

Abstract
Glial cell line–derived neurotrophic factor (GDNF) has great potential to treat Parkinson's disease (PD). However, constitutive expression of GDNF can over time lead to side effects. Therefore, it would be useful to regulate GDNF expression. Recently, a new gene inducible system using destabilizing domains (DD) from E. coli dihydrofolate reductase (DHFR) has been developed and characterized. The advantage of this novel DD is that it is regulated by trimethoprim (TMP), a well-characterized drug that crosses the blood–brain barrier and can therefore be used to regulate gene expression in the brain. We have adapted this system to regulate expression of GDNF. A C-terminal fusion of GDNF and a DD with an additional furin cleavage site was able to be efficiently regulated in vitro, properly processed and was able to bind to canonical GDNF receptors, inducing a signaling cascade response in target cells. In vivo characterization of the protein showed that it could be efficiently induced by TMP and it was only functional when gene expression was turned on. Further characterization in a rodent model of PD showed that the regulated GDNF protected neurons, improved motor behavior of animals and was efficiently regulated in a pathological setting.
Introduction
Parkinson's disease (PD) is a severe neurodegenerative disease that currently has no cure. Patients suffering from PD have limited treatment options available and conventional surgical or pharmacological treatments are palliative.1 Therefore, there is a need for therapies that can halt disease progression or reverse it. In contrast to current treatments, gene therapy approaches that directly deliver neuroprotective genes could potentially provide better treatments.2 Among the different genes tested in preclinical models, glial cell line–derived neurotrophic factor (GDNF) and related neurotrophic factors have shown the best promise so far.3,4 Although GDNF has very good effects in preclinical models of PD,5,6,7,8,9 it also has known side effects.7,10,11,12,13 When GDNF is constitutively expressed in the brain, it can lead to aberrant sprouting of axons, increased turnover of dopamine and downregulation of tyrosine hydroxylase, the rate-limiting enzyme involved in dopamine production. Taken together, preclinical and clinical trial findings suggest two things. First, that GDNF protein needs to be expressed in the basal ganglia. Second, GDNF protein or gene should be regulated so that the expression of GDNF can be turned on to protect or recover neuronal function and turned off to avoid side effects resulting from constitutive expression. However, current gene-regulation systems have limitations that may preclude their use for gene therapy in the brain.14 Therefore, there is a need to test new inducible systems to regulate therapeutic gene expression in the brain.
In 2006, Banaszynski and colleagues developed a new gene-regulation system based on targeted proteasomal degradation using destabilizing domains (DD), which are unstable mutated proteins prone to be degraded by the proteasome.15 The system operates by fusing a protein of interest to a DD. This fusion will lead the cellular machinery to recognize the fusion protein as unstable, thereby targeting the fusion protein for proteasomal destruction. The DD used in the original study was mutated from the protein FKBP12 and by developing a synthetic molecule that would serve as a ligand for the DD, the authors could specifically and reversibly regulate protein-DD stability in vivo.16
In 2010, a new DD based on dihydrofolate reductase (DHFR) from E. coli was developed.17 The DD was designed so that trimethoprim (TMP), a molecule that inhibits DHFR could be used to stabilize this novel DD. As TMP can cross the blood–brain barrier, the DD based on DHFR mutants could be used to regulate gene expression in the brain. Our group has recently characterized the kinetics of DD regulation when the DD is fused to yellow fluorescence protein (YFP-DD) in the brain.18 We also showed that the DD could be used to regulate GDNF expression in cell culture. However, the secretion rates of this first-generation GDNF DD were limited.
Due to the limitations of the first GDNF DD constructs, second-generation fusion proteins of GDNF to DD were designed and screened in vitro for secretion, functionality, processing and receptor binding ability. The optimal GDNF DD fusion was then tested in vivo, and it was shown that the fusion protein was regulated functional and had a negligible and nonfunctional expression when the DD was not stabilized by TMP. Moreover, the GDNF DD fusion protein was tested in a rodent model of PD, and it was observed that in addition to efficient regulation, the induced expression of GDNF DD fusion protein lead to improvements in motor behavior and protection of substantia nigra pars compacta neurons to levels comparable with wild-type GDNF.
Results
Design and validation of GDNF and DD fusion proteins
For the first-generation fusion proteins of GDNF and DD, the DD was placed either in the N or C terminus of the GDNF coding sequence. Although the N-terminal GDNF DD (DD-G) was able to secrete functional protein in vitro, a subsequent in vivo study where lentiviral vectors expressing DD-G were delivered to the striatum of intact animals revealed that there was impairment in DD-G secretion (data not shown). Signal IP4.0 signal peptide cleavage prediction tool19 was used to design an new N-terminal GDNF DD. The optimal signal peptide cleavage was achieved by placing the DD 9 residues downstream of the signal peptide cleavage site of GDNF (SP9-DD-G) (Figure 1). The optimal design was predicted to have signal peptide cleavage site efficiency comparable with wild-type GDNF.
Figure 1.

Design of first and second-generation DHFR DD and GDNF fusion proteins. Diagram of wild-type GDNF protein showing the signal peptide, precursor protein, furin cleavage site (F), and mature peptide. First-generation DD designs: N-terminal fusion of Y100I DD mutant to GDNF after signal peptide (DD-G) and C-terminal fusion of N18T DD mutant to GDNF (G-DD). Second-generation DD designs: N-terminal fusion of Y100I DD to GDNF 9 residues after signal peptide (SP9-DD-G) and C-terminal fusion of N18T DD mutant to GDNF after an additional furin cleavage site (G-F-DD). DD, destabilizing domains; DHFR, dihydrofolate reductase; GDNF, glial cell line–derived neurotrophic factor.
Previous data showed that only a very small amount of C-terminal GDNF DD (G-DD) was detected by ELISA in the medium of cells transduced with C-terminal DD when the cells were induced with TMP. As GDNF needs to be processed and dimerized before secretion, we hypothesized that placing the DD in the C terminus would disrupt the maturation and dimerization of G-DD. For secreted proteins, the proteasomal checkpoint occurs in the endoplasmic reticulum (ER),20 whereas subsequent modification and processing of secreted proteins with proteases such as furin takes place during trafficking through the Golgi apparatus.21 Therefore, placing an extra GDNF furin cleavage site between the GDNF and the DD moieties (G-F-DD) (Figure 1) could potentially enable DD regulation at the ER and further maturation of G-F-DD in the Golgi apparatus would remove the DD moiety and enable proper processing and folding of G-F-DD. Analysis with ProP 1.0 prediction tool22 indicated that the extra site was functional.
SP9-DD-G and G-F-DD coding sequences were cloned into lentiviral vectors. After production, lentiviral vectors encoding YFP-DD, GDNF (G), SP9-DD-G and G-F-DD were delivered to 293T cells (Figure 2a). After TMP induction, the amount of GDNF in cells (C) and in the culture medium (M) was analyzed by ELISA (Figure 2b). Analysis of intracellular fractions showed that in G, SP9-DD-G and G-F-DD, GDNF was readily detected inside cells. Moreover, there was an intracellular reservoir of SP9-DD-G and G-F-DD even when the samples were not induced by TMP. Analysis of culture media indicated that induction with TMP led to efficient secretion of 38 ng of SP9-G-DD and 22 ng G-F-DD. However, there was also secretion of 10 ng of SP9-DD-G in nontreated 293T cells. By contrast, there was a minimal secretion of 1 ng of G-F-DD in nontreated cells.
Figure 2.
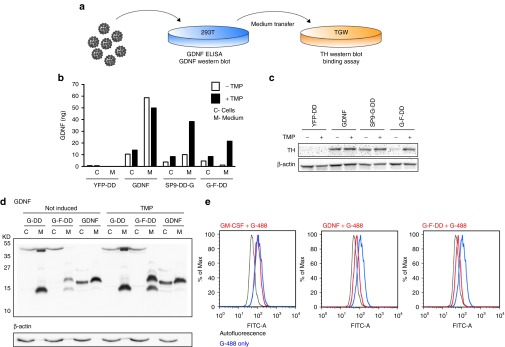
In vitro screening of second-generation DD. (a) To screen for regulation, activity, processing, and binding of second-generation DD, lentiviral vectors were used to transduce 293T cells and the medium was subsequently transferred to TGW cells. (b) GDNF ELISA of medium (M) or intracellular protein (C) from 293T cells transduced at an multiplicity of infection of 2.5 with lentiviral vectors expressing N-terminal fusion of DD to yellow fluorescence protein (YFP-DD), GDNF, SP9-DD-G, or G-F-DD. Five days after transduction, the cells were stimulated with 1 × 10−5 mol/l TMP for 24 hours. (c) Culture media from transduced 293T was transferred to TGW cells and 24 hours after the cells were processed for western blot and probed for TH, using β-actin as loading control. (d) Medium (M) and cells (C) from 293T cells transduced with lentiviral vectors transduced with lentiviral vectors expressing GDNF, G-DD, or G-F-DD and treated with TMP as described above was used for western blot and probed for GDNF. (e) Medium containing 1 × 10−8 mol/l granulocyte monocyte colony stimulating factor (GM-CSF), GDNF or G-F-DD was added to TGW cells together with 2 × 10−8 mol/l GDNF labeled with alexa 488 (G-488). Four hours after the proteins were added, the cells were analyzed by FACS. DD, destabilizing domains; GDNF, glial cell line–derived neurotrophic factor; YFP, yellow fluorescence protein.
To test for functionality, medium of transduced 293T cells was added to TGW cells (Figure 2a). This cell line expresses GDNF receptors endogenously.23 Consequently, treatment of TGW cells with GDNF leads to TH upregulation as shown in the GDNF controls (Figure 2c). Medium of 293T cells transduced with G-F-DD or SP9-DD-G and treated with TMP led to upregulation of TH expression in TGW cells (Figure 2c). These data indicate that SP9-DD-G and G-F-DD present in the medium of TMP treated cells were able to activate GDNF signaling. However, the amount of GDNF present in the medium of untreated 293T cells transduced with SP9-DD-G was also able to upregulate TH expression in TGW cells, indicating that the residual expression of SP9-G-DD when it was not induced is functional and leads to activation of downstream signaling cascades.
Due to the leakage observed with the SP9-DD-G and tight regulation observed with G-F-DD, subsequent analysis and in vitro screening focused on G-F-DD. To determine if the extra furin cleavage site was indeed functional, cells and culture medium of 293T cells expressing G-F-DD was analyzed by western blot (Figure 2d). In cells or medium of 293T cells transduced with G-DD, there is a band of ~40 kD present. Moreover, the medium fraction of these cells contains a band of low molecular weight that is smaller than wild-type GDNF monomer. The 40 kD band could be also readily observed inside of 293T cells transduced with G-F-DD. However, in the medium of G-F-DD cells, there was a band slightly higher than the GDNF monomer and the lower band observed also in G-DD. Comparing G-DD and G-F-DD medium lanes shows that the extra furin cleavage site led to efficient processing of G-F-DD as the 40 kD band corresponding to processed GDNF+DD moieties is substituted with a predominant 17–18 kD band. This is the expected size for the processed G-F-DD monomer containing the mature peptide, linker sequence, and residual furin cleavage site.
To confirm that G-F-DD is able to efficiently bind to GDNF receptors, we established a FACS-based binding assay using TGW cells.24 Treatment of TGW cells with medium containing 2 × 10−8 mol/l GDNF or 2 × 10−8 mol/l secreted G-F-DD from induced 293T cells resulted in a strong displacement of GDNF tagged with Alexa 488 (G-488), indicating that the processed G-F-DD was able to bind efficiently to endogenously expressed GDNF receptors (Figure 2e). The displacement was specific to GDNF and G-F-DD as incubation of TGW with another secreted protein using different receptors, granulocyte/monocyte colony stimulating factor, did not displace G-488 binding. The data obtained in vitro indicate that when G-F-DD expression was induced, G-F-DD was able to be efficiently processed, secreted, bind and elicit a functional signaling response in target cells.
G-F-DD is regulated and functional in vivo
The next step was to determine if G-F-DD could be regulated in the brain. Therefore, lentiviral vectors expressing YFP-DD were injected into the left striatum, whereas lentiviral vectors expressing GDNF (G) or G-F-DD were delivered to the right striatum of Sprague Dawley rats. Two days after lentiviral vector injection, one group of G-F-DD and YFP-DD rats were treated with 0.5 mg/ml TMP in the drinking water (ON) whereas the remaining YFP-DD and G-F-DD rats had normal drinking water (OFF). Three weeks after the treatment was initiated, the animals were euthanized. The brains of animals from all groups were processed for histology. Staining by immunohistochemistry of striatal sections for GDNF showed that in the G and G-F-DD ON group, GDNF could be readily detected in the striatum (Figure 3a). Conversely, the striatum from the G-F-DD OFF group showed only a few stained cells and most of the expression was located to cell soma, suggesting degradation of G-F-DD in the absence of TMP.
Figure 3.
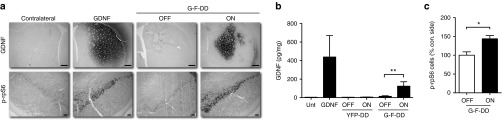
In vivo characterization of G-F-DD regulation and activity. Lentiviral vectors expressing either C-terminal fusion of YFP and DD (YFP-DD), GDNF or G-F-DD were delivered to the striatum of rats. One group of G-F-DD animals were treated with TMP. Three weeks after treatment, part of the YFP-DD and G-F-DD groups were processed for histology and the remaining animals for GDNF ELISA. (a) Immunohistochemistry for GDNF and p-rpS6. Scale bars: first row, 500 µm; second row, 100 µm. (b) GDNF ELISA for untransduced (Unt) GDNF, YFP-DD, and G-F-DD groups. Two-way ANOVA with Bonferroni multiple comparison test (**P < 0.01) (n = 5). (c) Relative quantification of the number of p-rpS6 cells, using the contralateral side as control. Unpaired t-test (*P < 0.02) (n = 3). DD, destabilizing domains; GDNF, glial cell line–derived neurotrophic factor; YFP, yellow fluorescence protein.
As the DD system operates at a posttranscriptional level, determining protein expression levels alone does not indicate efficient regulation, because there will always be a constant translation of mRNA into protein that will then be degraded. As others and we have observed,25 when regulating secreted proteins such as GDNF using DD, the intracellular protein levels do not reflect regulated protein secretion. To confirm G and G-F-DD function in vivo, histological sections containing substantia nigra pars compacta (SNpc) were stained for phosphorylated ribosomal protein S6 expression (p-rpS6), a protein that participates in GDNF signal transduction cascades.26 SNpc contain the population of neurons that project to the striatum and are responsive to GDNF, therefore functional G or G-F-DD expression results in increased phosphorylation of ribosomal protein S6 (Figure 3a).
The striata from G, YFP-DD and G-F-DD animals was dissected and analyzed by GDNF ELISA (Figure 3b). The striata of G animals contained 438 ± 232 pg/mg of GDNF in the brain parenchyma. The striata of YFP-DD OFF and YFP-DD ON animals contained 4.5 ± 0.6 pg/mg tissue and 6.2 ± 1.1 pg/mg tissue of GDNF, respectively. Conversely, the amount of GDNF present in striata of G-F-DD OFF or G-F-DD ON animals was 13.6 ± 5.8 pg/mg tissue and 124 ± 47.4 pg/mg tissue, respectively. Statistical analysis using two-way analysis of variance (ANOVA) indicated that there was a statistical significant increase in GDNF amount only in the striatum of G-F-DD ON animals. Statistical analysis of GDNF ELISA for YFP-DD and G-F-DD in ON and OFF conditions using two-way ANOVA indicated a significant difference in GDNF amounts and the difference was dependent on the type of vector (YFP-DD versus G-F-DD, F = 5.68, P < 0.05) and the treatment given to the animals (water versus TMP, F = 6.95, P < 0.05). Moreover, there was a statistically significant interaction between the type of vector and treatment (F = 5.08, P < 0.05), indicating that the increase in striatal GDNF was dependent on giving TMP to G-F-DD animals.
Moreover, the percentage of p-rpS6–positive cells was quantified in G-F-DD OFF and ON animals, using the contralateral side as control (Figure 3c). In G-F-DD OFF group, there were 100.0 ± 8.9% p-rpS6–positive cells when compared with the contralateral side. Conversely, in the G-F-DD ON group there were 143.8 ± 8.3% p-rpS6–positive cells when compared with the contralateral side. Statistical analysis using t-test showed that the increase in p-rpS6 cells was statistically significant (n = 3, P < 0.02). Densitometric analysis of sections stained for p-rpS6 indicated that in G or G-F-DD ON groups, p-rpS6 levels in SNpc neurons was increased (data not shown). Conversely, the SNpc on the side injected of G-F-DD OFF group showed only background levels of p-rpS6 expression. Histological analysis and GDNF ELISA suggested that G-F-DD expression and function were tightly controlled in vivo by TMP.
G-F-DD regulation in the 6-OHDA rat model of PD improves motor deficits
To determine if G-F-DD expression and induction could be neuroprotective (Figure 4), lentiviral vectors expressing YFP-DD, GDNF, or G-F-DD were delivered to the striatum of Sprague Dawley rats. Two days after vector delivery, the YFP-DD animals and half of the G-F-DD animals were given TMP. Three weeks after vector delivery, the animals were lesioned by striatal delivery of 3 × 7 µg 6-hydroxydopamine (6-OHDA). Behavioral assessments were performed at different timepoints and 6 weeks after lesion the animals were euthanized and their brains processed for histology.
Figure 4.
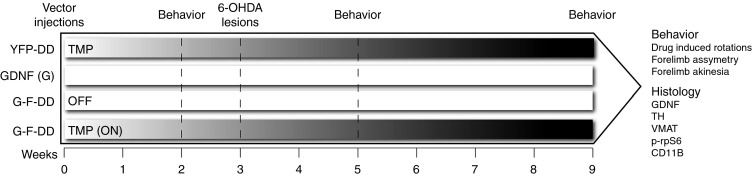
Experimental design of neuroprotection experiment. Lentiviral vectors expressing either YFP-DD, GDNF or G-F-DD were delivered to the striatum of animals. Two days after injection, YFP-DD and half of the G-F-DD group were given 0.5 mg/ml TMP in the drinking water. Two, six, and nine weeks after vector delivery, the behavior of animals was measured by drug-induced rotations. Moreover, 9 weeks after vector delivery, forelimb asymmetry and forelimb akinesia was measured. Three weeks after vector delivery, the animals were lesioned by striatal delivery of 6-OHDA (3 × 7 µg). Six weeks after lesion, the animals were euthanized and their brains analyzed by immunohistochemistry against GDNF, TH, vesicular monoamino transporter 2 (VMAT2) phosphorylated S6 ribosomal protein (p-rpS6 and CD11B expression). DD, destabilizing domains; GDNF, glial cell line–derived neurotrophic factor; YFP, yellow fluorescence protein.
Amphetamine-induced rotations measured throughout the experiment (Figure 5a) indicated that in the YFP-DD and G-F-DD OFF groups there was a significant and progressive degeneration at the last timepoint measured, resulting in 16.5 ± 1.3 rotations/minute and 19.6 ± 3.7 rotations/minute, respectively. Conversely, G and G-F-DD ON groups had 8.2 ± 2.1 rotations/minute and 7.6 ± 2.5 rotations/minute at the last timepoint, respectively. Statistical analysis indicated that there were statistically significant decreases in rotation bias when YFP-DD was compared with G and or G-F-DD ON groups. These findings suggest that there was protection from 6-OHDA of dopamine neurons in the SNpc only in the G and G-F-DD ON groups that was manifested in this behavior test as a rotational bias.
Figure 5.

Behavior tests. To screen for an effect of G-F-DD in the 6-OHDA rat model, several behavior tests were performed. Amphetamine-induced rotations were performed before lesion, 2 weeks after lesion and 6 weeks after lesion to determine progressive behavior impairments (a). At the end of the experiment, the effect of G-F-DD on spontaneous motor behavior was analyzed using forelimb asymmetry test (b) and forelimb akinesia test (c). In the forelimb akinesia test, both forehand and backhand adjustments were measured. One-way ANOVA with Bonferroni multiple comparison test. (n = 10) *P < 0.05, **P < 0.01. DD, destabilizing domains.
Spontaneous motor improvements were measured using the forelimb asymmetry test (Figure 5b) and forelimb akinesia test (Figure 5c) at the last timepoint. In the forelimb asymmetry test YFP-DD and G-F-DD OFF animals only used the left lesioned paw 14 ± 3.6% and 12 ± 2.7% when compared with total paw usage, respectively. Interestingly, G group animals also had a comparable left paw usage of 19 ± 4.5% of total paw usage. The G-F-DD animals had a statistically significant protection of left paw usage, reaching 31 ± 5.5% of total paw usage.
In the forelimb akinesia test, all groups had ~12.4 ± 0.2 backhand adjusting steps and 10.1 ± 0.4 forehand adjusting steps for the right paw. Despite variation, only G-F-DD ON had a statistical significant increase in left hand adjusting steps. In both tests, G-F-DD OFF and G groups showed motor deficits comparable with the YFP-DD group. Only the G-F-DD ON group showed statistically significant motor improvements in both tests.
G-F-DD regulation preserves SNpc neurons and striatal projections
Immunohistochemical analysis of coronal sections stained for TH and vesicular monoamino transporter 2 (VMAT2) (Figure 6a) indicated that although the cell soma of SNpc neurons was preserved only in the G and G-F-DD groups, TH expression in the striatum of G animals was low and comparable with YFP-DD and G-F-DD OFF animals, whereas TH expression in G-F-DD ON animals was higher when compared with the other groups. In contrast, VMAT2 expression in the striatum of both G and G-F-DD ON animals was significantly higher than VMAT2 expression in the YFP-DD and G-F-DD OFF groups.
Figure 6.

Immunohistochemistry, quantification, and densitometric analysis of neuroprotection experiment. Immunohistochemistry for TH and VMAT2 was performed in sections of striatum and substantia nigra pars compacta (SNpc) to estimate degree of neuroprotection 6 weeks after the animals were lesioned with 6-OHDA (a). Scale bars: first three rows, 500 µm; last row, 100 µm. Densitometry was performed on striatal sections to estimate the degree of protection of nigrostriatal neurites, using the contralateral uninjected side as control (b). Neuroprotection of cell bodies was estimated by quantifying he percentages of TH and VMAT2-positive cells in SNpc in comparison with the contralateral uninjected side (c). One-way ANOVA with Bonferroni multiple comparison test (n = 6) *P < 0.05, **P < 0.01, ***P < 0.001.
Densitometric analysis of striatal sections (Figure 6b) confirmed that in the G group, there was a downregulation of TH fiber density to 22 ± 6.2% of uninjected side that contrasted with maintenance of VMAT2 density up to 58 ± 8.8% of uninjected side. The discrepancy in TH versus VMAT2 striatal density indicated TH downregulation.7 In the G-F-DD ON group, TH and VMAT2 fiber density was 50 ± 8.2% and 64 ± 8.7%, respectively. Moreover, G-F-DD ON fiber densities were significantly higher than in the YFP-DD group, which were 17 ± 4.9% for TH and 25 ± 3.0% for VMAT2. Finally, loss of TH and VMAT2 fibers to 27 ± 3.1% and 43 ± 5.2% of uninjected side in the G-F-DD OFF group was comparable with the control YFP-DD group.
Quantification of TH and VMAT2-positive SNpc cells (Figure 6c) indicated that there was a severe loss of cells in the YFP-DD and G-F-DD OFF groups group. Only 25 ± 3.3% TH and 18 ± 2.2% VMAT2-positive cells remained in the YFP group. Comparable 22 ± 2.0% TH and 20 ± 2.7% VMAT2-positive cells remained in the G-F-DD OFF group. In contrast, there was a robust protection of SNpc cells of up to 80 ± 4.2% TH and 60 ± 4.3% VMAT2 in the G group. G-F-DD ON group had a level of protection of 60 ± 10.5% TH and 56 ± 10.5% VMAT2 cells that was comparable with the G group. Both the G and G-F-DD ON cell numbers were significantly higher than either the YFP-DD or the G-F-DD OFF groups, suggesting a significant neuroprotection only in the G and G-F-DD ON groups.
Tight regulation of G-F-DD in 6-OHDA rat model of PD
To determine if the functional protection given by G-F-DD on motor behavior, neuronal projections, and neuronal cell bodies was caused by efficient DD regulation of GDNF in vivo, histological analysis for GDNF and phosphorylated S6 protein was performed. Immunohistochemistry for GDNF (Figure 7a) showed a comparable striatal expression pattern to our previous experiment. Densitometry analysis of GDNF histological sections indicated that there was a significant 4 ± 1-fold increase of GDNF expression in G-F-DD ON group, when compared with the contralateral side. The densitometry analysis also revealed that there was limited 2 ± 1-fold increase in GDNF expression in the G-F-DD OFF group that appeared to be restricted to cell soma (Figure 7b). One-way ANOVA indicated that when comparing G-F-DD ON and G-F-DD OFF groups, only the G-F-DD ON group had a significant increase of GDNF expression (P < 0.05).
Figure 7.

Immunohistochemistry for GDNF expression in striatum. To evaluate the degree of G-F-DD expression and regulation in the 6-OHDA model, immunohistochemistry for GDNF was performed in striatal sections 6 weeks after 6-OHDA lesioning. Low magnification images of GDNF expression in striatum were performed to evaluate the general distribution of GDNF throughout the striatum (a). Scale bar: 500 µm. High magnification images were taken to highlighting the pattern of GDNF expression in the striatum of G-F-DD OFF and G-F-DD ON groups (b). Scale bar: 100 µm. DD, destabilizing domains; GDNF, glial cell line–derived neurotrophic factor.
To evaluate if G-F-DD regulation or expression led to inflammation in the brain, immunohistochemistry for CD11B and densitometry was performed on coronal sections (Supplementary Figure S1). In the YFP-DD group, there was a 1.6 ± 0.3-fold increase in CD11B expression when the lesioned hemisphere was compared with the intact hemisphere. In the G, G-F-DD ON, and G-F-DD OFF groups there was a 2.5 ± 0.3-, 2.5 ± 0.5-, and 2.6 ± 0.5-fold increase in CD11B expression in the ipsilateral intact side, respectively. None of the differences was statistically significant.
To determine if GDNF expression observed in the striatal sections of the different groups was functional under pathological conditions, SNpc sections were stained for p-rpS6. We hypothesized that to result in p-rpS6 staining the GDNF present in the G-F-DD ON or G-F-DD OFF groups had to bind to receptors and activate downstream signaling cascades (Figure 8a). Immunohistochemical analysis for p-rpS6 indicated a severe loss of p-rpS6 cells and consequent p-rpS6 signaling in the YFP-DD and G-F-DD OFF groups in 6-OHDA lesioned brains. Quantification of p-rpS6 cells (Figure 8b) showed that in the G and G-F-DD ON groups there were 144 ± 9% and 77 ± 13% cells remaining. In contrast, there was a significantly lower number of 22 ± 2% and 30 ± 3% p-rpS6–positive cells in the YFP-DD and G-F-DD OFF groups, respectively. The quantification suggested that there was a significant increase of GDNF signaling in the G and G-F-DD ON groups. The severe loss of p-rpS6 cells observed in the YFP-DD group that was comparable with the G-F-DD OFF group, indicated that the residual GDNF expression observed in the G-F-DD OFF group was not sufficient to elicit a functional response in a PD model. In summary, the histological analysis of striatal and nigral sections using several markers indicated that G-F-DD was able to protect neurons to levels comparable with wild-type GDNF and that G-F-DD expression and functional activity was tightly regulated in a PD disease model in vivo.
Figure 8.
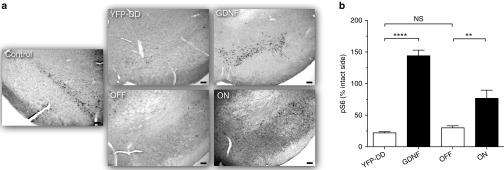
Immunohistochemistry and quantification of p-rpS6 cells in SNpc of a 6-OHDA model. Immunohistochemistry for p-rpS6–positive cells in SNpc was performed to determine if there was any leakage of G-F-DD under neuropathological conditions (a). Scale bars: 100 µm. Quantification of p-rpS6 cells in SNpc was performed, using the contralateral unlesioned side as control (b). One-way ANOVA with Bonferroni multiple comparison test (n = 6) **P < 0.01, ***P < 0.001. DD, destabilizing domains.
Discussion
In the current study, our group has used two DD based on the E. coli DHFR developed by Iwamoto and colleagues17 to design and optimize diverse fusion proteins of DD to GDNF and screened the resulting fusion proteins in vitro. The second-generation N-terminal construct, SP9-G-DD was able to solve the secretion problem observed in the first-generation N-terminal design. However, SP9-G-DD exhibited excessive secretion in the absence of induction that was sufficient to be biologically active, whereas G-F-DD did not. The YFP-DD control used confirmed that DD was functional in the assay conditions, indicating that the leakage was due to the specific SP9-G-DD design and not to a lack of DD regulation in general. As DD regulation is dependent on protein conformation,15 we postulated that the insertion of the DD 9 residues after the signal peptide induced conformational changes that decreased the recognition of SP9-G-DD by the proteasome, precluding its further use.
It was also observed that either SP9-G-DD or G-F-DD was readily detected inside cells using ELISA and western blot, regardless of induction. In a recent study, Sellmyer and colleagues have shown that there can be a basal accumulation of DD in the ER of cells expressing DD tagged with signal peptides.25 Nevertheless, the signal peptide-DD was still regulated by the proteasome and secretion of signal peptide-DD was efficiently regulated. The data obtained in our study are in line with these findings. Although G-F-DD was present inside cells regardless of TMP induction, the secretion of G-F-DD was efficiently regulated by TMP. Furthermore, only media from induced samples was able to upregulate TH expression in TGW cells, indicating that the intracellular expression of G-F-DD is not related to G-F-DD secretion or function.
Western blot media of induced G-F-DD 293T cells contained one band 2–3 kD higher and one band 2–3 kD lower than the corresponding band in the positive GDNF control. The higher band was not present in G-DD samples, indicating that it was a result of furin cleavage. Most likely, this band corresponds to the processed G-F-DD monomer that due to the presence of the glycin linker and residual furin recognition sequence should be 2–3 kD larger than a wild-type GDNF monomer. The lower molecular weight band is also present in G-DD medium, indicating that it is a product specific of G-F-DD or G-DD expression. Moreover, it is specifically detected by the antibody used that recognizes the C-terminal part of GDNF, indicating that the lower molecular weight band represent a truncated protein containing a C-terminal portion of GDNF. This truncated variant may be a product of cryptic splicing as no conventional splice sites are present in G-DD or G-F-DD coding sequences. However, culture media from G-DD–expressing cells that contains this truncated variant does not induce TH expression in TGW cells,18 suggesting that the low molecular weight product is not functional.
Due to the design of G-F-DD, the processed monomer contained small differences in primary structure that may affect posttranslational modifications and dimerization of G-F-DD. Although the glycosylation or dimerization status of secreted G-F-DD was not analyzed directly, data from TGW cells suggest that G-F-DD is dimerized. Although G-DD is present in the medium as unprocessed monomer, it is not functional in TGW cells,18 whereas G-F-DD is. Conversely, FACS binding assay and TH expression in TGW cells indicated that G-F-DD was binding and functional, when compared with glycosylated and dimerized GDNF.
The 6-OHDA protocol used for lesioning rats resulted in a more severe lesion than expected27 in terms of decreasing number of surviving SNpc neurons and increased deficits in behavior tests. Nevertheless, when compared with studies that used viral vectors and where a maximum 20% of SNpc neurons remained after 6-OHDA lesion,9,28 the GDNF group shows comparable levels of protection.
Other groups have used diverse systems to regulate GDNF in vivo. Tetracycline- and rapamycin-based inducible systems have previously been used to regulate GDNF expression in vivo.12,13,29,30,31,32,33,34 A comparison of our experiments with these studies suggests that the DD system has a lower dynamic range of induction when compared with tetracycline-based systems and a higher range of induction when compared with rapamycin-based systems. Nevertheless, the amount of induced G-F-DD in striatum was able to activate signaling pathways in SNpc and was comparable with therapeutic GDNF levels observed by others,6 indicating that the DD system could regulate GDNF to levels that are therapeutically relevant. The lower dynamic range observed for DD systems need to be taken into consideration for clinical applications. Ideally, the DD system can be applied to regulate therapeutic genes that require low levels of expression or have a long half-life such as neurotrophic factors. In the case of GDNF, it has been shown in primates that GDNF can elicit protective response with concentrations as low as 40 pg/mg.6 Moreover, the DD system could also be used to regulate proteins located upstream of signaling cascades that therefore require low levels of expression to mediate effects.
One of the concerns and shortcomings with inducible gene expression systems in the brain has been leaky gene expression when the system is not being induced.34 With GDNF this matter is of greater concern as it has been shown that constitutive expression of GDNF has side effects.7,10,11,12,13 Therefore, diverse markers and tests were used to determine if G-F-DD was leaky in vivo. Although GDNF ELISA indicated efficient GDNF regulation, immunohistochemistry for GDNF indicated basal expression that appeared restricted to cell soma, which was in line with the intracellular in vitro data and published studies.25 Therefore, p-rpS6 was used as a marker to ascertain if the basal G-F-DD OFF expression detected in GDNF immunohistochemistry was able to be secreted and activate GDNF signaling cascades in target neurons of SNpc. Analysis of p-rpS6–positive cells in SNpc showed that p-rpS6 the numbers of p-rpS6 cells in the G-F-DD OFF group were comparable with basal p-rpS6 levels observed in the contralateral side and lower than in the G and G-F-DD ON groups, suggesting that G-F-DD was efficiently regulated by TMP. Therefore, nonfunctional leakage was detected when G-F-DD was turned off.
G-F-DD was subsequently tested in a striatal 6-OHDA rat model of PD and its induction resulted in protection of SNpc neurons, striatal fiber output and motor skills comparable with wild-type GDNF.
It was also observed that despite efficient neuroprotection and behavior improvement in drug-induced rotations, there was no improvement in spontaneous motor behavior; in addition, there was downregulation of striatal TH expression in the G group. This has been previously observed in rats, where constitutively high expression of GDNF in the striatum leads to TH downregulation and that can lead to loss of behavioral improvements.7,10 It has been speculated that TH downregulation and associated side effects are most likely compensatory effects related to excessive expression of GDNF. In contrast, the G-F-DD ON group showed improvements in spontaneous motor behavior and no TH downregulation was observed. Despite efficient induction, G-F-DD levels obtained in the G-F-DD ON group may not have been sufficient to elicit a compensatory neuronal response and subsequent side effects. Immunohistochemical analysis of p-rpS6 in SNpc supports this hypothesis because p-rpS6 quantification showed that there was a stronger response in SNpc neurons of the G group when compared with the G-F-DD ON group.
A previous study by Chtarto and colleagues,29 where a variant of the tetracyclin-based inducible system was used to regulate GDNF in the 6-OHDA rat model of PD resulted in neuroprotection. However, there were also side effects such as TH downregulation and loss of behavioral improvements. Although there are differences in experimental setups, in our study, the G group exhibited the expected side effects whereas G-F-DD ON did not. Therefore, the differences between studies in terms of side effects of regulated GDNF cannot be explained solely by different experimental setups and may reflect intrinsic differences between DD and tetracycline-based systems. Although this discrepancy merits further investigation, it appears that the DD system may be inherently safer to regulate GDNF expression and possible side effects.
As the DD is regulated by the proteasome, and in addition, G-F-DD is accumulated in cells, it was important to determine if the regulation of G-F-DD could be maintained in vivo under pathological conditions. Therefore, GDNF expression and the number of p-rpS6 neurons were determined in the 6-OHDA model. It was observed that in the control G group, there appeared to be a higher number of p-rpS6 cells in the lesioned side than in the control side. However, TH and VMAT2 staining did not show such strong increase. Two things need to be considered. First, in the brain of unlesioned animals, wild-type GDNF expression in the striatum leads to strong activation of signaling pathways. Second, wild-type GDNF is expressed at higher levels than G-F-DD. Therefore, the excessive activation of signaling pathways in the G group may have increased p-rpS6 levels so that it appeared that there was a higher number of p-rpS6–positive SNpc cells. The excessive activation of signaling pathways in the G group may also be related to the side effects observed in the G group. Nevertheless, G-F-DD regulation could be maintained even in the presence of neurodegeneration and inflammation.
One of the main concerns when using inducible systems is the formation of an immune response. It has been shown that expression of bacterial Tet-controlled transcriptional activator can lead to an immune response in muscle tissue.35 As the DD used in the study was based on the bacterial DHFR, densitometric analysis of CD11B expression in the striatum was performed to determine if there was an inflammatory response to G-F-DD. Although there was an increase in inflammation in the lesioned brain hemisphere, no differences were found when G group was compared with YFP-DD, G-F-DD ON and G-F-DD OFF, suggesting that DD regulation did not lead increased inflammation. This is in line with previous studies using YFP-DD18 where YFP-DD regulation in the brain did not result in inflammation of brain parenchyma.
The data obtained suggested that there is an accumulation of G-F-DD at the ER level, both in ON and OFF groups. This accumulation may result in ER stress and be detrimental for the cell. Although the experiments reported here were not designed to evaluate ER response or toxicity in vivo, when the DD is targeted to the ER in cell-culture systems, it does not induce an unfolded protein response or stress.25 Moreover, expression and regulation of DD in the ON and OFF groups did not lead to increased CD11B expression when compared with the G group and the regulation of G-F-DD was maintained in under pathological conditions.
The experiments described in this study indicate that the DD system has several desired features to regulate therapeutic gene expression in the brain because the TMP is a well-characterized drug that crosses the blood–brain barrier,36,37 G-F-DD was induced to therapeutic levels and no G-F-DD leakage effects were observed when the system was not induced in the 6-OHDA rat model. Although further assessments are needed to fully characterize the DD system in vivo, the results presented in this study indicate that the DD system can potentially be used to regulate gene expression in the brain in a gene therapy setting.
Materials and Methods
Design of second-generation DD constructs. The R12Y/Y100I DHFR DD mutant was used to design the second-generation N-terminal GDNF (SP9-G-DD). The N-terminal DD was inserted in frame at an optimal distance (9 residues from signal peptide cleavage site) from the signal peptide cleavage site as determined by Signal IP4.0 prediction tool (http://www.cbs.dtu.dk/services/SignalP/). The second-generation C-terminal DD construct (G-F-DD) was designed placing the endogenous GDNF furin cleavage site surrounded with glycin linkers (Gly Gly Gly Ala Thr Ile Lys Arg Leu Lys Arg Ser Pro Asp Gly Gly Gly) in frame at the 3′ end of GDNF CDS and adding the N18T/A19V DHFR in frame after the furin-linker sequence. Functionality of the additional furin cleavage site was estimated using ProP prediction tool (http://www.cbs.dtu.dk/services/ProP/). Sequences for Gateway attachment sites attB1 and attB2 were inserted to flank both SP9-G-DD and G-F-DD coding sequences and the second-generation DD with respective attachment sites were synthesized into a pUC57 plasmid backbone (Genscript, Piscataway, NJ).
Cloning and lentiviral vector production. pUC57-SP9-G-DD and pUC57-G-F-DD were cloned into p221 plasmid using Gateway technology (Life Technologies Europe BV, Stockholm, Sweden) and resulting plasmids were subcloned into a destination pHG plasmid together with a plasmid containing human cytomegalovirus promoter (P4P1-CMV) to originate pHG-CMV-SP9-G-DD and pHG-CMV-G-F-DD. The pHG plasmid was created by insertion of a two-site gateway cassette into a pHR plasmid using ClaI and XhoI as restriction sites. Lentiviral vectors were made from pBG-CMV-G-DD, pBG-CMV-DD-G, pHR-CMV-GFP, pHG-CMV- SP9-DD-G pHG-CMV-G-F-DD, p2K7-CMV-YFP-DD, and pHR-CMV-GDNF plasmids,38 using standard protocols described previously.39 Titration of lentiviral vectors was performed by a combination of flow cytometry and quantitative PCR using lentiviral vectors containing pHR-CMV-GFP as a reference. For flow cytometry, 1 × 105 293T cells were seeded, and 6 hours later, these cells were transduced in a dilution series with pHR-CMV-GFP lentiviral vector. Seventy-two hours after seeding, cells were resuspended and fixed in phosphate-buffered saline (PBS) containing 1% formaldehyde (Sigma-Aldrich Sweden AB, Stockholm, Sweden) for 10 minutes. The number for GFP-positive fixed cells was determined using FACScalibur flow cytometer (BD Biosciences, San Jose, CA). The titers obtained for pHR-CMV-GFP lentiviral vectors were ~1–5 × 108 transducing units (TU)/ml. For quantitative PCR, 1 × 105 293T cells were seeded and 6 hours later, transduced with 0.3 µl, 1 µl, and 3 µl of lentiviral vector suspensions. Seventy-two hours after seeding, DNA from transduced cells was extracted using DNeasy blood and tissue kit (Qiagen AB, Sollentuna, Sweden) according to manufacturers instructions. Standard qPCR protocols were then performed using the following primers and probe to detect woodchuck hepatitis virus postregulatory element (WPRE): WPRE FP-GGCACTGACAATTCCGTGGT, WPRE RP-AGGGACGTAGCAGAAGGACG, WPRE probe- 5′Fam-ACGTCCTTTCCATGGCTGCTCGC -Tamra-3′. Human albumin was used as a standard: ALB FP-5′-TGAAACATACGTTCCCAAAGAGTTT-3′, ALB RP 5′-CTCTCCTTCTCAGAAAGTGTGCATAT-3′, ALB probe- 5′Fam-TGCTGAAACATTCACCTTCCATGCAGA-Tamra-3′. Relative quantification of WPRE and albumin content was performed by the ΔΔCT method. The resulting values were then used to calculate the estimated titer of each lentiviral vector relative to the pHR-CMV-GFP titer. Estimated titers ranged from 5 × 108 to 1 × 109 TU/ml for in vitro experiments. For in vivo experiments, the titers were 1 × 109 TU/ml.
Cell culture. TGW and 293T cells were cultured using DMEM (Life Technologies Europe BV) supplemented with 10% fetal calf serum (Saveen & Werner, Limhamn, Sweden) and penicillin/streptomycin cocktail (Life Technologies Europe BV). To determine expression, secretion and functionality of the second-generation DD, 1 × 105 293T cells were transduced at a multiplicity of infection of 2.5. One week after, transduced 293T cells were seeded at a density of 4 × 105 cells/well in 2 ml medium. Twenty-four hours later, 2 ml of medium containing 1 × 10−5 mol/l TMP (Sigma-Aldrich Sweden AB) was added to the transduced 293T cells and TGW cells were seeded at a density of 3 × 105 cells/well. Twenty-four hours later, 200 µl of conditioned medium from 293T cells was aliquoted to be analyzed by western blot or ELISA and the remaining 1.8 ml of medium was used to replace the medium in TGW cells. The transduced 293T cells were lysed and processed for western blot as described earlier.18 Twenty-four hours after medium change, TGW cells were lysed and processed for western blot.
To prepare culture media containing secreted G-F-DD, 1 × 105 293T cells were seeded and 6 hours later, transduced with lentiviral vector expressing G-F-DD at an multiplicity of infection of 60. The cells were allowed to propagate and 4 × 106 cells were seeded in a 10-cm dish (Nunc A/S, Kamstrup, Denmark) in 5 ml medium. Twenty-four hours after seeding, the medium was replaced with 4 ml culture medium containing 1 × 10−5 mol/l TMP. Twenty-four hours later, the media was aliquoted and stored at −20 °C for GDNF ELISA analysis.
GDNF ELISA. Samples were diluted in lysis buffer and the GDNF concentration was estimated using GDNF Emax Immunoassay (Promega Biotech AB, Nacka, Sweden) according to the manufacturers instructions.
Western blot. The western blot protocol used has been previously described in detail.18 The following antibodies were used: mouse anti-TH (1:5,000; Santa Cruz Biotechnology, Heidelberg, Germany), goat anti-GDNF (1:1,000; R&D Systems Europe, Abingdon, UK), mouse anti-β actin conjugated to horseradish peroxidase (HRP) (1:100,000; Sigma-Aldrich Sweden AB), goat anti-mouse-HRP (1:5,000; Santa Cruz Biotechnology), and donkey anti-goat (1:5,000; Santa Cruz Biotechnology).
FACS binding assay. The FACS binding assay has been described and validated elsewhere in more detail.24 Briefly, 50 µg of GDNF (recombinant human GDNF carrier free, R&D Systems Europe) was resuspended in 40 µl PBS and labeled with Alexa-488 using Alexa Fluor 488 Microscale Protein Labeling Kit (Life Technologies Europe BV) according to the manufacturers instructions. To perform the binding assay, 6 × 104 TGW cells/200 µl/well were seeded in 96-well plates. Five hours after seeding, the media was exchanged for medium containing 2 × 10−8 mol/l G-488 + 1 × 10−8 mol/l GDNF, 2 × 10−8 mol/l G-488 + 1 × 10−8 mol/l secreted G-F-DD or 2 × 10−8 mol/l G-488 + 1 × 10−8 mol/l GM-CSF (human recombinant GM-CSF, R&D Systems Europe). The cells were incubated for 4 hours at 37 °C, washed once in 1 ml staining buffer (Ca/Mg free PBS+ 2% FCS + 2 mmol/l EDTA), and resuspended in 150 µl staining buffer. The samples were analyzed in a FACSCanto (BD Biosciences).
Animals. All animals were housed and handled according to the principles of “Guide to the Care and Use of Experimental Animals.” All procedures have been approved and performed according to the guidelines established by the Ethical Committee for Use of Laboratory Animals at Lund University under the permit M09-10. Sprague Dawley female rats (Charles River, Sulzfeld, Germany) weighing 200–225 g were used for the experiments. TMP (TMP Oral suspension 10 mg/ml; Meda AB, Solna, Sweden) was diluted in water to a concentration of 0.5 mg/ml and given to the animals in their drinking water continuously throughout the experiment. A total of 66 animals were used for the experiments.
Stereotaxic surgeries. All anesthetic solutions and general procedures have been described in more detail previously.38 For the animal experiment testing secretion and function of G-F-DD in unlesioned animals, viral suspensions were delivered to two sets of coordinates: (i) Anteroposterior from bregma (AP) +1.2 mm, Mediolateral from bregma (ML) −2.5 mm, Dorsoventral from dura mater (DV) −5/−4 mm; and (ii) AP +0.6 mm, ML −2.5 mm, DV −5/−4 mm. The toothbar was set at 0. A total of 4 µl of lentiviral vector suspension (1 µl/ DV coordinate) was given. For the neuroprotection experiment, viral suspensions were delivered to three sets of coordinates: (i) AP +1.4 mm, ML −2.6 mm, DV −5/−4 mm; (ii) AP +0.4 mm, ML −3.8 mm, DV −5/−4 mm; and (iii) AP −0.8 mm, ML −4.4 mm, DV −5/−4 mm. Toothbar was set at 0 and a total of 6 µl of lentiviral vector suspension (1 µl/DV coordinate) was delivered. To lesion animals, a 3 × 7 µg striatal 6-OHDA lesion protocol was used. Briefly, 6-OHDA (Sigma-Aldrich Sweden AB) was resuspended to a concentration of 3.5 µg/µl in sterile 0.9% NaCl + 0.05% ascorbic acid (Sigma-Aldrich Sweden AB) and delivered to the following coordinates with the toothbar set at 0: (i) AP +1 mm, ML −3 mm, DV −5 mm; (ii) AP −0.1 mm, ML −3.7 mm, DV −5 mm; and (iii) AP −1.2 mm, ML −4.5 mm, DV −5 mm. Two microliter of 6-OHDA solution was delivered at each site. To minimize tissue damage in all surgeries, a glass capillary was attached to the needle of a 5 µl Hamilton syringe (Hamilton Bonaduz AG, Bonaduz, Switzerland).
Behavioral tests. The behavior of animals was measured at several timepoints. Drug-induced rotations caused by injection of D-amphetamine were measured 1 week before lesion, 2 and 6 weeks after lesion. Measurements of forelimb akinesia and forelimb use asymmetry were evaluated to assess protection on spontaneous motor behavior 6 weeks after 6-OHDA lesions. All tests used have been described extensively elsewhere.40
Striatal dissections. Animals were euthanized by intraperitoneal injection of 250 mg/kg pentobarbital (Apoteksbolaget, Lund, Sweden). Immediately after death, the animals were decapitated and their brains removed. The brain was placed in ice-cold 0.1 mol/l PBS for several minutes to facilitate slicing and then placed on an ice-cold brain slicer. A brain slice containing a coronal section 2 mm anterior to 0 mm relative to bregma was dissected. Striata were isolated by removing cortex, corpus callosum, septum, and area ventral to nucleus accumbens. Left and right hemispheres were further separated and snap frozen separately on dry ice for storage. The striatal slices were weighted and suspended in ice-cold radioimmunoprecipitation assay (RIPA) buffer (0.05 mol/l Tris pH 7.4, 0.15 mol/l NaCl, 1% Triton X) + proteinase inhibitor cocktail (PIC; Roche Applied Science, Bromma, Sweden) to a concentration of 1 mg sample/10 µl RIPA + PIC. The samples were sonicated on ice, incubated for 30 minutes on ice, and centrifuged at 10,000g at 4 °C for 10 minutes. The amount of protein was estimated as described previously.
Immunohistochemistry. Animals were euthanized and their brains processed for histology as described earlier.18 The brains were sectioned with starting point 2.5 mm anterior to bregma and ended 6.7 mm caudal to bregma, and a total of seven series of 35-µm coronal sections were prepared in a freezing stage microtome.
The following protocol was performed in free-floating sections. Samples were rinsed three times in 0.1 mol/l potassium PBS (KPBS) and incubated for 15 minutes in quenching solution (0.1 mol/l KBPS, 10% methanol (Merck KGaA, Darmstadt, Germany) and 3% H2O2 (Merck KGaA)).
The samples were then rinsed three times in 0.1 mol/l KPBS and incubated for 1 hour in 5% serum solution (0.1 mol/l KPBS + 5% horse or goat serum + 0.25% Triton X (Merck KGaA)).
Samples were then incubated overnight with primary antibody, diluted in 5% serum solution. The following primary antibodies were used: rabbit anti-TH (1:1,000, Merck KGaA), rabbit anti-VMAT2 (1:1,000, Abcam, Cambridge, UK), goat anti-GDNF (1:1,000, R&D Systems Europe), rabbit anti-p-rpS6 (1:400, Ser235/236, Cell Signaling Technology, Danvers, MA), and mouse anti-CD11B (1:200, Serotec, Raleigh, NC). On the next day, samples were washed two times in 0.1 mol/l KPBS, incubated 15 minutes in 5% serum solution; and incubated 1 hour with secondary antibodies diluted in 5% serum solution: biotinylated horse anti-mouse (1:200, Vector Labs, Peterborough, UK), biotinylated horse anti-goat (1:200, Vector Labs), and biotinylated goat anti-rabbit-biotin (1:200, Vector Labs). The samples were subsequently washed for three times with 0.1 mol/l KPBS, incubated 1 hour with 0.1 mol/l KPBS + ABC complex (Vectastain ABC, Vector Labs), and washed three times with 0.1 mol/l KPBS. The samples were incubated with 0.1 mol/l KPBS with 0.5 mg/ml 3,3-diaminobenzidine for 2 minutes. The reaction was then visualized by incubating the samples with 10 µl H2O2 solution (0.1 mol/l KPBS+ 0.9% H2O2) for 2–4 minutes. The samples were washed three times in 0.1 mol/l KPBS, mounted, and coverslipped using DPX mounting medium (Merck KGaA).
Quantification of histological sections. Densitometry for TH, VMAT2, and CD11B was measured using four coronal sections spanning the striatum (+1.2 mm, 0.8 mm, 0.0 mm, and −0.4 mm relative to bregma), densitometry for p-rpS6 was measured using two coronal sections immediately preceding the medial lemniscus and densitometry for GDNF expression was measured in the whole striatum. The images were obtained using an image scanner and processed using ImageJ software suite. Pixel densities were corrected for nonspecific background and data were expressed as a percentage of the intact contralateral side. Three coronal sections were used for relative quantification of TH, VMAT2, and p-rpS6–positive SNpc neurons: first coronal section containing medial lemniscus separating ventral tegmental area from SNpc (approximately −5 mm relative to bregma), adjacent cranial section, and adjacent caudal section. Data were presented as a percentage of neurons relative to the left intact side.
Statistical analysis. Graphpad Prism 5 software (Graphpad Software, La Jolla, CA) was used for statistical analysis. Two-way ANOVA with Bonferroni post hoc test was used to analyze in vivo GDNF ELISA. Unpaired t-test was used to compare p-rpS6 cell numbers. One-way ANOVA with Bonferroni post hoc test was used for statistical analysis of the rest of the data. Data are presented as mean ± SEM.
SUPPLEMENTARY MATERIAL Figure S1. Densitometric analysis of CD11B expression.
Acknowledgments
The authors want to thank Ulla Jarl, Bengt Matsson, Michael Sparrenius, and Aurélie Baudet for all the help during experiments. This work has been supported by the Swedish Research Council by grants #2010–4496 and #2007–8626, Strategic Research Area MultiPark funded by the Swedish government, Bagadilico Consortium, and Rapid Response Innovation Grant of the Michael J Fox Foundation for Parkinson's Research. The work was performed in Lund, Sweden.
Supplementary Material
References
- Meissner WG, Frasier M, Gasser T, Goetz CG, Lozano A, Piccini P, et al. Priorities in Parkinson's disease research. Nat Rev Drug Discov. 2011;10:377–393. doi: 10.1038/nrd3430. [DOI] [PubMed] [Google Scholar]
- Bjorklund T, Kordower JH. Gene therapy for Parkinson's disease. Mov Disord. 2010;25 Suppl 1:S161–S173. doi: 10.1002/mds.22785. [DOI] [PubMed] [Google Scholar]
- Even Ram S, Galun E. AAV2-GDNF gene therapy for Parkinson's disease. Hum Gene Ther. 2009;20:430–431. doi: 10.1089/hum.2009.1599. [DOI] [PubMed] [Google Scholar]
- Hoffer BJ, Harvey BK. Is GDNF beneficial in Parkinson disease. Nat Rev Neurol. 2011;7:600–602. doi: 10.1038/nrneurol.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowd E, Monville C, Torres EM, Wong LF, Azzouz M, Mazarakis ND, et al. Lentivector-mediated delivery of GDNF protects complex motor functions relevant to human Parkinsonism in a rat lesion model. Eur J Neurosci. 2005;22:2587–2595. doi: 10.1111/j.1460-9568.2005.04414.x. [DOI] [PubMed] [Google Scholar]
- Eslamboli A, Georgievska B, Ridley RM, Baker HF, Muzyczka N, Burger C, et al. Continuous low-level glial cell line-derived neurotrophic factor delivery using recombinant adeno-associated viral vectors provides neuroprotection and induces behavioral recovery in a primate model of Parkinson's disease. J Neurosci. 2005;25:769–777. doi: 10.1523/JNEUROSCI.4421-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgievska B, Kirik D, Björklund A. Aberrant sprouting and downregulation of tyrosine hydroxylase in lesioned nigrostriatal dopamine neurons induced by long-lasting overexpression of glial cell line derived neurotrophic factor in the striatum by lentiviral gene transfer. Exp Neurol. 2002;177:461–474. doi: 10.1006/exnr.2002.8006. [DOI] [PubMed] [Google Scholar]
- Ciesielska A, Mittermeyer G, Hadaczek P, Kells AP, Forsayeth J, Bankiewicz KS. Anterograde axonal transport of AAV2-GDNF in rat basal ganglia. Mol Ther. 2011;19:922–927. doi: 10.1038/mt.2010.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirik D, Rosenblad C, Bjorklund A, Mandel RJ. Long-term rAAV-mediated gene transfer of GDNF in the rat Parkinson's model: intrastriatal but not intranigral transduction promotes functional regeneration in the lesioned nigrostriatal system. J Neurosci. 2000;20:4686–4700. doi: 10.1523/JNEUROSCI.20-12-04686.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblad C, Georgievska B, Kirik D. Long-term striatal overexpression of GDNF selectively downregulates tyrosine hydroxylase in the intact nigrostriatal dopamine system. Eur J Neurosci. 2003;17:260–270. doi: 10.1046/j.1460-9568.2003.02456.x. [DOI] [PubMed] [Google Scholar]
- Hovland DN, Jr, Boyd RB, Butt MT, Engelhardt JA, Moxness MS, Ma MH, et al. Six-month continuous intraputamenal infusion toxicity study of recombinant methionyl human glial cell line-derived neurotrophic factor (r-metHuGDNF in rhesus monkeys. Toxicol Pathol. 2007;35:1013–1029. doi: 10.1177/01926230701481899. [DOI] [PubMed] [Google Scholar]
- Manfredsson FP, Tumer N, Erdos B, Landa T, Broxson CS, Sullivan LF, et al. Nigrostriatal rAAV-mediated GDNF overexpression induces robust weight loss in a rat model of age-related obesity. Mol Ther. 2009;17:980–991. doi: 10.1038/mt.2009.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredsson FP, Burger C, Rising AC, Zuobi-Hasona K, Sullivan LF, Lewin AS, et al. Tight Long-term dynamic doxycycline responsive nigrostriatal GDNF using a single rAAV vector. Mol Ther. 2009;17:1857–1867. doi: 10.1038/mt.2009.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredsson FP, Bloom DC, Mandel RJ. Regulated protein expression for in vivo gene therapy for neurological disorders: progress, strategies, and issues. Neurobiol Dis. 2012;48:212–221. doi: 10.1016/j.nbd.2012.03.001. [DOI] [PubMed] [Google Scholar]
- Banaszynski LA, Chen LC, Maynard-Smith LA, Ooi AG, Wandless TJ. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell. 2006;126:995–1004. doi: 10.1016/j.cell.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banaszynski LA, Sellmyer MA, Contag CH, Wandless TJ, Thorne SH. Chemical control of protein stability and function in living mice. Nat Med. 2008;14:1123–1127. doi: 10.1038/nm.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto M, Björklund T, Lundberg C, Kirik D, Wandless TJ. A general chemical method to regulate protein stability in the mammalian central nervous system. Chem Biol. 2010;17:981–988. doi: 10.1016/j.chembiol.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai K, Quintino L, Isaksson C, Gussing F, Lundberg C. Destabilizing domains mediate reversible transgene expression in the brain. PLoS ONE. 2012;7:e46269. doi: 10.1371/journal.pone.0046269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9:944–957. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol. 2002;3:753–766. doi: 10.1038/nrm934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckert P, Brunak S, Blom N. Prediction of proprotein convertase cleavage sites. Protein Eng Des Sel. 2004;17:107–112. doi: 10.1093/protein/gzh013. [DOI] [PubMed] [Google Scholar]
- Xiao H, Hirata Y, Isobe K, Kiuchi K. Glial cell line-derived neurotrophic factor up-regulates the expression of tyrosine hydroxylase gene in human neuroblastoma cell lines. J Neurochem. 2002;82:801–808. doi: 10.1046/j.1471-4159.2002.00993.x. [DOI] [PubMed] [Google Scholar]
- Quintino L, Baudet A, Larsson J, Lundberg C. FACS binding assay for analysing GDNF interactions. J Neurosci Methods. 2013;218:25–28. doi: 10.1016/j.jneumeth.2013.04.006. [DOI] [PubMed] [Google Scholar]
- Sellmyer MA, Chen LC, Egeler EL, Rakhit R, Wandless TJ. Intracellular context affects levels of a chemically dependent destabilizing domain. PLoS ONE. 2012;7:e43297. doi: 10.1371/journal.pone.0043297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decressac M, Kadkhodaei B, Mattsson B, Laguna A, Perlmann T, Björklund A. a-Synuclein-induced down-regulation of Nurr1 disrupts GDNF signaling in nigral dopamine neurons. Sci Transl Med. 2012;4:163ra156. doi: 10.1126/scitranslmed.3004676. [DOI] [PubMed] [Google Scholar]
- Kirik D, Rosenblad C, Björklund A. Characterization of behavioral and neurodegenerative changes following partial lesions of the nigrostriatal dopamine system induced by intrastriatal 6-hydroxydopamine in the rat. Exp Neurol. 1998;152:259–277. doi: 10.1006/exnr.1998.6848. [DOI] [PubMed] [Google Scholar]
- Georgievska B, Kirik D, Rosenblad C, Lundberg C, Björklund A. Neuroprotection in the rat Parkinson model by intrastriatal GDNF gene transfer using a lentiviral vector. Neuroreport. 2002;13:75–82. doi: 10.1097/00001756-200201210-00019. [DOI] [PubMed] [Google Scholar]
- Chtarto A, Yang X, Bockstael O, Melas C, Blum D, Lehtonen E, et al. Controlled delivery of glial cell line-derived neurotrophic factor by a single tetracycline-inducible AAV vector. Exp Neurol. 2007;204:387–399. doi: 10.1016/j.expneurol.2006.11.014. [DOI] [PubMed] [Google Scholar]
- Hadaczek P, Beyer J, Kells A, Narrow W, Bowers W, Federoff HJ, et al. Evaluation of an AAV2-based rapamycin-regulated glial cell line-derived neurotrophic factor (GDNF) expression vector system. PLoS ONE. 2011;6:e27728. doi: 10.1371/journal.pone.0027728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Mertens B, Lehtonen E, Vercammen L, Bockstael O, Chtarto A, et al. Reversible neurochemical changes mediated by delayed intrastriatal glial cell line-derived neurotrophic factor gene delivery in a partial Parkinson's disease rat model. J Gene Med. 2009;11:899–912. doi: 10.1002/jgm.1377. [DOI] [PubMed] [Google Scholar]
- Szulc J, Wiznerowicz M, Sauvain MO, Trono D, Aebischer P. A versatile tool for conditional gene expression and knockdown. Nat Methods. 2006;3:109–116. doi: 10.1038/nmeth846. [DOI] [PubMed] [Google Scholar]
- Chtarto A, Tenenbaum L, Velu T, Brotchi J, Levivier M, Blum D. Minocycline-induced activation of tetracycline-responsive promoter. Neurosci Lett. 2003;352:155–158. doi: 10.1016/j.neulet.2003.08.067. [DOI] [PubMed] [Google Scholar]
- Georgievska B, Jakobsson J, Persson E, Ericson C, Kirik D, Lundberg C. Regulated delivery of glial cell line-derived neurotrophic factor into rat striatum, using a tetracycline-dependent lentiviral vector. Hum Gene Ther. 2004;15:934–944. doi: 10.1089/hum.2004.15.934. [DOI] [PubMed] [Google Scholar]
- Latta-Mahieu M, Rolland M, Caillet C, Wang M, Kennel P, Mahfouz I, et al. Gene transfer of a chimeric trans-activator is immunogenic and results in short-lived transgene expression. Hum Gene Ther. 2002;13:1611–1620. doi: 10.1089/10430340260201707. [DOI] [PubMed] [Google Scholar]
- Gleckman R, Blagg N, Joubert DW. Trimethoprim: mechanisms of action, antimicrobial activity, bacterial resistance, pharmacokinetics, adverse reactions, and therapeutic indications. Pharmacotherapy. 1981;1:14–20. doi: 10.1002/j.1875-9114.1981.tb03548.x. [DOI] [PubMed] [Google Scholar]
- Tu YH, Allen LV, Jr, Fiorica VM, Albers DD. Pharmacokinetics of trimethoprim in the rat. J Pharm Sci. 1989;78:556–560. doi: 10.1002/jps.2600780709. [DOI] [PubMed] [Google Scholar]
- Wettergren EE, Gussing F, Quintino L, Lundberg C. Novel disease-specific promoters for use in gene therapy for Parkinson's disease. Neurosci Lett. 2012;530:29–34. doi: 10.1016/j.neulet.2012.09.059. [DOI] [PubMed] [Google Scholar]
- Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol. 1997;15:871–875. doi: 10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]
- Cederfjäll E, Sahin G, Kirik D, Björklund T. Design of a single AAV vector for coexpression of TH and GCH1 to establish continuous DOPA synthesis in a rat model of Parkinson's disease. Mol Ther. 2012;20:1315–1326. doi: 10.1038/mt.2012.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.