.0 Hz, 2H), 6.98 (t, J=8.0 Hz, 1H), 6.81 (d, J=8.0 Hz, 1H), 5.15 (s, 2H), 4.96-4.89 (m, 1H), 3.43-3.38 (m, 2H), 3.30-3.26 (m, 2H), 2.48 (s, 3H), 2.30-2.22 (m, 2H), 2.20-2.11 (m, 2H). 13C-NMR (CD3OD, 125 MHz): δ 159.96, 153.18, 148.70, 138.03, 130.54, 126.75, 122.40, 120.63, 116.18, 115.81, 106.33, 105.89, 69.55, 61.90, 41.98, 28.34, 10.50. Calculated exact mass for the protonated molecule (C21H24NO3): 338.1756; measured accurate mass (ESI): 338.1772. LC-MS purity: 100%, Rt = 12.44 min.
4-({2-[(Benzyloxy)methyl]-3-methyl-1-benzofuran-4-yl}oxy)-piperidine (21)
A solution of I-4 (30 mg, 0.06 mmol) and sodium hydride (60% w/w, 7.2 mg, 0.18 mmol) in anhydrous DMF was stirred at room temperature for 30 minutes, and then treated with benzyl bromide (10.7 μL, 0.09 mmol). The resulting mixture was further stirred at room temperature for 12 hours. After that, the reaction mixture was quenched with 100 μL of cold water, diluted with EtOAc (20 mL) and sequentially washed with water and brine (each for 20 mL). The organic layer was concentrated in vacuo and the residue was purified by column chromatography over silica gel, to give the N-trityl precursor.
The trityl-deprotection was carried out in DCM (1 mL), containing 0.1% of TFA and 0.2% of water, at room temperature for 12 hours. After that, the reaction mixture was concentrated in vacuo and the residue was purified by semi-preparative reverse phase HPLC, with no TFA in the mobile phases, to afford the title compound as colourless oil (2 mg, 10% overall yield). 1H-NMR (CD3OD, 400 MHz): δ 7.40-7.28 (m, 5H), 7.23 (t, J=8.0 Hz, 1H), 7.09 (d, J=8.0 Hz, 1H), 6.81 (d, J=8.0 Hz, 1H), 4.91-4.85 (m, 1H), 4.64 (s, 2H), 4.60 (s, 2H), 3.46-3.39 (m, 2H), 3.30-3.27 (m, 2H), 2.43 (s, 3H), 2.30-2.22 (m, 2H), 2.20-2.12 (m, 2H). 13C-NMR (CD3OD, 125 MHz): δ 156.06, 151.72, 148.30, 137.87, 128.02, 127.62, 127.45, 125.19, 119.22, 114.14, 104.84, 104.45, 71.65, 68.06, 61.44, 40.53, 26.90, 9.14. Calculated exact mass for the protonated molecule (C22H26NO3): 352.1913; measured accurate mass (ESI): 352.1909. LC-MS purity: 100%, Rt = 12.34 min.
(2E)-1-[3-Methyl-4-(piperidin-4-yloxy)-1-benzofuran-2-yl]-3-phenylprop-2-en-1-one (23)
Prepared from a solution of compound I-6 (190 mg, 1.0 mmol), t-Butyl 4-hydrxypiperidine-1-carboxylate (500 mg, 2.5 mmol), triphenylphosphine (657 mg, 2.5 mmol) and DIAD (480 μL, 2.5 mmol) in anhydrous THF (4 mL) according to the procedure of making compound I-1, t-Butyl 4-[(2-acetyl-3-methyl-1-benzofuran-4-yl)oxy]piperidine-1-carboxylate was obtained as a light yellow solid (318 mg).
Above solid (50 mg, 0.13 mmol) and benzaldehyde (15 μL, 0.15 mmol) in ethanol (1 mL) was added 0.25 M NaOH solution (1 mL). The mixture was stirred at room temperature for 3 hours, and then diluted with 20 mL EtOAc, followed by the wash with water and brine (20 mL each). The organic layer was dried over anhydrous sodium sulphate and concentrated in vacuo. The residue was purified by column chromatography over silica gel, to give the N-Boc precursor.
The N-Boc deprotection was carried out in DCM containing 5% TFA (2 mL) according to the preparation of compound 2, to afford the title compound as a yellow solid (14.7 mg, 24% overall yield). 1H-NMR (CD3OD, 400 MHz): δ 7.86-7.75 (m, 4H), 7.51-7.45 (m, 4H), 7.24 (d, J=8.0 Hz, 1H), 6.89 (d, J=8.0 Hz, 1H), 5.02-4.93 (m, 1H), 3.50-3.39 (m, 2H), 3.33-3.27 (m, 2H), 2.88 (s, 3H), 2.36-2.24 (m, 2H), 2.24-2.12 (m, 2H). 13C-NMR (CD3OD, 100 MHz): δ 183.19, 157.14, 154.95, 148.89, 145.04, 136.18, 131.91, 130.81, 130.14, 129.73, 127.50, 123.26, 120.94, 106.64, 106.38, 70.04, 41.99, 28.26, 12.17. Calculated exact mass for the protonated molecule (C23H24NO3): 362.1756; measured accurate mass (ESI): 362.1750. LC-MS purity: 97%, Rt = 13.17 min.
4-{[2-(Phenylmethane)sulfonyl-1-benzofuran-4-yl]oxy}piperidine(24)
A mixture of t-Butyl 4-{[2-(benzylsulfanyl)-1-benzofuran-4-yl]oxy} piperidine-1-carboxylate (20 mg, 0.046 mmol) and 3-chloroperbenzoic acid (77% w/w, 16 mg, 0.07 mmol) in DCM (1 mL) was stirred at room temperature for 1 hour. The reaction mixture was concentrated in vacuo, to give the N-Boc precursor without further purification.
The N-Boc deprotection was carried out in DCM containing 5% TFA (2 mL) according to the preparation of compound 2, to afford the title compound as pink oil (8.0 mg, 36% overall yield). 1H-NMR (CD3OD, 400 MHz): δ 7.52 (t, J=8.0 Hz, 1H), 7.46 (s, 1H), 7.35-7.21 (m, 6H), 6.99 (d, J=8.0 Hz, 1H), 4.98-4.92 (m, 1H), 4.72 (s, 2H), 3.48-3.39 (m, 2H), 3.29-3.19 (m, 2H), 2.28-2.18 (m, 2H), 2.14-2.07 (m, 2H). 13C-NMR (CD3OD, 100 MHz): δ 157.59, 151.61, 148.00, 130.60, 129.39, 128.55, 128.22, 127.77, 117.35, 112.47, 106.75, 105.11, 68.95, 60.60, 40.31, 26.78. Calculated exact mass for the protonated molecule (C20H22NO4S): 372.1270; measured accurate mass (ESI): 372.1263. LC-MS purity: 100%, Rt = 10.91 min.
BIOLOGY
Enzyme Inhibition Assay
Measurement of the ability of compounds to inhibit NMTs was performed in 96-well plates using a modification of the scintillation proximity assay platform:39 each well (in total 100 μL) contained variable amounts of an inhibitor, 9.4 ng of purified PfNMT (4.8 ng for PvNMT, 19.2 ng for PvNMTY211A, 5.1 ng for HsNMT1 and 20.4 ng for HsNMT1Y296A), 62.5 nM 3H-Myristoyl-CoA (8 Ci/mmol, purchased from Perkin Elmer) and 500 nM of the peptide substrate (GLYVSRLFNRLFQKK(Biotin)-NH2) in 4% DMSO buffer (30 mM Tris, pH 7.4, 0.5 mM EGTA, 0.5 mM EDTA, 2.5 mM DTT, 0.1% Triton X-100). Incubation was allowed to proceed for 30 min at 37°C, followed by the addition of 100 μL 1 mg/mL streptavidin PVT bead (purchased from Perkin Elmer) in a stop solution (200 mM phosphoric acid / NaOH, pH 4, 750 mM MgCl2, 0.05% (w/V) BSA, 0.01% (w/V) NaN3) to terminate the reaction. Overnight settling of the beads resulted in an accurate reading, with signal-to-noise ratio at least 20. The instrument used for counting is a Plate Chameleon multi-label reader (Hidex Oy, Finland). The readout of [3H]-myristoylated peptide in the reaction lacking an inhibitor was defined as 100% activity (positive control), and that from the reaction with no enzyme was defined as 0% (negative control). The effect of an inhibitor at the single concentration was calculated as a percentage of the enzyme activity left (% activity left = (readout-negative) / (positive-negative) × 100%). In terms of the IC50 determination, the inhibitory activity of an inhibitor was measured according to the method described above with the concentrations ranging from 500 μM to 2 nM and the 50% inhibitory concentration (IC50) of an inhibitor was calculated by a nonlinear regression analysis using GraFit 7.0.1 version (Erithacus Software Limited, UK). The assays were carried out in duplicate.
P. falciparum Inhibition Assay
Measurement of the ability of compounds to kill parasites was performed in 96-well plates using a modification of the fluorescent-activity cell sorting (FACS) assay platform:40, 41 each well (in total 100 μL, 0.5% DMSO) contained synchronous cultures of late trophozoite-stage parasites (1% parasitemia and 2% hematocrit) and variable concentrations of an inhibitor. The mixture was incubated at 37 °C for a full 48-hour growth cycle. Aliquots of 50 μL were removed from each well, added to 500 μL freshly diluted hydroethidine (HE, 1:200 dilution of 10 mg/mL DMSO stock in PBS) and incubated for 20 min at 37°C. Samples were then diluted with 1 mL PBS to enable appropriate cell counts (50,000) and stored on ice. Parasites cultured in the absence of an inhibitor and non-infected red blood cells were used as positive and negative controls respectively. Parasitemia was measured using a FACS Calibur flow cytometer. Growth inhibition at each concentration was calculated as % inhibition = [1-(readout-negative control) / (positive-negative control)] × 100%. To determine the EC50 of an inhibitor, its activity at a range of concentrations from 10 μM down to 0.31 μM and the 50% inhibitory concentration (EC50) of an inhibitor was calculated by a nonlinear regression analysis using GraFit 7.0.1 version (Erithacus Software Limited, UK). All assays were carried out in triplicate.
CRYSTALLOGRAPHY
Protein expression, purification and crystallization
For HsNMT1, a clone directing the synthesis of a protein without the first 108 amino acid residues corresponding to the NMT catalytic domain was expressed in E. coli and purified as described in reference34. PvNMT expression and purification followed protocols as described in reference38. Mutagenesis of PvNMT Y211 and HsNMT Y296 residues to alanine was performed using a QuikChange Lightning site-directed mutagenesis kit (Stratagene). Initial crystals of a binary complex with a non-hydrolysable cofactor (PvNMT-NHM) were grown from a solution of protein (6 mg/ml) containing 1/20th volume of S-(2-oxo) pentadecyl-coenzyme A (NHM, 10 mM in 50% DMSO) and an equal volume of IndexScreen (Hampton) solution F7 (0.2 M ammonium sulphate and 25% (w/v) PEG 3350 in 0.1 M Bis-Tris buffer, pH 6.5). The crystals belong to the orthorhombic space group P212121.The structure was solved by molecular replacement in the program MOLREP42 using the protein coordinates of NMT from L. donovani (PDB code 2WUU)43 as the search model. Three solutions (a trimer) were found, corresponding to a Matthew’s coefficient of 2.22 Å3/Da and a solvent content of 44.5%, and this trimer was used as the basis for structure determination of ternary complexes of PvNMT-NHM containing ligands (Table S5, Supporting information). For crystallization of the ligand complexes, PvNMT was incubated overnight at 4°C with NHM as above plus solid ligand (compounds 25 and 26) or 1/20th volume ligand (compounds 13 and 22,10 mM in 50% DMSO). Crystals were grown by the vapour diffusion method, typically from sitting drops consisting of 0.3 μL protein mixture (after centrifugation to pellet any solid material) and 0.3 μL of reservoir solution containing 0.2 M ammonium sulphate, 24-26% (w/v) PEG 3350, in 0.1 M Bis-Tris or MES buffer, pH 6.0-6.5 equilibrated against 0.1 mL reservoir solution in 48-well plates at 20°C. Crystals were vitrified directly in liquid nitrogen before storage.
Data collection, processing and refinement
X-ray diffraction data were collected on synchrotron beamlines at the Diamond Light Source (DLS), Oxford and processed using XDS44 and SCALA45 implemented within xia2.46 Data collection and refinement statistics are summarized in Supplementary Table S5. For Rfree calculations, 5% of the data were excluded and the Rfree flags for each ternary complex structure solution were copied from the binary complex structure (PvNMT-NHM). Rigid Body refinement using maximum likelihood methods implemented in REFMAC47 was followed by a cycle of model building and adjustment using COOT48 and five cycles of refinement using anisotropic temperature factors. Electron density maps from this early stage of refinement calculated using phases unbiased by contributions from ligand (‘omit’ maps) are presented in Figure S3, Supporting Information. Complete chains (corresponding to residues 26 - 410, numbering as in full-length protein) can be traced for two of the three molecules in the asymmetric unit. N-terminal residues (remaining from the tag) in all three chains and loop residues 227 - 238 in chain C have not been modeled and these are assumed to be disordered. The protein structure models display good geometry with 95.9% of the residues in the preferred region of the Ramachandran plot, with an additional 3.9% in allowed regions and 0.2% (corresponding to two residues in chain C) as outliers. For ligand co-crystal structures, three residues (F336 in all three chains) also appear in outlying regions of the plot. The coordinates and structure factor files have been deposited in the Protein Data Bank under the accession codes 4B10 (PvNMT-NHM), 4B11 (PvNMT-NHM-13), 4B12 (PvNMT-NHM-22), 4B13 (PvNMT-NHM-25) and 4B14 (PvNMT-NHM-26).
Supplementary Material
Table 1.
Activities of esters and amides against PfNMTa
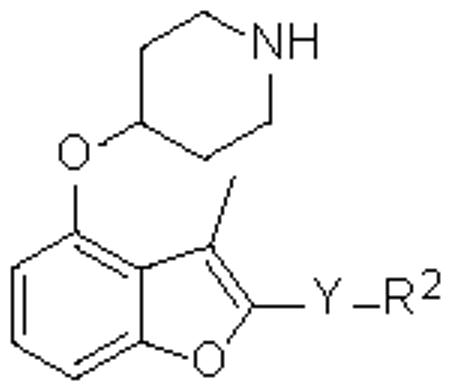
| No. | Y | R2 | IC50 (μM) |
|---|---|---|---|
| 2 | COO | Et | 16.5 |
| 3 | COO | Ph | 24.4 |
| 4 | COOCH2 | Ph | 0.27 |
| 5 | COOCH2 | α-napthyl | 1.4 |
| 6 | COOCH2 | cyclo-hex | 9.7 |
| 7 | COO(CH2)2 | Ph | 4.1 |
| 8 | COO(CH2)3 | Ph | 36.2 |
| 9 | CONH | i-Pr | >200 |
| 10 | CONH | cyclo-hex | >100 |
| 11 | CONH | Ph | 28.8 |
| 12 | CONHCH2 | Ph | 13.0 |
| 13 | CONHCH2 | α-napthyl | 2.3 |
The IC50(PfNMT) values were averaged from two independent dose-response curves, the standard deviation was within 30% of the resulting values.
ACKNOWLEDGEMENTS
This work was supported by the Wellcome Trust (grant number: 087792), the Medical Research Council (grant numbers: 0900278 and U117532067) and the EU F97 project MALSIG (HEALTH-F3-2009-223044). The authors thank Profs. J. Frearson and P. Wyatt of the Dundee Drug Discovery Unit for kindly providing a focused library of reported NMT inhibitors for the initial screening and Diamond Light Source for Synchrotron facilities.
ABBREVIATION USED
- NMT
N-myristoyltransferase
- myr-CoA
myristoyl-coenzyme A
- NHM
Non-Hydrolyzable Myristoyl-coenzyme A
- SAR
Structure-Activity Relationship
- PDB
Protein Data Bank
- Ph
phenyl
- i-Pr
iso-propyl
- Et
ethyl
- hex
hexane
- Boc
tert-butoxycarbonyl
- Tr
trityl
Footnotes
Supporting Information Available: Synthetic details for all compounds, additional compound biological data (Tables S1-S4), alignment of PvNMT and PfNMT (Figure S1) and X-ray crystallographic data (Table S5 and Figures S2, S3). This material is available free of charge via the internet at http://pubs.acs.org.
REFERENCES
- 1.World Malaria Report 2011. World Health Organization; Geneva: 2011. [Google Scholar]
- 2.Opar A. Quarter-century quest for malaria vaccine shows signs of success. Nat. Rev. Drug Discov. 2011;10:887–888. doi: 10.1038/nrd3611. [DOI] [PubMed] [Google Scholar]
- 3.Hyde JE. Drug - resistant malaria - an insight. FEBS J. 2007;274:4688–4698. doi: 10.1111/j.1742-4658.2007.05999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guidelines for the treatment of malaria - 2nd edition. World Health Organization; Geneva: 2010. pp. 13–47. [PubMed] [Google Scholar]
- 5.Dondorp AM, Nosten F, Yi P, Das D. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009;361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gamo F-J, Sanz LM, Vidal J, de Cozar C, Alvarez E, Lavandera J-L, Vanderwall DE, Green DVS, Kumar V, Hasan S, Brown JR, Peishoff CE, Cardon LR, Garcia-Bustos JF. Thousands of chemical starting points for antimalarial lead identification. Nature. 2010;465:305–310. doi: 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- 7.Plouffe D, Brinker A, McNamara C, Henson K, Kato N, Kuhen K, Nagle A, Adrian F, Matzen JT, Anderson P, Nam T.-g., Gray NS, Chatterjee A, Janes J, Yan SF, Trager R, Caldwell JS, Schultz PG, Zhou Y, Winzeler EA. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc. Natl. Acad. Sci. USA. 2008;105:9059–9064. doi: 10.1073/pnas.0802982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rottmann M, McNamara C, Yeung BKS, Lee MCS, Zou B, Russell B, Seitz P, Plouffe DM, Dharia NV, Tan J, Cohen SB, Spencer KR, Gonzalez-Paez GE, Lakshminarayana SB, Goh A, Suwanarusk R, Jegla T, Schmitt EK, Beck H-P, Brun R, Nosten F, Renia L, Dartois V, Keller TH, Fidock DA, Winzeler EA, Diagana TT. Spiroindolones, a Potent Compound Class for the Treatment of Malaria. Science. 2010;329:1175–1180. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Neill PM, Amewu RK, Nixon GL, Bousejra ElGarah F, Mungthin M, Chadwick J, Shone AE, Vivas L, Lander H, Barton V, Muangnoicharoen S, Bray PG, Davies J, Park BK, Wittlin S, Brun R, Preschel M, Zhang K, Ward SA. Identification of a 1,2,4,5-Tetraoxane Antimalarial Drug-Development Candidate (RKA 182) with Superior Properties to the Semisynthetic Artemisinins. Angew. Chem. Int. Ed. 2010;49:5693–5697. doi: 10.1002/anie.201001026. [DOI] [PubMed] [Google Scholar]
- 10.Meister S, Plouffe DM, Kuhen KL, Bonamy GMC, Wu T, Barnes SW, Bopp SE, Borboa R, Bright AT, Che J, Cohen S, Dharia NV, Gagaring K, Gettayacamin M, Gordon P, Groessl T, Kato N, Lee MCS, McNamara CW, Fidock DA, Nagle A, Nam T.-g., Richmond W, Roland J, Rottmann M, Zhou B, Froissard P, Glynne RJ, Mazier D, Sattabongkot J, Schultz PG, Tuntland T, Walker JR, Zhou Y, Chatterjee A, Diagana TT, Winzeler EA. Imaging of Plasmodium Liver Stages to Drive Next-Generation Antimalarial Drug Discovery. Science. 2011;334:1372–1377. doi: 10.1126/science.1211936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bowyer PW, Tate EW, Leatherbarrow RJ, Holder AA, Smith DF, Brown KA. N-Myristoyltransferase: a Prospective Drug Target for Protozoan Parasites. ChemMedChem. 2008;3:402–408. doi: 10.1002/cmdc.200700301. [DOI] [PubMed] [Google Scholar]
- 12.Duronio RJ, Towler DA, Heuckeroth RO, Gordon JI. Disruption of the yeast N-myristoyl transferase gene causes recessive lethality. Science. 1989;243:796–800. doi: 10.1126/science.2644694. [DOI] [PubMed] [Google Scholar]
- 13.Lodge JK, Jackson-Machelski E, Toffaletti DL, Perfect JR, Gordon JI. Targeted gene replacement demonstrates that myristoyl-CoA: protein N-myristoyltransferase is essential for viability of Cryptococcus neoformans. Proc. Natl. Acad. Sci. USA. 1994;91:12008–12012. doi: 10.1073/pnas.91.25.12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weinberg RA, McWherter CA, Freeman SK, Wood DC, Gordon JI, Lee SC. Genetic studies reveal that myristoylCoA:protein N-myristoyltransferase is an essential enzyme in Candida albicans. Mol. Microbiol. 1995;16:241–250. doi: 10.1111/j.1365-2958.1995.tb02296.x. [DOI] [PubMed] [Google Scholar]
- 15.Price HP, Menon MR, Panethymitaki C, Goulding D, McKean PG, Smith DF. Myristoyl-CoA:Protein N-Myristoyltransferase, an Essential Enzyme and Potential Drug Target in Kinetoplastid Parasites. J. Biol. Chem. 2003;278:7206–7214. doi: 10.1074/jbc.M211391200. [DOI] [PubMed] [Google Scholar]
- 16.Johnson DR, Bhatnagar RS, Knoll LJ, Gordon JI. Genetic and Biochemical Studies of Protein N-Myristoylation. Annu. Rev. Biochem. 1994;63:869–914. doi: 10.1146/annurev.bi.63.070194.004253. [DOI] [PubMed] [Google Scholar]
- 17.Wright M, Heal W, Mann D, Tate E. Protein myristoylation in health and disease. J. Chem. Biol. 2010;3:19–35. doi: 10.1007/s12154-009-0032-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rees-Channer RR, Martin SR, Green JL, Bowyer PW, Grainger M, Molloy JE, Holder AA. Dual acylation of the 45kDa gliding-associated protein (GAP45) in Plasmodium falciparum merozoites. Mol. Biochem. Parasitol. 2006;149:113–116. doi: 10.1016/j.molbiopara.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 19.Stafford WHL, Stockley RW, Ludbrook SB, Holder AA. Isolation, Expression and Characterization of the Gene for an ADP-Ribosylation Factor from the Human Malaria Parasite, Plasmodium Falciparum. Eur. J. Biochem. 1996;242:104–113. doi: 10.1111/j.1432-1033.1996.0104r.x. [DOI] [PubMed] [Google Scholar]
- 20.Leber W, Skippen A, Fivelman QL, Bowyer PW, Cockcroft S, Baker DA. A unique phosphatidylinositol 4-phosphate 5-kinase is activated by ADP-ribosylation factor in Plasmodium falciparum. Int. J. Parasitol. 2009;39:645–653. doi: 10.1016/j.ijpara.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 21.Green JL, Rees-Channer RR, Howell SA, Martin SR, Knuepfer E, Taylor HM, Grainger M, Holder AA. The Motor Complex of Plasmodium falciparum. J. Biol. Chem. 2008;283:30980–30989. doi: 10.1074/jbc.M803129200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pino P, Graindorge A, Kim A, Soldati-Favre D. Conditional system for the malaria parasites: functional dissection of N-myristoyltransferase and rhomboid 4 genes in Plasmodium berghei. Molecular Approaches to Malaria 2012; Lorne, Australia. 2012; Lorne, Australia, 2012. [Google Scholar]
- 23.Rudnick DA, McWherter CA, Rocque WJ, Lennon PJ, Getman DP, Gordon JI. Kinetic and structural evidence for a sequential ordered Bi Bi mechanism of catalysis by Saccharomyces cerevisiae myristoyl-CoA:protein N-myristoyltransferase. J. Biol. Chem. 1991;266:9732–9739. [PubMed] [Google Scholar]
- 24.Masubuchi M, Ebiike H, Kawasaki K, Sogabe S, Morikami K, Shiratori Y, Tsujii S, Fujii T, Sakata K, Hayase M, Shindoh H, Aoki Y, Ohtsuka T, Shimma N. Synthesis and biological activities of benzofuran antifungal agents targeting fungal N-myristoyltransferase. Bioorg. Med. Chem. 2003;11:4463–4478. doi: 10.1016/s0968-0896(03)00429-2. [DOI] [PubMed] [Google Scholar]
- 25.Kawasaki K, Masubuchi M, Morikami K, Sogabe S, Aoyama T, Ebiike H, Niizuma S, Hayase M, Fujii T, Sakata K, Shindoh H, Shiratori Y, Aoki Y, Ohtsuka T, Shimma N. Design and synthesis of novel benzofurans as a new class of antifungal agents targeting fungal N-myristoyltransferase. Part 3. Bioorg. Med. Chem. Lett. 2003;13:87–91. doi: 10.1016/s0960-894x(02)00844-2. [DOI] [PubMed] [Google Scholar]
- 26.Ebiike H, Masubuchi M, Liu PL, Kawasaki K, Morikami K, Sogabe S, Hayase M, Fujii T, Sakata K, Shindoh H, Shiratori Y, Aoki Y, Ohtsuka T, Shimma N. Design and synthesis of novel benzofurans as a new class of antifungal agents targeting fungal N-myristoyltransferase. Part 2. Bioorg. Med. Chem. Lett. 2002;12:607–610. doi: 10.1016/s0960-894x(01)00808-3. [DOI] [PubMed] [Google Scholar]
- 27.Yamazaki K, Kaneko Y, Suwa K, Ebara S, Nakazawa K, Yasuno K. Synthesis of potent and selective inhibitors of Candida albicans N-myristoyltransferase based on the benzothiazole structure. Bioorg. Med. Chem. 2005;13:2509–2522. doi: 10.1016/j.bmc.2005.01.033. [DOI] [PubMed] [Google Scholar]
- 28.Devadas B, Freeman SK, Zupec ME, Lu HF, Nagahashi S, Kishore NS, Lodge JK, Kuneman DW, McWherter CA, Vinjamoori DV, Getman DP, Gordon JI, Sikorski JA. Design and synthesis of novel imidazole-substituted dipeptide amides as potent and selective inhibitors of Candida albicans myristoyl-CoA:protein N-myristoyltransferase and identification of related tripeptide inhibitors with mechanism-based antifungal activity. J. Med. Chem. 1997;40:2609–2625. doi: 10.1021/jm970094w. [DOI] [PubMed] [Google Scholar]
- 29.Masubuchi M, Kawasaki K, Ebiike H, Ikeda Y, Tsujii S, Sogabe S, Fujii T, Sakata K, Shiratori Y, Aoki Y, Ohtsuka T, Shimma N. Design and synthesis of novel benzofurans as a new class of antifungal agents targeting fungal N-myristoyltransferase. Part 1. Bioorg. Med. Chem. Lett. 2001;11:1833–1837. doi: 10.1016/s0960-894x(01)00319-5. [DOI] [PubMed] [Google Scholar]
- 30.Gelb MH, Van Voorhis WC, Buckner FS, Yokoyama K, Eastman R, Carpenter EP, Panethymitaki C, Brown KA, Smith DF. Protein farnesyl and N-myristoyl transferases: piggy-back medicinal chemistry targets for the development of antitrypanosomatid and antimalarial therapeutics. Mol. Biochem. Parasitol. 2003;126:155–163. doi: 10.1016/s0166-6851(02)00282-7. [DOI] [PubMed] [Google Scholar]
- 31.Brand S, Cleghorn LAT, McElroy SP, Robinson DA, Smith VC, Hallyburton I, Harrison JR, Norcross NR, Spinks D, Bayliss T, Norval S, Stojanovski L, Torrie LS, Frearson JA, Brenk R, Fairlamb AH, Ferguson MAJ, Read KD, Wyatt PG, Gilbert IH. Discovery of a Novel Class of Orally Active Trypanocidal N-Myristoyltransferase Inhibitors. J. Med. Chem. 2012;55:140–152. doi: 10.1021/jm201091t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frearson JA, Brand S, McElroy SP, Cleghorn LAT, Smid O, Stojanovski L, Price HP, Guther MLS, Torrie LS, Robinson DA, Hallyburton I, Mpamhanga CP, Brannigan JA, Wilkinson AJ, Hodgkinson M, Hui R, Qiu W, Raimi OG, van Aalten DMF, Brenk R, Gilbert IH, Read KD, Fairlamb AH, Ferguson MAJ, Smith DF, Wyatt PG. N-myristoyltransferase inhibitors as new leads to treat sleeping sickness. Nature. 2010;464:728–732. doi: 10.1038/nature08893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Belanger PC, Dufresne C, Lau CK, Scheigetz J. An improved synthesis of ethyl 4-hydroxy-3-methylbenzofuran-2-carboxylate. Org. Prep. Proced. Int. 1988;20:299–302. [Google Scholar]
- 34.Goncalves V, Brannigan JA, Thinon E, Olaleye TO, Serwa R, Lanzarone S, Wilkinson AJ, Tate EW, Leatherbarrow RJ. A fluorescence-based assay for N-myristoyltransferase activity. Ana. Biochem. 2012;421:342–344. doi: 10.1016/j.ab.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farazi TA, Manchester JK, Waksman G, Gordon JI. Pre-Steady-State Kinetic Studies of Saccharomyces cerevisiae MyristoylCoA:Protein N-Myristoyltransferase Mutants Identify Residues Involved in Catalysis. Biochemistry. 2001;40:9177–9186. doi: 10.1021/bi0107997. [DOI] [PubMed] [Google Scholar]
- 36.Wu JT,Y, Zhang ML, Howard MH, Gutteridge S, Ding JP. Crystal Structures of Saccharomyces cerevisiae N-Myristoyltransferase with Bound Myristoyl-CoA and Inhibitors Reveal the Functional Roles of the N-terminal Region. J. Biol. Chem. 2007;282:22185–22194. doi: 10.1074/jbc.M702696200. [DOI] [PubMed] [Google Scholar]
- 37.Bell AS, Mills JE, Williams GP, Brannigan JA, Wilkinson AJ, Parkinson T, Leatherbarrow RJ, Tate EW, Holder AA, Smith DF. Selective Inhibitors of Protozoan Protein N-myristoyltransferases as Starting Points for Tropical Disease Medicinal Chemistry Programs. PLoS Negl. Trop. Dis. 2012;6:e1625. doi: 10.1371/journal.pntd.0001625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goncalves V, Brannigan JA, Whalley D, Ansell KH, Saxty B, Holder AA, Wilkinson AJ, Tate EW, Leatherbarrow RJ. Discovery of Plasmodium vivaxN-Myristoyltransferase Inhibitors: Screening, Synthesis, and Structural Characterization of their Binding Mode. J. Med. Chem. 2012;55:3578–3582. doi: 10.1021/jm300040p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Panethymitaki C, Bowyer PW, Price HP, Leatherbarrow RJ, Brown KA, Smith DF. Characterization and selective inhibition of myristoyl-CoA:protein N-myristoyltransferase from Trypanosoma brucei and Leishmania major. Biochem. J. 2006;396:277–285. doi: 10.1042/BJ20051886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bergmann-Leitner ES, Dundcan EH, Mullen GE, Burge JR, Khan F, Long CA, Angov E, Lyon JA. Critical evaluation of different methods for measuring the functional activity of antibodies against malaria bolld stage antigens. Am. J. Trop. Med. Hyg. 2006;75:437–442. [PubMed] [Google Scholar]
- 41.Moss DK, Remarque EJ, Faber BW, Cavanagh DR, Arnot DE, Thomas AW, Holder AA. Plasmodium falciparum 19-Kilodalton Merozoite Surface Protein 1 (MSP1)-Specific Antibodies That Interfere with Parasite Growth In Vitro Can Inhibit MSP1 Processing, Merozoite Invasion, and Intracellular Parasite Development. Infect. Immun. 2012;80:1280–1287. doi: 10.1128/IAI.05887-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vagin A, Teplyakov A. An approach to multi-copy search in molecular replacement. Acta Crystallogr. Sect. D. 2000;56:1622–1624. doi: 10.1107/s0907444900013780. [DOI] [PubMed] [Google Scholar]
- 43.Brannigan JA, Smith BA, Yu Z, Brzozowski AM, Hodgkinson MR, Maroof A, Price HP, Meier F, Leatherbarrow RJ, Tate EW, Smith DF, Wilkinson AJ. N-Myristoyltransferase from Leishmania donovani: Structural and Functional Characterisation of a Potential Drug Target for Visceral Leishmaniasis. J. Mol. Biol. 2010;396:985–999. doi: 10.1016/j.jmb.2009.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kabsch W. XDS. Acta Crystallogr. Sect. D. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Evans P. Scaling and assessment of data quality. Acta Crystallogr. Sect. D. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 46.Winter G. xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 2010;43:186–190. [Google Scholar]
- 47.Murshudov GN, Vagin AA, Dodson EJ. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallogr. Sect. D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 48.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. Sect. D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.