

Abstract
Antiangiogenic therapy is a promising new treatment modality for cancer, but it generally produces only transient tumor regression. We have previously devised a tumor-targeted nanosystem, in which a pentapeptide, CGKRK, delivers a proapoptotic peptide into the mitochondria of tumor blood vessel endothelial cells and tumor cells. The treatment was highly effective in glioblastoma mouse models completely refractory to other antiangiogenic treatments. Here, we identify p32/gC1qR/HABP, a mitochondrial protein that is also expressed at the cell surface of activated (angiogenic) endothelial cells and tumor cells, as a receptor for the CGKRK peptide. The results demonstrate the ability of p32 to cause internalization of a payload bound to p32 into the cytoplasm. We also show that nardilysin, a protease capable of cleaving CGKRK, plays a role in the internalization of a p32-bound payload. As p32 is overexpressed and surface displayed in breast cancers, we studied the efficacy of the nanosystem in this cancer. We show highly significant treatment results in an orthotopic model of breast cancer. The specificity of cell surface p32 for tumor-associated cells, its ability to carry payloads to mitochondria, and the efficacy of the system in important types of cancer make the nanosystem a promising candidate for further development.
Introduction
Nanotechnology shows great promise in improving the performance of existing drugs and in creating new therapies, particularly in cancer.1,2,3 Some nanoparticle drugs (NP; Abraxane and doxorubicin liposomes (Doxil)) are already in the clinic for cancer treatment.4,5 However, these clinically used NPs are simple carriers that lack advanced functions, such as target seeking, controlled payload release, and combined treatment and imaging (theranostic NPs).
We recently developed a theranostic nanosystem for cancer treatment and tested it in mouse glioblastoma (GBM) models.6 The system consists of elongated iron oxide NPs,7 which are coated with a chimeric peptide through a polyethylene glycol linker. One branch of the peptide, CGKRK, is a tumor-specific vascular homing element8 and the other branch is D[KLAKLAK]2, a membrane-perturbing proapoptotic D-amino acid peptide,9 which serves as a drug.10 The proapoptotic peptide in soluble form is effective for tumor treatment but causes significant systemic toxicity.10,11 We showed that the NP-bound D[KLAKLAK]2 was ~100–300 times more potent in killing cultured cells than the soluble form.6 The increased potency and high tumor specificity of the NP-bound proapoptotic peptide made it possible to use the peptide at lower dose, which reduced general toxicity. Moreover, the efficacy of the targeting improved because the dose was more compatible with the limited number of CGKRK receptors in the tumor.12 Another novel feature of the CGKRKD[KLAKLAK]2 system is that the homing peptide directs the proapoptotic D[KLAKLAK]2 peptide to mitochondria, the subcellular organelle that D[KLAKLAK]2 acts on. We found CGKRK nanoworms (NWs) colocalizing with mitochondria in target cells and showed that CGKRK directly binds to mitochondria.6 The mitochondrial localization indicates that CGKRK is capable of penetrating into the cytoplasm and taking the NWs with it. In tumor-bearing mice, the CGKRK-NWs efficiently homed to tumor vessels but did not significantly enter into the extravascular tumor tissue. We used a tumor-penetrating peptide, iRGD (CRGDK/RGPD/EC) (internalizing-RGD peptide), to increase the capability of the system to reach tumor tissue.13,14
We validated the CGKRKD[KLAKLAK]2 nanosystem in orthotopic GBM models. It eradicated the tumors in one model and significantly extended the life span of mice in another model with a more aggressive disease.6 These results encouraged us to identify the cell surface molecule (receptor) responsible for the specific binding of CGKRK to tumor-associated cells.
Here, we report the identity of the dominant receptor for the CGKRK peptide and validate the CGKRK-delivered nanosystem in breast cancer, the tumor type that most consistently expresses high levels of the receptor.
Results
The target molecules (receptors) for CGKRK
The earlier treatment results in GBM models6 prompted us to study the nature of the molecule(s) CGKRK recognizes on tumor vessels and tumor cells. Because CGKRK accumulates at the mitochondria of the target cells,6 we started our study by fractionating mitochondrial extracts on a CGKRK affinity matrix. Elution with soluble CGKRK peptide produced a band at ~32 kDa, which was not seen in the column washes or eluates obtained with a control peptide, CREKA (Figure 1a and Supplementary Figure S1a). This band was identified by mass spectrometry as the mitochondrial/multicompartment protein p32/gC1qR/HABP, which binds the C1q complement component hyaluronic acid, and nucleic acid.15 This protein will be referred to as p32 hereafter. The identification of the CGKRK-binding protein as p32 was confirmed by immunoblotting (Figure 1b). We have previously shown that p32 is the receptor for another tumor-homing peptide, LyP-1, and that the specificity of LyP-1 as a tumor-homing peptide is based on the expression of p32 on the surface of tumor cells and other activated cells in tumors, but not in normal cells.16
Figure 1.

Identification of a CGKRK-binding protein in mitochondrial protein extracts. Protein extracts prepared from mouse liver mitochondria were fractionated by affinity chromatography on CGKRK-coupled columns. Bound proteins were eluted with 2 mmol/l CGKRK peptide solution and separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. (a) Silver-stained gel. The arrow points to the band identified as p32 by mass spectrometry, and (b) immunoblotting detection of p32.
To ascertain that p32 is the target molecule for CGKRK in tumor tissue, we fractionated 005 mouse GBM tumor extracts on CGKRK affinity matrix. We found p32 in the eluates, but other specifically eluted proteins were also identified by mass spectrometry: nuclear ribonucleoprotein A3/2/1 (HNRNPA3/2/1), cytoskeleton-associated protein 4 (CKAP4), nucleolin, and nardilysin-1 (NRD1; Figure 2a and Supplementary Figure S1b and Supplementary Table S1). The identification of the proteins was confirmed by immunoblotting (Figure 2b). Similar results were obtained in two tumor models: MCF10CA1a and MDA-MB-435 (Figure 2c). The MDA-MB-435 human tumor cell line was used even though it is of unclear origin because it is a strong p32 expressor.6
Figure 2.

Identification of CGKRK-binding proteins in brain tumor protein extracts. Protein extracts prepared from 005 mouse brain tumors were fractionated on CGKRK affinity columns. Bound proteins were eluted with 2 mmol/l CGKRK peptide solution and separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. (a) Silver-stained gel. The arrows point to the bands subjected to mass spectrometry for protein identification. The proteins identified are shown on the left side of the arrows. (b) Immunoblot with anti-p32, anti-CKAP4, anti-nucleolin, and anti-nardilysin (NRD). (c) Protein extracts prepared from MDA-MB-435 and MCF10CA1a tumors were fractionated on a CGKRK affinity column. Bound proteins were eluted with 2 mmol/l CGKRK solution. Results of immunoblotting with anti-p32, anti-CKAP4, anti-nucleolin, and anti-NRD are shown.
To determine the relative importance of the CGKRK-binding proteins obtained by affinity chromatography in cell binding and internalization of CGKRK, we determined the affinities of these proteins for CGKRK, changes in their expression during neoplastic progression, and the effects of knocking down or blocking them in cells. In addition to p32, we primarily focused on NRD1 and nucleolin because these proteins are expressed at the cell surface;17,18 in the case of nucleolin, the cell surface expression is specific for activated cells, such as angiogenic endothelial cells and tumor cells,18 and it serves as the receptor for one of our tumor-homing peptides.18
Affinity measurements by saturation binding to purified proteins gave an average binding affinity constant for CGKRK interaction of Kd = 0.4 ± 0.2 µmol/l for p32, Kd = 24.5 ± 5.5 µmol/l for NRD1, Kd = 61 ± 13 µmol/l for nucleolin, and Kd = 116 ± 7 mmol/l for CKAPA4 (Figure 3a). An antibody against p32 reduced the binding of 5(6)-carboxyfluorescein (FAM)-labeled CGKRK peptide to purified p32 protein in a concentration-dependent manner by up to 60% (Supplementary Figure S2).
Figure 3.

CGKRK binding to candidate receptor proteins. (a) Binding of FAM–CGKRK to purified p32, NLC3, CKAP4, and nardilysin1 (NRD1). Binding of increasing amounts of FAM–CGKRK peptide and a control FAM–CREKA peptide to immobilized p32, NLC3, CKAP4, and NRD1 proteins was detected by fluorescence and normalized to nonspecific binding to the plastic plate. One representative binding curve out of three independent experiments with similar results is shown. The Kd was calculated from all three experiments using Prism software. (b) Binding and internalization of CGKRK phage particles in a series of MCF10 human breast tumor cells. MCF10 cells were incubated with phage displaying CGKRK or a polyglycine control peptide, CG7C, overnight at 4 °C or for 1 hour at 37 °C. To assess internalization, phages bound at the cell surface were removed by washing the cells with an acid buffer before phage titration. Statistical analyses were performed with Student's t-test. n = 3; error bars, mean ± SEM; **P < 0.01. (c) Total expression levels of NRD-1, NLC3, CKAP4, and p32 in the MCF10 cells assessed by immunoblotting.
We next used cell lines that were derived from the spontaneously immortalized, but otherwise normal, MCF10A cells to study CGKRK binding and the expression of the candidate CGKRK receptors in tumor progression. MCF10AT cells are premalignant in that they are prone to neoplastic transformation.19 MCF10CA1a cells are fully malignant and metastatic.20 CGKRK phage showed progressively increasing binding with increasing degree of malignancy from the nonmalignant to the malignant and metastatic cell lines (Figure 3b). The expression of the candidate receptors p32, nucleolin, and NRD1, but not CKAP4, positively correlated with the malignancy of the MCF10 series (Figure 3c). The low affinities of nucleolin and CKAPA4, and the lack of correlation of CKAP4 expression with CGKRK binding to cells, do not support a significant role for these proteins in CGKRK binding and internalization. Therefore, we decided to focus on p32 and NRD1 in further studies.
The presence of p32 at the cell surface in several kinds of cancer, such as breast and prostate cancer, has been reported,16 but not in GBM. Fluorescence-activated cell sorting analysis demonstrated surface expression of p32 in GBM tumor-initiating cells (005) and transdifferentiated endothelial cells derived from 005 cells (T3) (Figure 4a).21 Staining of sections from 005 GBM tumors for p32 revealed high expression of total p32 in the tumor tissue compared with that in the normal brain (Figure 4b). To validate the interaction between p32 and CGKRK, we studied the effect of knocking down p32 on CGKRK binding. We previously generated MDA-MB435 cells with stable p32 knockdown.16 The binding and internalization of CGKRK phage to cells with nearly complete knockdown of p32 (Figure 4c) was reduced by >50% (Figure 4d). To generate p32-knockdown GBM cells, we used a lentivirus shp32 vector expressing a sequence homologous to the human sequence used in the MDA-MB-435 cells. We were unable to generate 005 cells lacking p32, probably because p32 is crucial to the survival of these cells.16 However, we were successful in knocking down p32 in T3 cells to an undetectable level (Figure 4e). The knockdown reduced the binding and internalization of the CGKRK phage by 60–75% in these cells (Figure 4f).
Figure 4.

p32-dependent binding of CGKRK to tumor cells. (a) Cell-surface expression of p32 on 005 brain tumor cells, T3 brain tumor endothelial cells, and human umbilical vein endothelial cells assessed by flow cytometry. (b) Localization of p32 in 005 brain tumors. Frozen sections of mouse brain containing green fluorescent protein–expressing 005 brain tumor cells (green) were stained for p32 (magenta) and nuclei (blue) and were imaged under a confocal microscope. Scale bars, 100 µm. (c–f) Effect of p32 knockdown on CGKRK binding. p32 expression levels were determined by immunoblot on whole-cell lysates from MDA-MB-435 cells (c) or T3 cells (e) stably expressing p32 short hairpin RNA (shRNA) or control shRNA. β-actin was used as a loading control. Cells with varying p32 expression levels were incubated with phages displaying CGKRK or CG7C overnight at 4 °C or for 1 hour at 37 °C. To assess internalization, phages bound at the cell surface were removed by washing the cells with an acid buffer before phage titration. Note that the p32 knockdown resulted in reduced CGKRK binding. Statistical analysis was performed with Student's t-test;**P < 0.01; ***P < 0.001; n = 3; error bars, mean ± SEM.
Transfection with an NRD1 small interfering RNA markedly reduced NRD1 expression in p32-knockdown MDA-MB-435 cells (Figure 5a). The NRD1 knockdown had little effect in the assay that measures cell surface binding of CGKRK phage but reduced the residual phage internalization by ~45% (Figure 5b). Therefore, cell surface NRD1 minimally contributes to the binding of CGKRK to cells but may play a role in the internalization of the peptide. These results show that the p32 protein is the main receptor for CGKRK. High expression of cell surface p32 is found in many types of cancer but appears to be particularly consistent in breast cancer.16 The prevalence of p32 expression in breast cancer prompted us to study the efficacy of the CGKRKD[KLAKLAK]2-NWs in this cancer.
Figure 5.

Effect of nardilysin (NRD) knockdown on CGKRK phage binding and internalization into cells. (a) MDA-MB-435 cells were transiently transfected with NRD small interfering RNA (siRNA) or control siRNA. After 48 hours, NRD expression levels were determined by immunoblotting. β-actin was used as a loading control. The results are representative of three independent experiments. (b) The cells transfected with NRD siRNA or control siRNA were incubated with CGKRK phage for 1 hour at 37 °C to assess internalization into cells or overnight at 4 °C to assess cell binding. In internalization assays, the phages bound at the cell surface were removed by washing the cells with an acid buffer before phage titration. n = 3; error bars, mean ± SEM.
Breast cancer homing of CGKRK peptide and CGKRK-coated NWs
We evaluated the potential of the CGKRKD[KLAKLAK]2-NW therapy in breast cancer in two different models: MCF10CA1a (orthotopic human breast cancer xenografts) and Tg(c3-1-TAg)Jeg (transgenic breast mice). Furthermore, we also used xenografts generated with the MDA-MB-435 human cancer cells. We initially tested the ability of CGKRK to recognize these tumors. Intravenously injected FAM-labeled CGKRK peptide strongly accumulated in each of the tumors but not in normal tissues, with the exception of the kidneys, from where the peptides are excreted (Figure 6a and Supplementary Figure S3). The peptide distribution within the tumors was patchy, presumably reflecting the presence of tumor microdomains, as has been previously observed with LyP-1.22 Nuclear staining revealed normal nuclei in the areas containing CGKRK, indicating that these areas were viable (Figure 6a, row on the right). Unlike the CGKRK peptide, which was present within the extravascular tumor tissue, CGKRK-NWs were localized in tumor vessels, presumably because the large size of the NPs prevented extravasation (Figure 6b). No fluorescence from the CGKRK-NW was observed in normal tissues of the tumor-bearing mice, with the exception of the liver and the spleen, which nonselectively take up NPs (Supplementary Figure S4).
Figure 6.

Homing of CGKRK peptide and CGKRK-nanoworms (NWs) to breast tumors. Mice bearing MCF10CA1a, MDA-MB-435, or MMTV-PyMT orthotopic tumors were intravenously injected with (a) 200 µg of FAM-CGKRK peptide or (b) 5 mg iron/kg of FAM-CRGDK-coated NWs. Circulation time was 3 hours for the peptide and 5–6 hours for the NWs. The mice were perfused through the heart with PBS, and tissues were collected and processed for immunofluorescence. Representative confocal microscopy images from three mice per tumor type are provided. Arrows in panel b point to tumor blood vessels targeted by CGKRK-NW. Green, (a) FAM-CGKRK and (b) CGKRK-NWs; red, CD31; blue, nuclei; Scale bars, (a) 200 μm and (b) 100 μm.
Therapeutic efficacy of CGKRKD[KLAKLAK]2-NWs
The homing results above indicated that both of the breast cancers studied and the MDA-MB-435 tumors would be suitable targets for CGKRKD[KLAKLAK]2-NW treatment. We chose the MCF10CA1a model for a treatment study. Although the primary target of these NWs is the tumor vasculature, the tumor cells may also be targeted. Indeed, the MCF10CA1a cells were highly susceptible to the CGKRK-targeted NWs, more so than to the untargeted D[KLAKLAK]2-NWs (Supplementary Figure S5). Treatment of mice bearing orthotropic MCF10CA1a tumors with intravenous injections of CGKRKD[KLAKLAK]2-NWs given every other day resulted in significant (P < 0.001) inhibition of tumor growth (Figure 7). We also combined these NWs with the tumor-penetrating peptide iRGD because the combination had shown enhanced efficacy in an earlier GBM study.6 The inclusion of iRGD yielded an additional increment of tumor growth inhibition (Figure 7). CGKRK-NWs alone showed no significant effect. Untargeted D[KLAKLAK]2-NW slightly inhibited tumor growth (P < 0.02), and its activity was also augmented by coinjection with iRGD (P < 0.001).
Figure 7.

CGKRKD[KLAKLAK]2-nanoworm (NW) treatment in MCF10CA1a tumor mice. Mice bearing MCF10CA1a orthotopic tumor xenografts were intravenously treated with peptide-coated NWs every other day for 3 weeks at a dose of 5 mg/kg. In some groups, 100 µg of iRGD was coinjected with the NWs to enhance extravasation and tissue penetration of the particles. PBS, n = 7; CGKRK-NW, n = 5;D[KLAKLAK]2-NWs, n = 6; [KLAKLAK]2-NWs+iRGD, n = 6; CGKRKD[KLAKLAK]2-NWs, n = 8; CGKRKD[KLAKLAK]2-NWs+iRGD, n = 8. a, Tumor growth was followed throughout the treatment and for an additional 14 days posttreatment period in the groups, where the size of the tumors stayed below the size limit allowed by the institutional review committee (in the Materials and Methods section). The experiment was done twice. Error bars, mean ± SD. Statistical analyses were performed with ANOVA; n.s., not significant; *P < 0.05; ***P < 0.001.
Analysis of tumors treated with a single NW injection showed extensive fibrin accumulation 24 and 48 hours after a single injection of CGKRKD[KLAKLAK]2-NWs (Supplementary Figure S7a,b). TUNEL-positive areas demonstrated damage to the vasculature at these same time points (Supplementary Figure S7c). There was also a significant reduction in number of blood vessels in the CGKRKD[KLAKLAK]2-NW-treated tumors at the end of the treatment, with remaining blood vessels mainly present in tumor periphery in reduced numbers (Supplementary Figure S7d). These results show that the treatment causes vascular disruption.
Confocal microscopy at the end of the tumor treatment showed that many of the blood vessels in the tumors treated with CGKRKD[KLAKLAK]2-NWs contained NWs and that the NWs were extravascular in the tumors of the iRGD-coinjected mice (Figure 8a). Small amounts of D[KLAKLAK]2-NWs were observed in tumor vessels, and coinjection with iRGD also resulted in spreading of these untargeted NWs into the extravascular tumor tissue (Figure 8a). This latter result may explain the observation that the D[KLAKLAK]2-NW plus iRGD combination inhibited tumor growth more effectively than D[KLAKLAK]2-NWs alone. Ki67 staining identified a high proliferation rate in the phosphate-buffered saline (PBS) group compared with both CGKRKD[KLAKLAK]2-NWs groups (Figure 8b). The absence of Ki67 was particularly prominent in areas close to blood vessels and in extravascular areas that contained NWs coinjected with iRGD (Figure 8b). Analysis of nontumor tissues from the mice at the end of the treatment revealed some fluorescence from the CGKRKD[KLAKLAK]2-NWs in the liver and spleen, and more substantial quantities were present in the kidneys (Supplementary Figure S6). The liver and the spleen are known to nonselectively take up NPs. The fluorophor is attached to the D[KLAKLAK]2 part of the NW construct, so the fluorescence in the kidneys is probably because this protease-resistant D-amino acid peptide is released from the NWs and is cleared through the kidneys. However, in agreement with our earlier GBM study,6 no overt signs of toxicity were seen in these tissues. These results indicate that the nanosystem is efficacious in treating tumors that express cell surface p32.
Figure 8.
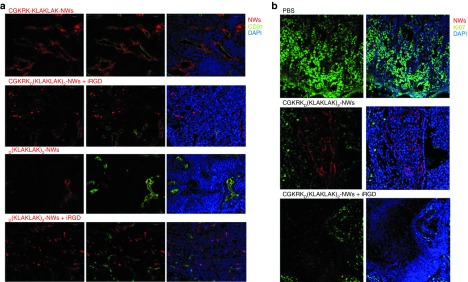
Nanoworm (NW) distribution and Ki67 staining in tumors of treated mice. At the end of a 3-week treatment of MCF10CA1a tumor in a study similar the one shown in Figure 7, the tumors were collected and processed for immunofluorescence. Confocal microscopy images of tumors from four representative animals in each group are shown. Red, NWs; green, (a) CD31 and (b) Ki67; blue, nuclei. Scale bars, 100 μm.
Discussion
We report here the identification of the target molecule (receptor) responsible for the specific homing of the CGKRK peptide to tumors as the mitochondrial protein p32, which is expressed at the cell surface specifically in tumor cells and other activated cells. Cell surface expression of p32 is characteristic of breast cancer, and we show that a tumor-targeting theranostic nanosystem, which uses the CGKRK peptide as a homing device and a proapoptotic peptide (D[KLAKLAK]2) as a drug, provides effective treatment in an orthotopic breast cancer model. The system was particularly effective when coadministered with the tumor-penetrating peptide iRGD.
A search of a receptor for CGKRK by affinity chromatography of tumor extracts on matrix-attached CGKRK revealed five proteins capable of binding this peptide. Two of them were known cell surface markers of angiogenic endothelial cells and tumor cells, namely, p3216,23 and nucleolin.18,24,25 Three of the proteins either had a low affinity for CGKRK or have not been observed at the cell surface. Binding and internalization assays conducted on knockdown cells showed that p32 is the predominant receptor for both functions, while NRD1 may play a role in the internalization of the peptide. The multiple binding activities of CGKRK bring up the possibility that it might be feasible to select for a peptide with a substantial affinity for multiple tumor-specific receptors. The number of any given receptor suitable for drug targeting in tumor tissue is limited, picomoles per gram of tumor, and often not sufficient to allow specific delivery of a drug to a tumor at high enough doses for a therapeutic effect.12 Multiple receptors for a single peptide would increase the number of receptors available for specific targeting.
The ability of CGKRK to become internalized into the target cell and to the mitochondria is likely to be a property of p32. In activated cells, p32 is present in mitochondria and at the cell surface.16 It may be that this protein cycles between these two localizations, as has been proposed for nucleolin,25 and can take a payload with it. We have described another tumor-homing peptide, a cyclic 9-amino acid peptide LyP-1, which binds to p32 and internalizes into its target cells.16,26 The differences between CGKRK and LyP-1 are interesting. While CGKRK mostly homes to tumor vasculature,6,27 LyP-1 accumulates in tumor macrophages, tumor lymphatics, and to a lesser degree in tumor cells.16,22,26 The main difference between these two peptides is that LyP-1 contains a cryptic sequence that binds to neuropilin-1 after Lyp-1 has been cleaved by a cell surface protease.28 The R/KXXR/K neuropilin-1-binding motif,13,29 which has to be C-terminal in a peptide to bind to neuropilin-1, is not present in CGKRK. This sequence, dubbed C-end Rule (CendR) sequence, activates an endocytic pathway that transports payloads through the vascular wall and through tumor tissue. CGKRK lacks the CendR function, and as a consequence, it mainly targets tumor blood vessels. In contrast, very little LyP-1 stays in the blood vessels, and it mostly accumulates in tumor macrophages.16 The effects on tumor macrophages may have contributed to the antitumor effects that we observed with the iRGD combination. As normal leukocyte cells do not express cell surface p32,16 they would not be affected. To combine the advantages of CGKRK and the CendR system, we made use of the ability of CendR peptides to promote extravasation and tumor penetration of coinjected payloads that are not coupled to the peptide.14 Indeed, coinjecting the CGKRKD[KLAKLAK]2-NWs with the tumor-specific cell-penetrating peptide iRGD14 enhanced the efficacy of the nanosystem, apparently because the tumor cells had now become a target in addition to tumor vessels.
The effectiveness of the CGKRKD[KLAKLAK]2-NW system has now been demonstrated in two tumor types, GBM6 and breast cancer (this study). The therapy in an aggressive GBM model significantly postponed the death of the animals. Rather than using survival as the endpoint, the accessible breast cancer tumors allowed us to monitor tumor size. We terminated the experiment shortly after the control tumors had reached the maximum size allowed by the institutional animal committee. Projecting tumor growth from the end of the experiment indicates that the extension of survival would have been as significant as in the GBM model. There are reasons to believe that the nanosystem will be broadly applicable to the treatment of additional tumor types. First, the CGKRK peptide homes to the blood vessels and binds to tumor cells in many, but not all, tumor types.27 Second, the receptor for CGKRK, p32, is overexpressed as a mitochondrial protein and is located on the cell surface in many types of tumors, whereas it remains exclusively intracellular in normal cells.16 Breast cancers in particular are almost universally positive for the cell surface form that makes p32 a tumor-specific target.16 Third, the D[KLAKLAK]2 peptide has a proapoptotic effect in a variety of mammalian cell types.6,10,30
Regarding the mechanisms of the antitumor activity, CGKRKD[KLAKLAK]2-NWs induced cell death by apoptosis in the same manner as monovalent peptide conjugates of D[KLAKLAK]2 alone.6,10 Consistent with an apoptotic mechanism, CGKRKD[KLAKLAK]2-NWs and D[KLAKLAK]2-NWs, but not CGKRK-NWs, induced annexin V expression, and the cell death was caspase dependent.6 As [KLAKLAK]2 acts by destroying mitochondria, the ability of CGKRK to take its payload to mitochondria through the p32 receptor makes this peptide a particularly effective tool for D[KLAKLAK]2 delivery and the ensuing apoptosis. As expected from the specificity of CGKRK, the damage appears to be primarily to the vasculature, as evidenced by the early–time point fibrin accumulation in the vasculature and reduction in the number of blood vessels later in the treatment. Why some of the vessels remain unaffected is an intriguing question. Answers to this question could well shed light to the somewhat disappointing clinical performance of antiangiogenic therapies. Regarding the increased efficacy of the CGKRKD[KLAKLAK]2-NWs in combination with iRGD, the likely mechanism is that the penetration of the NWs outside the blood vessels makes the tumor cells an additional target for the NWs, augmenting the effect of loss of vascular support on the tumors.
The D[KLAKLAK]2 peptide is particularly effective when presented on a NP, which is the case in our nanosystem.6 This high efficacy allowed us to reduce the dose of the D[KLAKLAK]2 peptide to such an extent that the pronounced toxicity encountered in treatment studies using homing peptide–targeted soluble D[KLAKLAK]210 was essentially eliminated.6 In agreement with the earlier GBM study,6 no histological evidence of tissue damage was seen in the present study in the liver, spleen, or kidneys of the treated animals, and their body weight remained at the same level. In the earlier study, we also monitored the potential toxicity of the CGKRKD[KLAKLAK]2 NWs by measuring L-Alanine-2-oxoglutarate aminotransferase levels in serum, used as an indicator of liver toxicity and found only minor elevation, which was transient. Finally, the CGKRK peptide, and any payload attached to it, is internalized into the target cells and transported to mitochondria.6 As D[KLAKLAK]2 acts on mitochondria,10 the subcellular targeting provided by CGKRK is a highly useful feature that further enhances the efficacy of the system in cancer treatment and reduces toxicity to normal cells because they are not targeted by CGKRK. Therefore, although substantial toxicity limited the use of soluble D[KLAKLAK]2 to proof-of-principle studies, NP-bound D[KLAKLAK]2 is a viable candidate for further preclinical and clinical development.
Materials and Methods
Cell lines and tumors. MDA-MB-435 human tumor cells were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum and 1% Glutamine-Pen-Strep (Omega scientific, Tarzana, CA) at 37 °C/5% CO2. The MCF10A cell line was obtained from the American Type Culture Collection (Manassas, VA), while the MCF10AT and MCF10CA1a cells were from The Barbara Ann Karmanos Cancer Institute (Detroit, MI). Cells were maintained in Dulbecco's Modified Eagle Medium supplemented with 10% fetal bovine serum, 1% Glutamine-Pen-Strep and 100 ng/ml human epidermal growth factor (Sigma-Aldrich, St. Louis, MI). The FVB-Tg(c3-1-TAg)Cjeg/JegJ mice (Jackson Laboratory, Bar Harbor, Maine), which develop spontaneous breast cancers, were maintained in the animal facility at Sanford-Burnham Medical Research Institute. Human umbilical vein endothelial cells (HUVEC; Lonza Walkersville, Walkersville, MD) were cultured using EBM-2 medium with endothelial cell growth supplement (Lonza Walkersville). Mouse GBM-initiating 005 cells31 were maintained in N2 medium, which contains Dulbecco's modified Eagle medium/F-12 (Omega Scientific), 1% N-2 supplement (Invitrogen, Carlsbad, CA), 20 ng/ml human fibroblast growth factor-2 (Prepotech, Rocky Hill, NJ), 20 ng/ml human epidermal growth factor(Promega, Madison, WI), and 40 µg/ml heparin (Sigma-Aldrich). T3 cells were obtained by induction of differentiation in 005 cells21 cultured in EGM-2 (Lonza Walkersville).
To produce MDA-MB-435 and MCF10CA1a tumors, BALB/c nude mice were orthotopically injected into the mammary fat pad with 2 × 106 cells suspended in 100 µl of PBS. Mouse GBM-initiating 005 cells were transplanted into brains of NOD–SCID mice. A total of 3 × 105 cells were suspended in 1.5 µl of PBS and injected stereotaxically in the right hippocampus. Animal experimentation was performed according to the procedures approved by the Animal Research Committee at the University of California, Santa Barbara; the Sanford-Burnham Medical Research Institute; and The Salk Institute for Biological Research, San Diego.
Peptide synthesis. Peptides were synthesized using an automatic microwave-assisted peptide synthesizer (Liberty; CEM, Matthews, NC) using standard solid-phase Fmoc/t-Bu chemistry with 2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphatem ethanaminium (Anaspec, San Jose, CA) as the coupling reagent. During synthesis, the peptides were labeled with FAM or rhodamine (Sigma-Aldrich) with a 6-aminohexanoic acid spacer separating the dye from the sequence. The peptides were cleaved from the resin using 95% trifluoroacetic acid (Sigma-Aldrich) with 2.5% water and triisopropylsilane (Sigma-Aldrich). Subsequent purification by high-performance liquid chromatography (Gilson, Middleton, WI) gave peptides with >90% purity.
Protein isolation and affinity chromatography. Mitochondria were isolated from livers of Balb/c mice using differential centrifugation with buffers from a Pierce mitochondrial isolation kit for tissue according to the manufacturer's instructions (Pierce Biotechnology, Rockford, IL).
Mitochondria/tumors were lysed in PBS containing 400 mmol/l n-octyl-β-D-glucopyranoside (Calbiochem, La Jolla, CA), and clarified lysates were incubated with CGKRK-coated Sulfolink-beads (Pierce Biotechnology). After washing, bound proteins were eluted with lysis buffer containing 2 mmol/l free CGKRK peptide and separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Gel bands excised from silver-stained gels were analyzed by matrix-assisted laser desorption-ionization time-of-flight mass spectrometry (MALDI-TOF) mass spectrometry at the Burnham Institute for Medical Research Proteomics Resource.
Immunoblot analysis. The lysates were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. After transfer of the proteins onto nitrocellulose membranes for 2 hours at 200 mA, the membranes were treated for 1 hour at room temperature with Tris-buffered saline–0.05% Tween 20 containing 5% milk and incubated with 1 µg/ml anti-p32 (R&D system, Minneapolis, MN), followed by anti-goat (R&D system), anti-nucleolin (Abcam, Cambridge, MA), anti-nardilysin (GeneTex, Irvine, CA), or anti-CKAP4 (Novus, Littleton, CO), followed by anti-rabbit IgG (Millipore, Billerica, MA), or anti-actin (Sigma-Aldrich), followed by anti-mouse (Invitrogen) antibodies.
Affinity measurements. The affinity of CGKRK for p32 was measured by an enzyme-linked immunosorbent assay-based experiment. Wells in 96-well plates were coated with 3–5 µg/ml of purified nucleolin, CKAP4, nardilysin, or p32 protein (Abcam) and incubated for 2 hours at 37 °C with various concentrations of FAM-CGKRK peptide (or FAM-CREKA as control) in PBS (100 µl/well). After washing with PBS containing 0.2 mol/l NaCl2 and 0.05% Tween 20, peptide binding to protein was quantified with a fluorescence plate reader (Spectra Max Gemini, San Diego, CA). Wells with various concentrations of FAM–peptide and without protein were used to determine background binding. Kd values were calculated using the Prism software.
Flow cytometry. Cells were harvested and stained using a phycoerythrin conjugate anti-p32 (Santa Cruz Biotechnology, Santa Cruz, CA) and analyzed on a BD LSR II flow cytometer (Becton Dickinson, Franklin Lakes, NJ).
In vitro phage binding and penetration assays. Suspended cells (106 cells in Dulbecco's modified Eagle medium containing 1% bovine serum albumin) were incubated with 109 plaque-forming units/ml of T7 phage overnight at 4 °C. The cells were washed four times with the binding buffer, lysed with lysogeny broth containing 1% NP-40, and titrated. Phage penetration assays used the same procedure, except that the cells were incubated with phage for 1 hour at 37 °C and an acidic buffer (500 mmol/l sodium chloride, 0.1 mol/l glycine, 1% bovine serum albumin, pH 2.5) was substituted for the binding buffer in the second wash to remove the phage bound to the cell surface.
Lentiviral short hairpin RNA infection. The short hairpin RNA hairpin-targeting mouse p32 was cloned into the NheI site of the p156RRLsin vector (a third-generation lentiviral vector (Ref. 1). The short hairpin-targeting p32 oligo sequence used in this study was: 5′-GGATGAGATTGGTCACGAAGA-3′. Lentivirus was prepared as described previously (Ref. 2).32,33
Small interfering RNA transfection. Oligonucleotide duplexes for transient small interfering RNA (siRNA) knockdown of nardilysin (Nrd1RSS303733 Stealth RNAi) and negative control duplexes (Stealth RNAi control, low GC and medium GC) were purchased from Invitrogen. Seventy-five thousand cells were transfected with 10–90 nmol/l NRD1 small interfering RNAs using lipofectamine RNAiMAX reagent (Invitrogen). Forty-eight hours after transfection, the cells were harvested and analyzed by immunoblotting with an antibody against NRD1 (GeneTex), to confirm the downregulation of NRD1, and in phage binding assays as described above.
Preparation of NWs. NWs coated with peptides were prepared as described earlier.7,14 Briefly, aminated NWs were pegylated with maleimide-5KPEG-NHS (Jenkem Technology, Allen, TX). Peptides were conjugated to the NPs through a thioether bond between the cysteine thiol of the peptide sequence and the maleimide on the functionalized particles.
In vivo peptide homing. FAM-labeled CGKRK (200 µg) was intravenously injected into mice with orthotopic breast tumors and allowed to circulate for 3 hours. The mice were perfused with PBS through the heart under anesthesia, and tissues were collected and processed for fluorescence analysis.
In vivo NW injections. Mice bearing orthotopic breast tumors were injected into the tail vein with NWs (5 mg/kg body weight of iron). In homing experiments, the mice were euthanized 5–6 hours after the injection by cardiac perfusion with PBS under anesthesia, and organs were dissected and analyzed for NWs. In tumor treatment experiments, tumor-bearing mice were intravenously treated with NWs in 150 µl PBS, or PBS as a control, every other day for 3 weeks. Mice with breast tumors were also intravenously treated with CGKRKD[KLAKLAK]2-NW (5 mg of iron/kg) in combination with 4 µmol/kg of the tumor-penetrating peptide iRGD. Tumor volume was calculated using the following formula: volume = (d2 × D)/2, where d and D are the smallest and the largest tumor diameters, respectively.
Histology and immunohistology. Tissues were fixed in 4% paraformaldehyde overnight at 4 °C, cryoprotected in 30% sucrose overnight, and frozen in optimum cutting temperature embedding medium. Tissue sections (7 µm) were cut and stained with hematoxylin and eosinor processed for immunostaining. For CD31, Ki67, or p32 staining, sections were first incubated for 1 hour at room temperature with 10% serum from the species in which the secondary antibody was generated, followed by incubation with monoclonal anti-rat CD31 (BD Pharmingen, San Jose, CA) and Alexa 488 goat anti-rat secondary antibody (1:1,000; Molecular Probes, Eugene, OR); anti-goat fibrinogen (Acris Antibodies, San Diego, CA), followed by incubation with Alexa 488 donkey anti-goat secondary antibody; anti-rabbit Ki67 (Abcam) and Alexa 488 goat anti-rabbit secondary antibody (1:1,000; Molecular Probes); or anti-rabbit p3216 and Alexa 647 goat anti-rabbit secondary antibody (1:1,000; Molecular Probes). Each staining experiment included sections stained with the secondary antibody only as a negative control. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay for apoptosis detection was conducted with Click-iT TUNEL 647 Alexa Fluor Imaging Assay (Invitrogen) according to the manufacturer's instruction. Nuclei were counterstained with diamidino-2-phenylindole (5 mg/ml; Molecular Probes). The sections were mounted in Gel/Mount mounting medium (Biomeda, Foster City, CA) and viewed under a Fluoview 500 confocal microscope (Olympus America, Center Valley, PA).
Statistical analysis. Data were analyzed by two-tailed Student's unpaired t-test or one-way analysis of variance (ANOVA), followed by suitable post hoc test. P values <0.05 were considered statistically significant.
SUPPLEMENTARY MATERIAL Figure S1. Detection of nonspecific protein binding to CGKRK affinity columns. Figure S2. Inhibition of FAM-CGKRK binding to purified p32 by anti-p32. Figure S3. CGKRK peptide distribution in normal mouse organs. Figure S4. CGKRKD[KLAKLAK]2-NW accumulation in normal mouse organs after a single injection. Figure S5. Distribution of CGKRK-NWs and D[KLAKLAK]2-NWs in tumor tissue after long-term treatment. Figure S6. Distribution of CGKRKD[KLAKLAK]2-NWs in normal mouse organs at the end of long-term treatment. Figure S7. Effects of CGKRKD[KLAKLAK]2-NW treatment on tumors. Table S1. Identification of CGKRK-binding 005 brain tumor proteins by mass spectrometry.
Acknowledgments
We thank Dr Inder Verma (Salk Institute) for comments and Dr Michael J Sailor (University of California, San Diego) and Dr Ji-Ho Park (Korea Advanced Institute of Science and Technology) for advice on the synthesis of iron oxide nanoparticles. This work was supported by grants W81XWH-10-1-0198 and W81XWH-08-1-0727 from the Department of Defense, grants CA152327 and CA012442 from the NIH/NCI, and Cancer Center Support Grants CA030199 and CA014195 from the NCI. VRK, KNS, and ER have ownership interest (including patents) in CendR Therapeutics. ER is the founder, Chairman of the board, consultant/advisory board member, and major shareholder of CendR Therapeutics and has ownership interest (including patents) in the same. No potential conflicts of interest were disclosed by the other authors.
The views and opinions of authors expressed on OER web sites do not necessarily state or reflect those of the US Government, and they may not be used for advertising or product endorsement purposes.
The opinions expressed herein are those of the author(s) and are not necessarily representative of those of the Uniformed Services University of the Health Sciences (USUHS), the Department of Defense (DOD); or the United States Army, Navy, or Air Force.
Supplementary Material
References
- Ferrari M. Frontiers in cancer nanomedicine: directing mass transport through biological barriers. Trends Biotechnol. 2010;28:181–188. doi: 10.1016/j.tibtech.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N, Alitalo K. Clinical applications of angiogenic growth factors and their inhibitors. Nat Med. 1999;5:1359–1364. doi: 10.1038/70928. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E. Peptides as targeting elements and tissue penetration devices for nanoparticles. Adv Mater Weinheim. 2012;24:3747–3756. doi: 10.1002/adma.201200454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradishar WJ, Tjulandin S, Davidson N, Shaw H, Desai N, Bhar P, et al. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J Clin Oncol. 2005;23:7794–7803. doi: 10.1200/JCO.2005.04.937. [DOI] [PubMed] [Google Scholar]
- Haley B, Frenkel E. Nanoparticles for drug delivery in cancer treatment. Urol Oncol. 2008;26:57–64. doi: 10.1016/j.urolonc.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Agemy L, Friedmann-Morvinski D, Kotamraju VR, Roth L, Sugahara KN, Girard OM, et al. Targeted nanoparticle enhanced proapoptotic peptide as potential therapy for glioblastoma. Proc Natl Acad Sci USA. 2011;108:17450–17455. doi: 10.1073/pnas.1114518108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, von Maltzahn G, Zhang L, Derfus AM, Simberg D, Harris TJ, et al. Systematic surface engineering of magnetic nanoworms for in vivo tumor targeting. Small. 2009;5:694–700. doi: 10.1002/smll.200801789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce JA, Laakkonen P, Bernasconi M, Bergers G, Ruoslahti E, Hanahan D. Stage-specific vascular markers revealed by phage display in a mouse model of pancreatic islet tumorigenesis. Cancer Cell. 2003;4:393–403. doi: 10.1016/s1535-6108(03)00271-x. [DOI] [PubMed] [Google Scholar]
- Javadpour MM, Juban MM, Lo WC, Bishop SM, Alberty JB, Cowell SM, et al. De novo antimicrobial peptides with low mammalian cell toxicity. J Med Chem. 1996;39:3107–3113. doi: 10.1021/jm9509410. [DOI] [PubMed] [Google Scholar]
- Ellerby HM, Arap W, Ellerby LM, Kain R, Andrusiak R, Rio GD, et al. Anti-cancer activity of targeted pro-apoptotic peptides. Nat Med. 1999;5:1032–1038. doi: 10.1038/12469. [DOI] [PubMed] [Google Scholar]
- Arap W, Haedicke W, Bernasconi M, Kain R, Rajotte D, Krajewski S, et al. Targeting the prostate for destruction through a vascular address. Proc Natl Acad Sci USA. 2002;99:1527–1531. doi: 10.1073/pnas.241655998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruoslahti E, Bhatia SN, Sailor MJ. Targeting of drugs and nanoparticles to tumors. J Cell Biol. 2010;188:759–768. doi: 10.1083/jcb.200910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Girard OM, et al. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell. 2009;16:510–520. doi: 10.1016/j.ccr.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Greenwald DR, et al. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science. 2010;328:1031–1035. doi: 10.1126/science.1183057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krainer AR, Mayeda A, Kozak D, Binns G. Functional expression of cloned human splicing factor SF2: homology to RNA-binding proteins, U1 70K, and Drosophila splicing regulators. Cell. 1991;66:383–394. doi: 10.1016/0092-8674(91)90627-b. [DOI] [PubMed] [Google Scholar]
- Fogal V, Zhang L, Krajewski S, Ruoslahti E. Mitochondrial/cell-surface protein p32/gC1qR as a molecular target in tumor cells and tumor stroma. Cancer Res. 2008;68:7210–7218. doi: 10.1158/0008-5472.CAN-07-6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdreich-Epstein A, Shimada H, Groshen S, Liu M, Metelitsa LS, Kim KS, et al. Integrins alpha(v)beta3 and alpha(v)beta5 are expressed by endothelium of high-risk neuroblastoma and their inhibition is associated with increased endogenous ceramide. Cancer Res. 2000;60:712–721. [PubMed] [Google Scholar]
- Christian S, Pilch J, Akerman ME, Porkka K, Laakkonen P, Ruoslahti E. Nucleolin expressed at the cell surface is a marker of endothelial cells in angiogenic blood vessels. J Cell Biol. 2003;163:871–878. doi: 10.1083/jcb.200304132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basolo F, Elliott J, Tait L, Chen XQ, Maloney T, Russo IH, et al. Transformation of human breast epithelial cells by c-Ha-ras oncogene. Mol Carcinog. 1991;4:25–35. doi: 10.1002/mc.2940040106. [DOI] [PubMed] [Google Scholar]
- Santner SJ, Dawson PJ, Tait L, Soule HD, Eliason J, Mohamed AN, et al. Malignant MCF10CA1 cell lines derived from premalignant human breast epithelial MCF10AT cells. Breast Cancer Res Treat. 2001;65:101–110. doi: 10.1023/a:1006461422273. [DOI] [PubMed] [Google Scholar]
- Soda Y, Marumoto T, Friedmann-Morvinski D, Soda M, Liu F, Michiue H, et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc Natl Acad Sci USA. 2011;108:4274–4280. doi: 10.1073/pnas.1016030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laakkonen P, Akerman ME, Biliran H, Yang M, Ferrer F, Karpanen T, et al. Antitumor activity of a homing peptide that targets tumor lymphatics and tumor cells. Proc Natl Acad Sci USA. 2004;101:9381–9386. doi: 10.1073/pnas.0403317101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamzah J, Kotamraju VR, Seo JW, Agemy L, Fogal V, Mahakian LM, et al. Specific penetration and accumulation of a homing peptide within atherosclerotic plaques of apolipoprotein E-deficient mice. Proc Natl Acad Sci USA. 2011;108:7154–7159. doi: 10.1073/pnas.1104540108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said EA, Krust B, Nisole S, Svab J, Briand JP, Hovanessian AG. The anti-HIV cytokine midkine binds the cell surface-expressed nucleolin as a low affinity receptor. J Biol Chem. 2002;277:37492–37502. doi: 10.1074/jbc.M201194200. [DOI] [PubMed] [Google Scholar]
- Shibata Y, Muramatsu T, Hirai M, Inui T, Kimura T, Saito H, et al. Nuclear targeting by the growth factor midkine. Mol Cell Biol. 2002;22:6788–6796. doi: 10.1128/MCB.22.19.6788-6796.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laakkonen P, Porkka K, Hoffman JA, Ruoslahti E. A tumor-homing peptide with a targeting specificity related to lymphatic vessels. Nat Med. 2002;8:751–755. doi: 10.1038/nm720. [DOI] [PubMed] [Google Scholar]
- Hoffman JA, Giraudo E, Singh M, Zhang L, Inoue M, Porkka K, et al. Progressive vascular changes in a transgenic mouse model of squamous cell carcinoma. Cancer Cell. 2003;4:383–391. doi: 10.1016/s1535-6108(03)00273-3. [DOI] [PubMed] [Google Scholar]
- Roth L, Agemy L, Kotamraju VR, Braun G, Teesalu T, Sugahara KN, et al. Transtumoral targeting enabled by a novel neuropilin-binding peptide. Oncogene. 2012;31:3754–3763. doi: 10.1038/onc.2011.537. [DOI] [PubMed] [Google Scholar]
- Teesalu T, Sugahara KN, Kotamraju VR, Ruoslahti E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc Natl Acad Sci USA. 2009;106:16157–16162. doi: 10.1073/pnas.0908201106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standley SM, Toft DJ, Cheng H, Soukasene S, Chen J, Raja SM, et al. Induction of cancer cell death by self-assembling nanostructures incorporating a cytotoxic peptide. Cancer Res. 2010;70:3020–3026. doi: 10.1158/0008-5472.CAN-09-3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marumoto T, Tashiro A, Friedmann-Morvinski D, Scadeng M, Soda Y, Gage FH, et al. Development of a novel mouse glioma model using lentiviral vectors. Nat Med. 2009;15:110–116. doi: 10.1038/nm.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikawa M, Tanaka N, Kao WW, Verma IM. Generation of transgenic mice using lentiviral vectors: a novel preclinical assessment of lentiviral vectors for gene therapy. Mol Ther. 2003;8:666–673. doi: 10.1016/s1525-0016(03)00240-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.