

Abstract
Cadmium is a known human lung carcinogen. Here, we attempt to develop an in vitro model of cadmium-induced human lung carcinogenesis by chronically exposing the peripheral lung epithelia cell line, HPL-1D, to a low level of cadmium. Cells were chronically exposed to 5 μM cadmium, a noncytotoxic level, and monitored for acquired cancer characteristics. By 20 weeks of continuous cadmium exposure, these chronic cadmium treated lung (CCT-LC) cells showed marked increases in secreted MMP-2 activity (3.5-fold), invasion (3.4-fold), and colony formation in soft agar (2-fold). CCT-LC cells were hyperproliferative, grew well in serum-free media, and overexpressed cyclin D1. The CCT-LC cells also showed decreased expression of the tumor suppressor genes p16 and SLC38A3 at the protein levels. Also consistent with an acquired cancer cell phenotype, CCT-LC cells showed increased expression of the oncoproteins K-RAS and N-RAS as well as the epithelial-to-mesenchymal transition marker protein Vimentin. Metallothionein (MT) expression is increased by cadmium, and is typically overexpressed in human lung cancers. The major MT isoforms, MT-1A and MT-2A were elevated in CCT-LC cells. Oxidant adaptive response genes HO-1 and HIF-1A were also activated in CCT-LC cells. Expression of the metal transport genes ZNT-1, ZNT-5, and ZIP-8 increased in CCT-LC cells culminating in reduced cadmium accumulation, suggesting adaptation to the metal. Overall, these data suggest that exposure of human lung epithelial cells to cadmium causes acquisition of cancer cell characteristics. Furthermore, transformation occurs despite the cell’s ability to adapt to chronic cadmium exposure.
Keywords: Cadmium, Human lung cells, Transformation, Adaptation, Lung cancer, Epithelial-to-mesenchymal transition
Introduction
Cadmium is a widespread pollutant metal with clear carcinogenic potential in both humans and rodents (IARC, 2012; WHO, 2008). Its biological half-life in humans is measured in decades (WHO, 2008). As such, it is essentially a cumulative toxicant making carcinogenic potential of particular concern (IARC, 2012). Human exposure occurs mainly through inhalation and ingestion (IARC, 2012). Exposure to the metal can result from environment or via occupational settings. Cigarette smoking can be a major source of cadmium exposure (Huff et al., 2007).
Lung cancer is the second leading cause of cancer-related deaths in the United States, with one of the highest mortality rates of any malignancy in both men and women (ACS, 2012). Recent data indicate that pulmonary cancer accounts for an estimated 29% and 26% of cancer deaths for males and females, respectively within the United States (ACS, 2012). The association between cadmium and lung cancer is well established in humans and rodents (IARC, 1993, 2012; NTP, 2011). Cadmium has also been recently shown to transform human bronchial epithelial cells (Jing et al., 2012). The lung absorbs relatively high amounts of cadmium after inhalation (Beveridge et al., 2010; Bruske-Hohlfeld, 2009). With cadmium, lung cancer is most frequently linked to occupational exposure although some data indicate that increased lung cancer risk may be associated with environmental exposure as well (IARC, 2012; NTP, 2011).
It is thought that the metallothionein (MT), a high affinity metal binding protein, can bind large amounts of cadmium and is at least partially responsible for the long biological half-life and accumulation of cadmium (Klaassen et al., 2009; NTP, 2011). This bioaccumulation is likely due, at least in part, to sequestration of cadmium by MT which reduces free intracellular cadmium but in turn results in increased levels of the toxic metal in cells (Klaassen et al., 2009). Since cadmium does not biodegrade, binding to MT may constitute a biological “strategy” to reduce its immediate toxic potential. However, MT-bound cadmium actually permits large amounts of the metal to accumulate within target cells, which may well eventually become toxic (Klaassen et al., 2009).
Oxidative stress can be another challenge for cells during cadmium exposure and in some cells, it is thought to drive acquired malignant phenotype (Jing et al., 2012). In other cells, the role of oxidative stress appears to have a more limited role in cadmium-induced malignant transformation, as tolerance generated to reactive oxygen species (ROS) develops long before transformation and altered oncogene expression that appears more to be of an important factor in acquired malignant characteristics (Qu et al., 2005). Thus some cells adapt to cadmium during chronic exposure by activating genes that limit the impact of oxidative stress (He et al., 2008; Liu et al., 2002; Qu et al., 2005) or various other toxic responses. This can also be seen on the whole animal level as a resistance to acute cadmium intoxication (Amara et al., 2008). How adaptation might impact malignant transformation is incompletely defined but appears cell specific (Jing et al., 2012; Qu et al., 2005).
It is critical to maintain cellular homeostasis of essential trace elements. Transport of cations across membrane barriers is often accomplished by members of the Zrt/Irt-like family of proteins, or ZIPs (Eide, 2006). ZIPs belong to the SLC39A gene family (Eide, 2004; Guerinot, 2000; Liuzzi and Cousins, 2004). ZIPs primarily transport essential transition elements such as zinc (Liuzzi and Cousins, 2004), and appear to aid in internalization of cadmium, although transporters for other essential elements such as magnesium, calcium, manganese, and iron, have also been implicated in cadmium influx into mammalian cells (Himeno et al., 2009). Toxic metal cations like cadmium can displace essential metals in part due to the broad specificity of SLC39A binding (Guerinot, 2000). It is believed that ZIP-8 protein is located on the apical surfaces of lung alveolar cells (He et al., 2009) making it readily accessible to inhaled cadmium. In fact, the lung is the site of the greatest expression of ZIP-8 (He et al., 2009) which would possibly account for the high levels of cadmium uptake after inhalation. Indeed, high ZIP-8 activity greatly sensitizes cells to cadmium-induced cell death (Dalton et al., 2005). Thus, the association between ZIP-8 and cadmium-induced human cancer has been proposed to be critical (He et al., 2009), although this link has not actually been tested.
With regard to the lung as a target of carcinogenesis, the HPL-1D cells are an immortalized, human peripheral lung epithelial cell line that was developed to study molecular events involved with the malignant transformation of normal lung cells in vitro (Masuda et al., 1997). HPL-1D cells made it possible for us to investigate the effects of chronic low-level exposure to cadmium on human lung epithelial cells. Thus, in the present study we attempted to develop an in vitro model of cadmium-induced lung carcinogenesis. Here we provide results that indicate chronic cadmium exposure caused acquisition of lung cancer cell characteristics in HPL-1D cells. Adaptation to various aspects of cadmium toxicity occurred concurrently, but this appeared to have little effect on transformation.
Materials and methods
Chemicals and reagents
Cadmium chloride (CdCl2), p-iodonitrotetrazolium (INT), bovine insulin, hydrocortisone and triiodothyronine were all purchased from Sigma Chemical Company (St. Louis, MO). Other chemicals and sources included: HEPES buffer (Gibco/Invitrogen, Carlsbad, CA); human transferrin (Calbiochem/EMD Chemicals, San Diego, CA); antibiotic/antimycotic solution (Gibco/Invitrogen); Ham’s F-12 media (PromoCell, Heidelberg, Germany); fetal bovine serum (Gibco/Invitrogen, Carlsbad, CA); CellTiter 96 Aqueous ONE Solution Cell Proliferation Assay [3-(4,5-dimethyl-thiazol-2yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS assay)] reagent (Promega, Madison, WI).
Cells, culture conditions and cadmium exposure
HPL-1D cells were established by Dr. Takashi Takahashi, Laboratory of Ultrastructure Research (Japan) and were graciously provided by Dr. Lucy Anderson, National Cancer Institute. Cells were maintained in Ham’s F-12 medium buffered with 15 mM HEPES (pH 7.3) and supplemented with 5 μg/ml bovine insulin, 5 μg/ml human transferrin, hydrocortisone 10−7 M, 2 × 10−10 M triode thyronine, 1% antibiotic/antimycotic, and 1% fetal bovine serum. Cells were passaged weekly and fed every 2–3 days. For LC50 assessment cells were plated in 96-well plates and exposed to varying concentrations of cadmium (0 to 45 μM) for 72 h. The MTS assay was used to determine cell viability. The LC50 was determined to be 13 μM and cells were continuously exposed to 5 μM (100% cell viability at 72 h) cadmium, a non-toxic level, for up to 20 weeks. In some experiments, cells were grown without the 1% FBS to determine ability to grow in serum deprived media.
Cell proliferation and autonomous growth
Cell proliferation was determined by plating 6 × 105 control or cadmium treated cells (20 weeks) in triplicate using T-75 cell culture flasks for a period of seven days. Cells were counted using Trypan blue and represent the mean ± SEM. Autonomous growth was measured using control or cadmium treated cells (20 weeks) plated at a density of 2.0 × 104 cells per well in 96-well plates (n = 8). Cells were grown in standard Ham’s F-12 culture medium with reduced serum concentrations (from 1 to 0% fetal bovine serum) in both control and cadmium treated cells. Cells were incubated at 37 °C. Following 1, 3, 5 and 7 days to each well was added with 100 μl of fresh media. The MTS assay was performed following the manufacturer’s instructions and absorbances were measured at a wavelength of 490 nm using iMark Microplate Reader (Bio-Rad, Hercules, CA) to determine cell number.
Transcript expression
Relative gene expression levels were determined using quantitative real-time reverse transcriptase polymerase chain reaction (qRT-PCR) as described by Tokar et al. (2010). Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA) and purified using RNeasy Mini Kit columns (Qiagen, Valencia, CA) according to the manufacturer’s instructions. The purified RNA was reverse transcribed with MuLV (Moloney murine leukemia virus) (ABgene, Rockford, IL) reverse transcriptase and oligo-DT primers (ABgene, Rockford, IL). Absolute SYBR Green ROX Mix (ABgene, Rockford, IL) was used to measure mRNA levels. The cycle threshold times (CT) were normalized to β-actin from the same sample based on the control representing 100%. Primers were designed using Primer Express 3.0 software (Carlsbad, CA) and synthesized by Sigma Chemical Company (St. Louis, MO). To examine the cDNA we used the Bio-Rad MyiQ qRT-PCR system and quantitated the relative gene expression using the comparative CT method (2−ΔΔCT) (Schmittgen and Livak, 2008). A list of gene and primer sequences used for real time RT-PCR can be found in Supplementary Table 1.
Zymographic analysis
Increased matrix metalloproteinase (MMP) activity often reflects the acquisition of a malignant phenotype. Secreted MMP-2 activity was measured by a standard zymographic method adapted from Tokar et al. (2010). Cells were cultured in basal medium (without serum or supplements) for 48 h from which conditioned medium was collected and assayed for MMP-2 activity. All groups were assessed in triplicate.
Colony formation
Soft agar colony formation was assayed by the method of Masuda et al. (1997). A 0.5% solution of Difco Noble agar (BD, Franklin Lakes, NJ) was allowed to solidify in 35-mm sterile culture dishes. A 1 ml suspension of HPL-1D cells, 1.25 × 104 cells/ml, was then layered on top of the solidified agar in a concentration of 0.33% agar. The colonies were counted using Interscience Scan 300 colony counter after an incubation period of 21 days at 37 °C. All groups were assessed in triplicate.
Invasion
Invasiveness of cells was quantified using a modified Boyden chamber assay adapted from Tokar et al. (2010). A total of 2 × 105 cells were layered onto Matrigel-coated membranes in basal serum free media. A 10% concentration of fetal bovine serum in the culture media was used as the chemo-attractant. The cells were incubated at 37 ° C for 48 h. Following incubation, membranes were fixed and stained using the Hema 3 Stain set (Fisher Scientific, Pittsburg, PA). The stain was extracted using 0.1% HCl. The absorbance of the extracted stain was measured using an iMark microplate absorbance reader (Hercules, CA) at a wavelength of 595 nm. All groups were assessed in triplicate.
Western blot
The M-PER extraction reagent (Pierce, Rockford, IL) was used to isolate cellular protein. The proteins were electrophoresed using NuPAGE 4–12% Bis-Tris gels (Invitrogen, Carlsbad, CA), transferred onto PVDF, polyvinyl difluoride membranes, and immunoblotted with p16, K-RAS, N-RAS (Santa Cruz Biotechnology, Santa Cruz, CA), Vimentin and β-actin (Sigma, St. Louis, MO), and SLC38A3 (Abcam, Boston, MA), antibodies followed by horseradish peroxidase-conjugated anti-mouse, anti-rabbit or anti-goat secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) as appropriate. Densitometry of individual bands was carried out using ImageJ (version 1.24; National Institutes of Health 2007). PVDF membranes were limited to three stripping applications prior to immunoblotting with β-actin for quantitation.
Oxidative DNA damage (ODD) using IST
ODD was measured in the HPL-1D cells at 20 weeks by the IST method (Ramirez et al., 2007). The IST method measures DNA radical formation in situ. Cells are exposed to DMPO which results in the conversion of DNA radicals to stable nitrone adducts, followed by DNA isolation and finally quantification of DNA adducts.
Immunofluorescence
HPL-1D cells were washed in DPBS and fixed with acetone and methanol (1:1). The cells were stained, blocked with normal horse serum, and followed by incubations in primary and secondary antibodies for Vimentin. The secondary antibody was conjugated with Alexa Fluor 488. Cells were then stained with DAPI. An automated Olympus inverted fluorescence microscope was used to capture photomicrographs. CellSens software (Olympus Corporation) was used to process the images for all groups.
Cadmium biokinetics
Cellular accumulation of cadmium was measured in control and cadmium treated HPL-1D cells. Treated and passaged-matched cells from weeks 4, 12 and 20 were thawed from liquid nitrogen stocks and plated at a concentration of 2 × 106 and allowed to grow for 1 week in cadmium-free media. The medium was changed three times to ensure removal of any residual cadmium. To measure cadmium accumulation cells were re-suspended and then, 1 × 106 cells were then plated and incubated in fresh cadmium-free medium for 24 h. After 24 h, cells were exposed to 5 μM cadmium for an additional 24 h. At the end of incubation, medium was removed and cells were rapidly washed 3 times with DPBS to remove loosely bound surface cadmium and harvested, digested and processed for internal cadmium accumulation assessment. Nitric acid was used to digest the cell suspensions followed by heating at 70 °C overnight. Cadmium was determined in diluted digests using graphite furnace atomic absorption spectrophotometry (Perkin-Elmer AAnalyst100, Norwalk, CT) and data were adjusted to pg/106 cells.
Statistical analysis
Data represent mean values ± SEM. A Student’s t-test was used to quantify differences between cadmium treated cells compared with control cells in all cases. The tests were two-tailed with significance set at p ≤ 0.05. All groups represent a minimum of three or more separate samples.
Results
Cytolethality of cadmium
HPL-1D cells were exposed to varying concentrations of cadmium to determine the lethal concentration in 50% of the cells (LC50) over 72 h. The LC50 for cadmium in HPL-1D cells was determined to be 13 μM. Thus 5 μM was selected for chronic testing, which was non-toxic.
HPL-1D cells exhibit cancer cell characteristics after chronic cadmium treatment
In an attempt to develop an in vitro model of cadmium-induced lung carcinogenesis, HPL-1D cells were exposed chronically to 5 μM cadmium. MMP-2, an enzyme commonly secreted by tumor cells to degrade the extracellular matrix and assist in local invasion or metastasis, was assessed as a marker of acquired oncogenic phenotype. Significant elevations of secreted MMP-2 activity occurred as a consequence of chronic cadmium exposure (Fig. 1A). After 20 weeks of cadmium exposure, secreted MMP-2 activity was increased over 3.5-fold of control. Increases in the levels of MMP activity of this magnitude are commonly associated with cadmium-induced acquired oncogenic phenotype (Achanzar et al., 2001; Benbrahim-Tallaa et al., 2009; Qu et al., 2012b). Increased in vitro invasive capacity of cells is also commonly associated with an acquired cancer phenotype. The invasive capacity of these chronic cadmium treated human lung cells was also markedly increased by more than 3-fold after 20 weeks of low-level cadmium exposure (Fig. 1B). We also examined the ability of the HPL-1D cells to form anchorage-independent colonies in soft agar (Fig. 1C). There was a 1.7-fold increase in colony formation induced by chronic exposure to cadmium. Enhanced colony formation is typically associated with acquisition of cancer phenotype (Tokar et al. 2010). Based on elevated secreted MMP-2 activity, increased invasive capacity, and colony formation it appeared that by 20 week cadmium treatment had induced multiple cancer characteristics in these lung epithelial cells. To acknowledge this likelihood and distinguish these cells from control cells, these cells exposed to cadmium for 20 weeks are henceforth designated as chronic cadmium treated lung cells (CCT-LC).
Fig. 1.
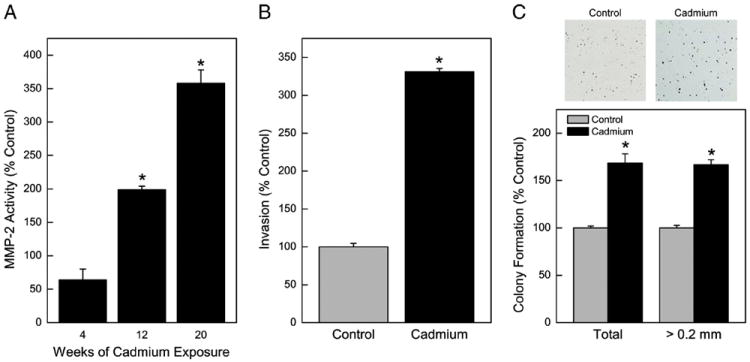
Chronic exposure to cadmium (5 μM) and acquired a cancer malignant phenotype in HPL-1D cells. (A) Zymographic analysis of secreted MMP-2 activity over time. (B) Invasiveness at 20 weeks of exposure. (C) Colony formation at 20 weeks of exposure. Note top: Picture of stained colonies. Data represent the mean ± SEM (n = 3). Treated data were compared in all cases with passage-matched controls. A p ≤0.05 was considered significant. An asterisk (*) indicates significance from control.
Indeed, when cell proliferation was assessed over one week, CCT-LC cells showed marked increases at later time points (Fig. 2A). CCT-LC cells also grew better in serum free media, i.e., in the absence of exogenous growth factors (Fig. 2B). CCT-LC cells overexpressed cyclin D1, (Fig. 2C), a gene associated with cell proliferation commonly over-expressed in tumors. Thus, CCT-LC cells showed physical and genetic hallmarks of hyperproliferation and autonomous growth that are commonly observed in cancer cells.
Fig. 2.
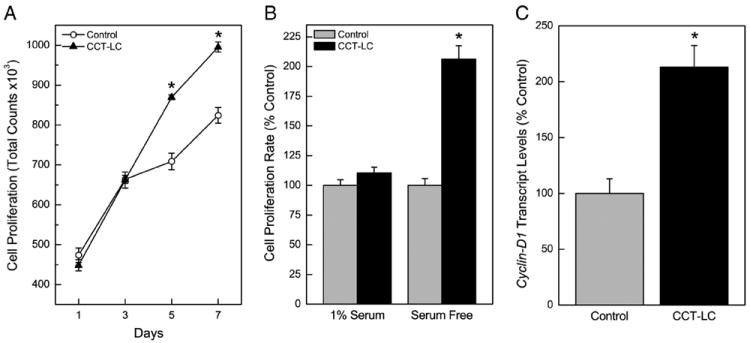
Cell proliferation and cyclin D1 expression in CCT-LC or control cells. (A) Cell proliferation over seven days. (B) Cell proliferation in serum free media. (C) Relative expression of Cyclin D1 transcript. Data represent the mean ± SEM (n = 3). Treated data were compared in all cases with passage-matched controls. A p ≤0.05 was considered significant. An asterisk (*) indicates significance from control.
Selected tumor suppressors and lung adenocarcinoma oncogenes in CCT-HL cells
The LUCA (lung cancer) region of chromosome 3p21.31 contains a protein encoded by the SLC38A3 gene which is considered to be a tumor suppressor gene (Ding et al., 2008; Kholodnyuk et al., 2006). Expression of the SLC38A3 protein was markedly reduced in CCT-LC cells by over 50% (Fig. 3A). Similarly, p16 protein expression was reduced in CCT-LC cells to only 34% of control. The p16 gene serves a critical role by preventing abnormal growth and is similarly considered a tumor suppressor gene (Liggett and Sidransky, 1998). The inactivation of p16 is a common event in lung oncogenesis. In CCT-HL cells p16 transcript was markedly suppressed to 39% of control levels (not shown), as would be consistent with a lung cell acquiring a malignant phenotype. Likewise, K-RAS and N-RAS proteins, translation products of oncogenes that regulate cell proliferation, showed robust increases in CCT-LC cells of up to 476% compared to control cells (Fig. 3B). Epithelial-to-mesenchymal (EMT) transition is common when epithelial cells undergo malignant transformation and can be detected morphologically as well as by altered gene expression (Kalluri and Weinberg, 2009; Thiery et al., 2009; van Zijl et al., 2009). EMT appeared to occur in CCT-LC cells based on expression of the mesenchymal marker, Vimentin protein (Fig. 3C) at levels (~350%) much higher than control cells by quantitative western blot. This corresponded with morphological evidence of EMT (Fig. 3C; Top) of long spindle-shaped cells in CCT-LC cells. Immunostaining for Vimentin confirmed it was primarily cytoplasmic and showed intense expression in CCT-LC cells (Fig. 3D). Thus, cadmium exposure causes acquisition of molecular characteristics typical in general for cancer cells and often seen specifically with lung cancer cells.
Fig. 3.
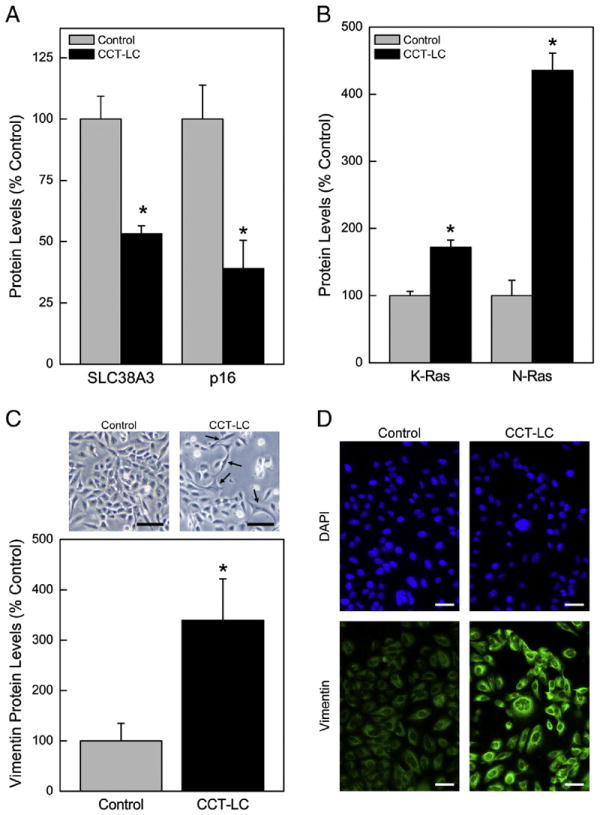
Levels of cancer relevant gene expression in CCT-LC or control cells. (A) Quantitative expression of tumor suppressor gene proteins SLC38A3 and p16. (B) Expression of the oncoproteins KRAS and NRAS. (C) Expression of the EMT marker Vimentin in CCT-LC cells. Inset: Cell morphology at 20 weeks, Bar, 50 μM. (D) Immunostaining of Vimentin, Bar, 50 μM. A p ≤0.05 was considered significant. An asterisk (*) indicates significance from control.
MT, oxidant stress genes, and oxidative DNA damage
MTs avidly bind cadmium, and will provide protection at least from acute toxicity. It also appears MT may be generally over-expressed in lung cancers. Following low-level, chronic cadmium exposure, the major MT isoforms, MT-1A and MT-2A were measured and shown to be elevated in CCT-LC cells more than 1.5-fold above control levels (Fig. 4A). The overabundance of MT-1A and MT-2A suggests that MTs were produced in response to cellular cadmium exposure as an adaptive response.
Fig. 4.
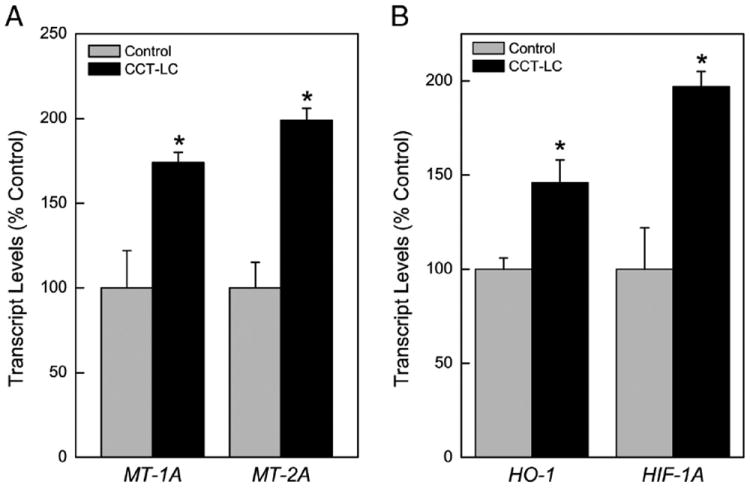
Expression of MT isoforms and Oxidant Stress Response Genes. (A) MT isoforms MT-1A and MT-2A. (B) HO-1 and HIF-1α. Values represent the mean ± SEM (n = 3) as determined by quantitative real time RT-PCR normalized to β-actin. All cells were compared to passage-matched control cells with p ≤0.05 considered significant. An asterisk (*) indicates significance from control.
The exposure of cells to cadmium can induce oxidative stress which in turn will induce oxidant stress response genes. Both Heme Oxygenase-1 (HO-1) and Hypoxia Inducible Factor-1A (HIF-1A) mRNA levels were measured to assess the oxidative stress response in CCT-LCs. HO-1 was modestly (~50%) increased in CCT-LC cells (Fig. 4B). HIF-1A was increased in CCT-LC cells by nearly 2-fold (Fig. 4B). CCT-LC cells did not show any evidence of increased oxidative DNA damage during acquisition of cancer phenotype (not shown).
Cadmium accumulation in CCT-LC cells
Cadmium accumulation in weeks 4, 12 and 20 over a 24 hour period was determined in cells using atomic absorption spectroscopy. Cadmium accumulation was highest in the earlier weeks when compared to control cells (Fig. 5A). This ability of cells to bioaccumulate less cadmium once transformed at 20 weeks occurred despite the fact that these cells express more MT at this point which would bind cadmium (see Fig. 4A). Taken together these data suggest that the transformed CCT-LC cells may have developed biokinetic alterations in such a way as to reduce cadmium intoxication, but despite this become transformed.
Fig. 5.
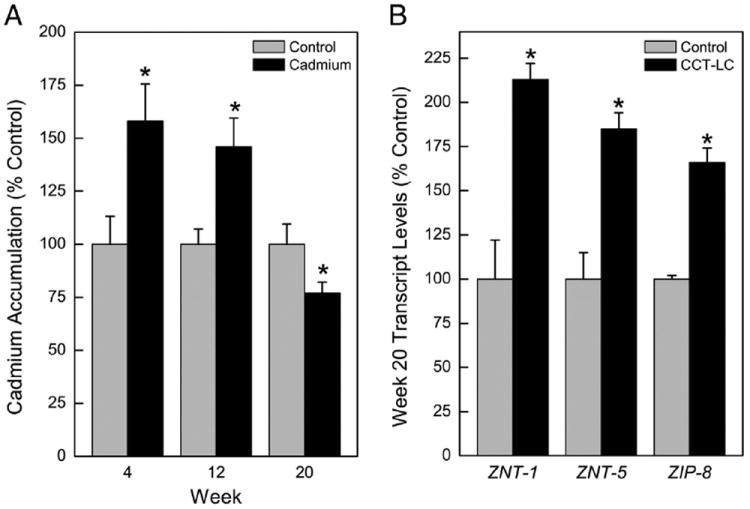
Determination of metal accumulation and transporters in CCT-LCs and control cells. (A) Accumulation of cadmium. See Methods for details. (B) Expression of metal transporters ZNT-1, ZIP-5, and ZIP-8. Data represent the mean ± SEM (n = 3). Real time RT-PCR data were normalized to β-actin. A p ≤0.05 was considered significant. An asterisk (*) indicates significance from control.
The metal ion transporter SLC30 (ZNT) family is widely known to facilitate the efflux of zinc (Palmiter and Huang, 2004; Sekler et al., 2007) and functions to maintain metal homeostasis in the cell either by transporting zinc ions directly out of the cell from the cytoplasm (ZNT-1) or within the cell between intracellular compartments (other ZNTs) (Urani et al., 2010). ZNTs can be induced by cadmium and aid in cellular resistance to metal accumulation (Urani et al., 2010). Thus, ZNT-1 and ZNT-5 expression levels were assessed in CCT-LC cells and found to be elevated by 2.0 and 1.6-fold, respectively (Fig. 5B). Moreover, the SLC39 family members (ZIPs) facilitate zinc influx and Zrt/Irt-like Protein-8 (ZIP8), enhances cadmium cytolethality by greatly increasing cadmium influx (Dalton et al., 2005). ZIP-8 showed a 1.7-fold increase in CCT-LC cells (Fig. 5B). However, overall, the pattern of expression of these influx/efflux transporters did not clearly account for reduced acute accumulation of cadmium in CCT-LC cells (see Fig. 5A).
Discussion
The mechanisms of cadmium-induced pulmonary carcinogenesis are incompletely defined and the development of target specific cellular models will greatly aid in defining the mode of action. In the present study, exposure to chronic, low-level cadmium induced multiple physical and molecular tumor cell characteristics in a human lung epithelial cell line producing the CCT-LC cell line. These cadmium-induced characteristics included increased MMP-2 activity, invasion, colony formation in soft agar, autonomous growth, EMT, and hyperproliferation, all of which are qualities that are consistent with a epithelial cancer cell phenotype (Achanzar et al., 2001; Benbrahim-Tallaa et al., 2009; Qu et al., 2012b; Tokar et al., 2010). In addition, CCT-LC cells showed a loss of p16 expression, and an increase in cyclin D1 expression, which are common in rapidly proliferating tumor cells (Demirhan et al., 2010; Jin et al., 2001) and typical for lung cancers (Suzuki et al., 2012). CCT-LC cells also showed over-expression of K-RAS, N-RAS, Vimentin, loss of SLC38A3 which again are either typical for a general cancer phenotype and/or typical specifically for lung cancers (Ding et al., 2008; Kholodnyuk et al., 2006; Suzuki et al., 2012). The over-expression of major MT isoforms is associated with both cadmium (Qu et al., 2006) and lung cancers (Eckschlager et al., 2009). These results mesh nicely with recent studies showing that human immortalized bronchial epithelial cells can be transformed by cadmium (Jing et al., 2012; Zhou et al., 2012). Thus, these present data further support the association between lung cancer and prolonged exposure to cadmium, and provide a model to study events in human lung cancer at the cellular level. It is thought that adenocarcinomas of the lung, that are common with cadmium exposure (IARC, 1993, 2012), arise from the epithelia of the peripheral lung (Masuda et al., 1997), as would be consistent with the cell model developed in the present study.
Lung cancer is one of the most prevalent cancers in the world (ACS, 2012), and evidence has shown that the lung is a key target of cadmium carcinogenesis (IARC, 1993, 2012). The evidence linking cadmium to human lung cancer is supported by epidemiological and experimental animal studies which support cadmium causation of malignant lung tumor formation (IARC, 1993, 2012). Importantly, in rodent inhalation studies, a variety of cadmium compounds can produce dose-dependent increases in pulmonary adenocarcinomas (IARC, 1993, 2012). Based on the carcinogenic activity of a wide variety of cadmium compounds, it is thought that it is the ionic form of cadmium that is the active, carcinogenic species (IARC, 2012). Thus, the carcinogenic potential of a given cadmium compound would depend on the degree to which that compound releases ionic cadmium under the conditions of exposure. Smoking and/or inhaled pollutants contaminated with cadmium will create a significant lung target cell burden in humans (Verougstraete et al., 2003), and the portion of this burden that is bio-available is critical to oncogenic outcome. It is clear from the present study that despite significant induction of adaptive mechanisms that would likely reduce “free” cellular cadmium, such as MT (via sequestration) or efflux transporters or adaptations that would deal with the consequences of free cadmium ion (such as oxidative stress), sufficient metal was still bioavailable to stimulate acquisition of cancer cell characteristics.
Matrix metalloproteinases are zinc-dependent enzymes that function in degradation of extracellular matrix components and basement membranes (Stetler-Stevenson et al., 1996). There are clear correlations between tumor development or cellular malignant transformation and increased secretion of various MMPs, including MMP-2 (Achanzar et al., 2001; Benbrahim-Tallaa et al., 2009; Coussens et al., 2002; Qu et al., 2012b; Stetler-Stevenson et al., 1996). This includes increased MMP-2 secretion and malignant transformation by cadmium in various human epithelial cells such as prostate (Achanzar et al., 2001), breast (Benbrahim-Tallaa et al., 2009), and pancreas (Qu et al., 2012b). A meta-analysis of numerous cohort studies of lung cancer patients concluded that lung cancer patients with high MMP-2 activity showed decreased survival (Wang and Cai, 2012), indicating that MMP-2 is linked to lung tumor aggressiveness.
MMP-2 activity can also increase as a result of oxidant stress (Cucu et al., 2011). This is consistent with the present data indicating an oxidant stress response occurred with cadmium in CCT-LC cells. In rats, MMP-2 activity is increased in the lung interstitium and alveolar epithelium following exposure to 100% oxygen, indicating a possible role in lung pathogenesis following an oxidative stress event (Pardo et al., 1996). Recently we have found that cadmium is able to produce oxidative DNA damage in cells in a fashion similar to chemical oxidants, particularly in sensitive cells such as those unable to produce the major isoforms of MT (Qu et al., 2012a). Our present findings demonstrate that HO-1 and HIF-1A are both activated when the CCT-LC cells have acquired multiple tumor cell characteristics, potentially indicating adaptive responses to oxidative conditions. HO-1 is considered an antioxidative protein and is inducible by cadmium exposure (Cucu et al., 2011). HIF-1 can be induced by cadmium through production of reactive oxygen species (Jing et al., 2012). Others have found that HIF-1 expression occurs through cadmium-induced reactive oxygen species formation with transformation of bronchial epithelial cells (Jing et al., 2012). Thus, evidence indicates that oxidant stress occurred during the acquisition of cancer cell characteristics induced by cadmium in lung cells, although the absence of oxidative DNA damage in the present study indicates that this did not have a direct effect on genetic material. Indeed, the work of Zhou et al. (2012) showing that aberrant methylation of DNA repair genes is a key factor in cadmium-induced transformation of bronchial epithelial cells indicating that epigenetic factors play an important role at least in some cases of cadmium transformation.
MT has been noted as a prognostic biomarker in various malignancies such as breast (Pedersen et al., 2009; Surowiak et al., 2005) and prostate (Elghany et al., 1990; Pedersen et al., 2009) as well as in lung cancer (Werynska et al., 2011). MT expression is frequently observed in lung carcinoma tissue samples from human patients (Eckschlager et al., 2009). It has previously been shown that epithelial barrier disruption following low-dose exposure to cadmium in airway epithelia results in elevated MT expression (Forti et al., 2010). Elevated MT levels are observed in the present study in CCT-LC cells when they acquired cancer cell characteristics with chronic cadmium exposure, supporting the association between MT and malignant lung phenotype. It is also well known that cadmium can directly stimulate production of MT both in vivo and in vitro. Pulmonary adaptation to cadmium is thought to play a role in cadmium carcinogenesis (Hart et al., 2001). It has been suggested that while cadmium adaption via MT up-regulation may seem protective, it may also actually predispose the tissue to cancer (Hart et al., 2001). In essence, MT, by sequestering cadmium in a tissue or target cell, would provide a significant long-term residual pool of cadmium within the cell for malignant transformation as the proteins go through multiple cycles of degradation and re-induction (Hart et al., 2001). Thus, what seems to be adaptive may actually contribute to carcinogenesis for cadmium. In point of fact, many cells that we have found sensitive to cadmium-induced transformation nonetheless are quite competent in production of MT (Benbrahim-Tallaa et al., 2009; Qu et al., 2005, 2006).
Transporters such as ZIP-8 which normally influx essential divalent cations, like zinc, have been implicated in cadmium influx (Himeno et al., 2009). In addition, when ZIP-8 is silenced cellular cadmium is markedly reduced (Fujishiro et al., 2012). Our results show that when cadmium-induced an acquired cancer cell phenotype, ZIP-8 levels were markedly increased. However, this presumed increase in ZIP-8 influx of cadmium did not appear to predominate overall cadmium accumulation in CCT-LC cells. Rather, overall cadmium accumulation was reduced with acquired cancer cell characteristics. This could potentially be due to enhanced efflux from the ZNT-1 efflux transporter, which was over-expressed in CCT-LC cells. Therefore, an aspect of cadmium adaptation in CCT-LC cells could be based in altered cadmium biokinetics. Despite these cellular adaptations, the cells still acquired a cancer phenotype. It is important to note that these assessments of ZIP-8, ZNT-1 and cadmium accumulation were carried out in cells after they had acquired multiple cancer characteristics. Derangements in transport that would have occurred during the earlier parts of the transformation process could have been quite distinct from what occurred at end of transformation. It does appear, however, that CCT-LC cells adapted to cadmium concurrent with transformation and part of this adaptation was potentially biokinetic in nature.
Expression of the tumor suppressor gene, p16, is frequently altered in various cancers. For example, in a clinical study of lung tumors, more than 60% showed low p16 levels (Gonzalez-Quevedo et al., 2002). In addition, p16 expression is markedly decreased by cadmium-induced malignant transformation of human prostate epithelial cells (Benbrahim-Tallaa et al., 2007). In the present study CCT-LC cells also showed a marked suppression of p16 expression similar to that seen in lung cancers (Liggett and Sidransky, 1998). The tumor suppressor gene SLC38A3 exhibited similar decreases in protein expression following chronic cadmium exposure (Kholodnyuk et al., 2006). These molecular events support CCT-LC cells as a model for cadmium induction of human lung cancer.
In summary, the present work demonstrates that chronic exposure to low-level cadmium was associated with acquisition of multiple tumor cell characteristics in human peripheral lung epithelial cells, some of which are commonly observed in human lung cancer (i.e., p16 suppression). These data suggest that cadmium may directly impact lung epithelial cells inducing malignant transformation. CCT-LC cells could provide a valuable model for the study of cadmium-induced lung cancer.
Supplementary Material
Acknowledgments
The authors wish to thank Drs. K. Pelch and Y. Sun for their critical comments and Mr. Matt Bell for assistance with figure preparation. This article may be the work product of an employee or group of employees of the National Institute of Environmental Health Sciences (NIEHS), National Institutes of Health (NIH), however, the statements, opinions or conclusions contained therein do not necessarily represent the statements, opinions or conclusions of NIEHS, NIH or the United States government.
Abbreviations
- BSA
bovine serum albumin
- CCT-LC
chronic cadmium treated-lung cells
- DAPI
4′,6-diamidino-2-phenylindole
- DMPO
5,5-dimethyl-1-pyrroline N-oxide
- HPL
human peripheral lung
- HIF-1A
hypoxia inducible factor-1 alpha
- HO-1
heme oxygenase-1
- INT
p-iodonitro-tetrazolium violet
- IST
immuno-spin trapping
- LC50
lethal concentration 50
- LUCA
lung cancer
- MMP-2
matrix metalloproteinase-2
- MT-1A
metallothionein-1A
- MT-2A
metallothionein-2A
- PBS
phosphate buffered saline
- qRT-PCR
quantitative real time reverse transcription polymerase chain reaction
- ZIP
Zrt/Irt-like Proteins
- ZNT
Zinc Transporter
Footnotes
Conflict of interest
No competing of interests.
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.taap.2013.06.013.
References
- Achanzar WE, Diwan BA, Liu J, Quader ST, Webber MM, Waalkes MP. Cadmium-induced malignant transformation of human prostate epithelial cells. Cancer Res. 2001;61:455–458. [PubMed] [Google Scholar]
- ACS. American Cancer Society Cancer Facts and Figures. Atlanta: 2012. [January 10, 2013]. www.cancer.org/research/cancerfactsfigures/acspc-031941. [Google Scholar]
- Amara S, Abdelmelek H, Garrel C, Guiraud P, Douki T, Ravanat JL, Favier A, Sakly M, Ben Rhouma K. Preventive effect of zinc against cadmium-induced oxidative stress in the rat testis. J Reprod Dev. 2008;54:129–134. doi: 10.1262/jrd.18110. [DOI] [PubMed] [Google Scholar]
- Benbrahim-Tallaa L, Liu J, Webber MM, Waalkes MP. Estrogen signaling and disruption of androgen metabolism in acquired androgen-independence during cadmium carcinogenesis in human prostate epithelial cells. Prostate. 2007;67:135–145. doi: 10.1002/pros.20479. [DOI] [PubMed] [Google Scholar]
- Benbrahim-Tallaa L, Tokar EJ, Diwan BA, Dill AL, Coppin JF, Waalkes MP. Cadmium malignantly transforms normal human breast epithelial cells into a basal-like phenotype. Environ Health Perspect. 2009;117:1847–1852. doi: 10.1289/ehp.0900999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beveridge R, Pintos J, Parent ME, Asselin J, Siemiatycki J. Lung cancer risk associated with occupational exposure to nickel, chromium VI, and cadmium in two population-based case–control studies in Montreal. Am J Ind Med. 2010;53:476–485. doi: 10.1002/ajim.20801. [DOI] [PubMed] [Google Scholar]
- Bruske-Hohlfeld I. Environmental and occupational risk factors for lung cancer. Methods Mol Biol. 2009;472:3–23. doi: 10.1007/978-1-60327-492-0_1. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- Cucu D, D’Haese PC, De Beuf A, Verhulst A. Low doses of cadmium chloride and methallothionein-1-bound cadmium display different accumulation kinetics and induce different genes in cells of the human nephron. Nephron Extra. 2011;1:24–37. doi: 10.1159/000330069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton TP, He L, Wang B, Miller ML, Jin L, Stringer KF, Chang X, Baxter CS, Nebert DW. Identification of mouse SLC39A8 as the transporter responsible for cadmium-induced toxicity in the testis. Proc Natl Acad Sci U S A. 2005;102:3401–3406. doi: 10.1073/pnas.0406085102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirhan O, Tastemir D, Hasturk S, Kuleci S, Hanta I. Alterations in p16 and p53 genes and chromosomal findings in patients with lung cancer: fluorescence in situ hybridization and cytogenetic studies. Cancer Epidemiol. 2010;34:472–477. doi: 10.1016/j.canep.2010.03.018. [DOI] [PubMed] [Google Scholar]
- Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, Fulton L, Fulton RS, Zhang Q, Wendl MC, Lawrence MS, Larson DE, Chen K, Dooling DJ, Sabo A, Hawes AC, Shen H, Jhangiani SN, Lewis LR, Hall O, Zhu Y, Mathew T, Ren Y, Yao J, Scherer SE, Clerc K, Metcalf GA, Ng B, Milosavljevic A, Gonzalez-Garay ML, Osborne JR, Meyer R, Shi X, Tang Y, Koboldt DC, Lin L, Abbott R, Miner TL, Pohl C, Fewell G, Haipek C, Schmidt H, Dunford-Shore BH, Kraja A, Crosby SD, Sawyer CS, Vickery T, Sander S, Robinson J, Winckler W, Baldwin J, Chirieac LR, Dutt A, Fennell T, Hanna M, Johnson BE, Onofrio RC, Thomas RK, Tonon G, Weir BA, Zhao X, Ziaugra L, Zody MC, Giordano T, Orringer MB, Roth JA, Spitz MR, Wistuba II, Ozenberger B, Good PJ, Chang AC, Beer DG, Watson MA, Ladanyi M, Broderick S, Yoshizawa A, Travis WD, Pao W, Province MA, Weinstock GM, Varmus HE, Gabriel SB, Lander ES, Gibbs RA, Meyerson M, Wilson RK. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckschlager T, Adam V, Hrabeta J, Figova K, Kizek R. Metallothioneins and cancer. Curr Protein Pept Sci. 2009;10:360–375. doi: 10.2174/138920309788922243. [DOI] [PubMed] [Google Scholar]
- Eide DJ. The SLC39 family of metal ion transporters. Pflugers Arch. 2004;447:796–800. doi: 10.1007/s00424-003-1074-3. [DOI] [PubMed] [Google Scholar]
- Eide DJ. Zinc transporters and the cellular trafficking of zinc. Biochim Biophys Acta. 2006;1763:711–722. doi: 10.1016/j.bbamcr.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Elghany NA, Schumacher MC, Slattery ML, West DW, Lee JS. Occupation, cadmium exposure, and prostate cancer. Epidemiology. 1990;1:107–115. doi: 10.1097/00001648-199003000-00005. [DOI] [PubMed] [Google Scholar]
- Forti E, Bulgheroni A, Cetin Y, Hartung T, Jennings P, Pfaller W, Prieto P. Characterisation of cadmium chloride induced molecular and functional alterations in airway epithelial cells. Cell Physiol Biochem. 2010;25:159–168. doi: 10.1159/000272060. [DOI] [PubMed] [Google Scholar]
- Fujishiro H, Yano Y, Takada Y, Tanihara M, Himeno S. Roles of ZIP8, ZIP14, and DMT1 in transport of cadmium and manganese in mouse kidney proximal tubule cells. Metallomics. 2012;4:700–708. doi: 10.1039/c2mt20024d. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Quevedo R, Iniesta P, Moran A, de Juan C, Sanchez-Pernaute A, Fernandez C, Torres A, Diaz-Rubio E, Balibrea JL, Benito M. Cooperative role of telomerase activity and p16 expression in the prognosis of non-small-cell lung cancer. J Clin Oncol. 2002;20:254–262. doi: 10.1200/JCO.2002.20.1.254. [DOI] [PubMed] [Google Scholar]
- Guerinot ML. The ZIP family of metal transporters. Biochim Biophys Acta. 2000;1465:190–198. doi: 10.1016/s0005-2736(00)00138-3. [DOI] [PubMed] [Google Scholar]
- Hart BA, Potts RJ, Watkin RD. Cadmium adaptation in the lung — a double-edged sword? Toxicology. 2001;160:65–70. doi: 10.1016/s0300-483x(00)00436-4. [DOI] [PubMed] [Google Scholar]
- He X, Chen MG, Ma Q. Activation of Nrf2 in defense against cadmium-induced oxidative stress. Chem Res Toxicol. 2008;21:1375–1383. doi: 10.1021/tx800019a. [DOI] [PubMed] [Google Scholar]
- He L, Wang B, Hay EB, Nebert DW. Discovery of ZIP transporters that participate in cadmium damage to testis and kidney. Toxicol Appl Pharmacol. 2009;238:250–257. doi: 10.1016/j.taap.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himeno S, Yanagiya T, Fujishiro H. The role of zinc transporters in cadmium and manganese transport in mammalian cells. Biochimie. 2009;91:1218–1222. doi: 10.1016/j.biochi.2009.04.002. [DOI] [PubMed] [Google Scholar]
- Huff J, Lunn RM, Waalkes MP, Tomatis L, Infante PF. Cadmium-induced cancers in animals and in humans. Int J Occup Environ Health. 2007;13:202–212. doi: 10.1179/oeh.2007.13.2.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IARC. International Agency for Research on Cancer. Cadmium and cadmium compounds. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Vol. 58. Lyon, France: 1993. pp. 119–237. [PMC free article] [PubMed] [Google Scholar]
- IARC. International Agency for Research on Cancer. A review of human carcinogens: arsenic, metals, fibres, and dusts. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. 100C. Lyon, France: 2012. pp. 121–141. [PMC free article] [PubMed] [Google Scholar]
- Jin M, Inoue S, Umemura T, Moriya J, Arakawa M, Nagashima K, Kato H. Cyclin D1, p16 and retinoblastoma gene product expression as a predictor for prognosis in non-small cell lung cancer at stages I and II. Lung Cancer. 2001;34:207–218. doi: 10.1016/s0169-5002(01)00225-2. [DOI] [PubMed] [Google Scholar]
- Jing Y, Liu LZ, Jiang Y, Zhu Y, Guo NL, Barnett J, Rojanasakul Y, Agani F, Jiang BH. Cadmium increases HIF-1 and VEGF expression through ROS, ERK, and AKT signaling pathways and induces malignant transformation of human bronchial epithelial cells. Toxicol Sci. 2012;125:10–19. doi: 10.1093/toxsci/kfr256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kholodnyuk ID, Kozireva S, Kost-Alimova M, Kashuba V, Klein G, Imreh S. Down regulation of 3p genes, LTF, SLC38A3 and DRR1, upon growth of human chromosome 3-mouse fibrosarcoma hybrids in severe combined immunodefficiency mice. Int J Cancer. 2006;119:99–107. doi: 10.1002/ijc.21794. [DOI] [PubMed] [Google Scholar]
- Klaassen CD, Liu J, Diwan BA. Metallothionein protection of cadmium toxicity. Toxicol Appl Pharmacol. 2009;238:215–220. doi: 10.1016/j.taap.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liggett WH, Jr, Sidransky D. Role of the p16 tumor suppressor gene in cancer. J Clin Oncol. 1998;16:1197–1206. doi: 10.1200/JCO.1998.16.3.1197. [DOI] [PubMed] [Google Scholar]
- Liu J, Kadiiska MB, Corton JC, Qu W, Waalkes MP, Mason RP, Liu Y, Klaassen CD. Acute cadmium exposure induces stress-related gene expression in wild-type and metallothionein-I/II-null mice. Free Radic Biol Med. 2002;32:525–535. doi: 10.1016/s0891-5849(01)00826-7. [DOI] [PubMed] [Google Scholar]
- Liuzzi JP, Cousins RJ. Mammalian zinc transporters. Annu Rev Nutr. 2004;24:151–172. doi: 10.1146/annurev.nutr.24.012003.132402. [DOI] [PubMed] [Google Scholar]
- Masuda A, Kondo M, Saito T, Yatabe Y, Kobayashi T, Okamoto M, Suyama M, Takahashi T, Takahashi T. Establishment of human peripheral lung epithelial cell lines (HPL1) retaining differentiated characteristics and responsiveness to epidermal growth factor, hepatocyte growth factor, and transforming growth factor beta1. Cancer Res. 1997;57:4898–4904. [PubMed] [Google Scholar]
- NTP. National Toxicology Program, 12th Report on Carcinogens, Cadmium and Cadmium Compounds. 12. Department of Health and Human Services, Research; Triangle Park, NC: 2011. pp. 80–83. [Google Scholar]
- Palmiter RD, Huang L. Efflux and compartmentalization of zinc by members of the SLC30 family of solute carriers. Pflugers Arch. 2004;447:744–751. doi: 10.1007/s00424-003-1070-7. [DOI] [PubMed] [Google Scholar]
- Pardo A, Selman M, Ridge K, Barrios R, Sznajder JI. Increased expression of gelatinases and collagenase in rat lungs exposed to 100% oxygen. Am J Respir Crit Care Med. 1996;154:1067–1075. doi: 10.1164/ajrccm.154.4.8887609. [DOI] [PubMed] [Google Scholar]
- Pedersen MO, Larsen A, Stoltenberg M, Penkowa M. The role of metallothionein in oncogenesis and cancer prognosis. Prog Histochem Cytochem. 2009;44:29–64. doi: 10.1016/j.proghi.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Qu W, Diwan BA, Reece JM, Bortner CD, Pi J, Liu J, Waalkes MP. Cadmium-induced malignant transformation in rat liver cells: role of aberrant oncogene expression and minimal role of oxidative stress. Int J Cancer. 2005;114:346–355. doi: 10.1002/ijc.20736. [DOI] [PubMed] [Google Scholar]
- Qu W, Fuquay R, Sakurai T, Waalkes MP. Acquisition of apoptotic resistance in cadmium-induced malignant transformation: specific perturbation of JNK signal transduction pathway and associated metallothionein overexpression. Mol Carcinog. 2006;45:561–571. doi: 10.1002/mc.20185. [DOI] [PubMed] [Google Scholar]
- Qu W, Pi J, Waalkes MP. Metallothionein blocks oxidative DNA damage in vitro. Arch Toxicol. 2012a;87:311–321. doi: 10.1007/s00204-012-0927-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu W, Tokar EJ, Kim AJ, Bell MW, Waalkes MP. Chronic cadmium exposure in vitro causes acquisition of multiple tumor cell characteristics in human pancreatic epithelial cells. Environ Health Perspect. 2012b;120:1265–1271. doi: 10.1289/ehp.1205082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez DC, Gomez-Mejiba SE, Mason RP. Immuno-spin trapping analyses of DNA radicals. Nat Protoc. 2007;2:512–522. doi: 10.1038/nprot.2007.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband WS. ImageJ. Bethesda, MD: U.S. National Institutes of Health; 2013. [July 8, 2013]. 1997-2005. http://rsb.info.nih.gov/ij/ [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Sekler I, Sensi SL, Hershfinkel M, Silverman WF. Mechanism and regulation of cellular zinc transport. Mol Med. 2007;13:337–343. doi: 10.2119/2007-00037.Sekler. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetler-Stevenson WG, Hewitt R, Corcoran M. Matrix metalloproteinases and tumor invasion: from correlation and causality to the clinic. Semin Cancer Biol. 1996;7:147–154. doi: 10.1006/scbi.1996.0020. [DOI] [PubMed] [Google Scholar]
- Surowiak P, Matkowski R, Materna V, Gyorffy B, Wojnar A, Pudelko M, Dziegiel P, Kornafel J, Zabel M. Elevated metallothionein (MT) expression in invasive ductal breast cancers predicts tamoxifen resistance. Histol Histopathol. 2005;20:1037–1044. doi: 10.14670/HH-20.1037. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Oonishi T, Kudo T, Doi H. LKB1, TP16, EGFR, and K-RAS somatic mutations in lung adenocarcinomas from a Chiba Prefecture, Japan cohort. Drug Discov Ther. 2012;6:24–30. [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Tokar EJ, Diwan BA, Waalkes MP. Arsenic exposure transforms human epithelial stem/progenitor cells into a cancer stem-like phenotype. Environ Health Perspect. 2010;118:108–115. doi: 10.1289/ehp.0901059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urani C, Melchioretto P, Gribaldo L. Regulation of metallothioneins and ZnT-1 transporter expression in human hepatoma cells HepG2 exposed to zinc and cadmium. Toxicol in Vitro. 2010;24:370–374. doi: 10.1016/j.tiv.2009.11.003. [DOI] [PubMed] [Google Scholar]
- van Zijl F, Zulehner G, Petz M, Schneller D, Kornauth C, Hau M, Machat G, Grubinger M, Huber H, Mikulits W. Epithelial-mesenchymal transition in hepatocellular carcinoma. Future Oncol. 2009;5:1169–1179. doi: 10.2217/fon.09.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verougstraete V, Lison D, Hotz P. Cadmium, lung and prostate cancer: a systematic review of recent epidemiological data. J Toxicol Environ Health B Crit Rev. 2003;6:227–255. doi: 10.1080/10937400306465. [DOI] [PubMed] [Google Scholar]
- Wang J, Cai Y. Matrix metalloproteinase 2 polymorphisms and expression in lung cancer: a meta-analysis. Tumour Biol. 2012;33:1819–1828. doi: 10.1007/s13277-012-0441-0. [DOI] [PubMed] [Google Scholar]
- Werynska B, Pula B, Muszczynska-Bernhard B, Piotrowska A, Jethon A, Podhorska-Okolow M, Dziegiel P, Jankowska R. Correlation between expression of metallothionein and expression of Ki-67 and MCM-2 proliferation markers in non-small cell lung cancer. Anticancer Res. 2011;31:2833–2839. [PubMed] [Google Scholar]
- WHO. World Health Organization guidelines for drinking water quality, incorporating 1st and 2nd addenda. Recommendations. 3. World Health Organization; 2008. pp. 317–319. [Google Scholar]
- Zhou ZH, Lei YX, Wang CX. Analysis of aberrant methylation in DNA repair genes during malignant transformation of human bronchial epithelial cells induced by cadmium. Toxicol Sci. 2012;125:412–417. doi: 10.1093/toxsci/kfr320. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.