

Abstract
Mutations in superoxide dismutase 1 (SOD1) are linked to familial amyotrophic lateral sclerosis (ALS) resulting in progressive motor neuron death through one or more acquired toxicities. Involvement of wild-type SOD1 has been linked to sporadic ALS, as misfolded SOD1 has been reported in affected tissues of sporadic patients and toxicity of astrocytes derived from sporadic ALS patients to motor neurons has been reported to be reduced by lowering the synthesis of SOD1. We now report slowed disease onset and progression in two mouse models following therapeutic delivery using a single peripheral injection of an adeno-associated virus serotype 9 (AAV9) encoding an shRNA to reduce the synthesis of ALS-causing human SOD1 mutants. Delivery to young mice that develop aggressive, fatal paralysis extended survival by delaying both disease onset and slowing progression. In a later-onset model, AAV9 delivery after onset markedly slowed disease progression and significantly extended survival. Moreover, AAV9 delivered intrathecally to nonhuman primates is demonstrated to yield robust SOD1 suppression in motor neurons and glia throughout the spinal cord and therefore, setting the stage for AAV9-mediated therapy in human clinical trials.
Amyotrophic lateral sclerosis (ALS) is an adult-onset, rapidly progressive, and fatal neurodegenerative disease, characterized by selective degeneration of both upper and lower motor neurons. ALS is the most prominent motor neuron disease, responsible for one in every 2,000 deaths. Most of the cases have no clear genetic linkage and are referred to as sporadic but in 10% of the instances, disease is familial with dominant inheritance. Twenty percent of the familial cases are caused by mutations in superoxide dismutase 1 (SOD1), with over 140 distinct mutations identified to date.1,2 Many efforts to identify how mutations alter the function of SOD1 have produced a consensus view that SOD1 mutants acquire one or more toxicities, whose nature still remains controversial.3 However, there is clear evidence that a proportion of mutant SOD1 is misfolded and subsequently aggregates.4,5 SOD1 aggregates are, in fact, one of the histological hallmarks of SOD1-related ALS cases.4
In the past 20 years, multiple animal models expressing mutant forms of human SOD1 have been generated. These models recapitulate the hallmarks of ALS, developing age-dependent motor axon degeneration and accompanying muscle denervation, glial inflammation, and subsequent motor neuron loss. Selective gene excision experiments have determined that mutant SOD1 expression within motor neurons themselves contributes to disease onset and early disease progression,6 as does mutant synthesis in NG2+ cells7 that are precursors to oligodendrocytes. However, mutant SOD1 protein expression in microglia and astrocytes significantly drives rapid disease progression,6,8 findings which have led to the conclusion that ALS pathophysiology is noncell autonomous.3
Furthermore, astrocytes have been found to be toxic to motor neurons in multiple in vitro models where mutant forms of human SOD1 were overexpressed.9,10,11 A recent study by our group derived astrocytes from postmortem spinal cords of ALS patients with or without SOD1 mutations. In all cases, astrocytes from sporadic ALS patients were as toxic to motor neurons as astrocytes carrying genetic mutations in SOD1, but neither sporadic nor familial ALS glia were toxic to GABAergic neurons.12 Even more strikingly, reduction of SOD1 in astrocytes derived from both sporadic and familial ALS patients decreased astrocyte-derived toxicity toward motor neurons. This finding, along with the reports that misfolded SOD1 inclusions are found in the spinal cords of familial as well as some sporadic ALS patients,13,14,15 has provided strong evidence for a pathogenic role of wild-type SOD1 in sporadic ALS.
Despite the insights that SOD1-mutant–expressing animal models have provided for understanding the mechanisms involved in motor neuron degeneration, their utility for the development of therapeutic approaches has been questioned,16 as no drug with a reported survival benefit in mutant SOD1G93A mice has been effective in clinical trials with sporadic ALS patients. We note, however, that in all but one case, the drugs taken to human trial had been reported only to extend mutant SOD1 mouse survival when applied presymptomatically and even then to provide a survival benefit solely by delaying disease onset with no benefit in slowing disease progression. The one exception to this was riluzole, which similar to the human situation, modestly extended survival of mutant SOD1G93A mice and did so by slowing disease progression.17 Recognizing that success at human trial will require slowing of disease progression, the SOD1-mutant mice have perfectly predicted the success of riluzole and the failure of efficacy of each other drug attempted in human trial. What have been missing are additional therapies that affect disease progression in these mice.
Previous studies have established that adeno-associated virus 9 (AAV9) can cross the blood–brain barrier and efficiently target neurons and astrocytes in the brain and spinal cord when injected systemically.18,19 We hypothesized that these attributes of AAV9 could be used to deliver SOD1 shRNA to slow disease progression in models of ALS. We have now tested this hypothesis in two mouse models using a single, peripheral injection of AAV9-encoding SOD1 shRNA. Furthermore, we also tested if intrathecal delivery of AAV9-encoding SOD1 shRNA and green flourescent protein (GFP) into nonhuman primates could efficiently target all levels of the spinal cord and significantly reduce the levels of SOD1.
Results
AAV9 transduction pattern and persistence in SOD1G93A mice
We first evaluated the efficiency of AAV9 transduction in the SOD1G93A mouse model that develops fatal paralytic disease. Animals were injected intravenously at postnatal day 1 or day 21 (to be referred to as P1 and P21, respectively) with self-complementary AAV9-expressing GFP from the cytomegalovirus enhancer/β-actin (CB) promoter (AAV9-CB-GFP; n = 3 per group). Three weeks postinjection, animals were killed and spinal cords were examined for GFP expression (Figure 1a–u). Transduction efficiency was high in SOD1G93A astrocytes with GFP expressed in 34 ± 2 and 54 ± 3% of P1- and P21-injected spinal gray matter astrocytes (defined by immunoreactivity for glial fibrillary acidic protein (GFAP)). This efficiency was similar to our previous report of 64 ± 1% in P21-injected wild-type animals.18 Motor neurons were a prominent cell type transduced at all levels of the spinal cords of P1-injected SOD1G93A animals (62 ± 1%), compared with significantly lower targeting to motor neurons in P21-injected animals (8 ± 1%).
Figure 1.

AAV9 transduction pattern and persistence in SOD1G93A mice. SOD1G93A mice were injected intravenously with AAV9-CB-GFP at P1 and P21 and euthanized 21 days postinjection (n = 3 per time point). Spinal cords were examined for GFP, ChAT (motor neuron marker), and GFAP (astrocyte marker) expression. (a,f,k,p) Temporal vein injection of AAV9-CB-GFP at P1 resulted in efficient transduction of motor neurons and glia in SOD1G93A mice. (b,g,l,q) Tail vein injection at P21 predominantly targeted astrocytes with few GFP-positive motor neurons. To test the persistence of transduced cells, AAV9-CB-GFP was intravenously injected at P1 and P21 in SOD1G93A animals that were killed at end stage (~P130). (c,d,h,i,m,n,r,s) Immunofluorescence analysis of lumbar ventral horn demonstrated that GFP expression was maintained in astrocytes throughout the disease course. To determine whether SOD1-mediated inflammation and damage would affect AAV9 transduction, we intravenously injected SOD1G93A mice at P85 and harvested their spinal cords at end stage. There was no difference observed in the transduction pattern of SOD1G93A mice treated at P21 or P85. Insets in (r–t) show colocalization between GFP and GFAP signal. (u) Quantification of transduced cells in ALS spinal cords (for each group tissues were analyzed from three animals). GFP, ChAT,and GFAP columns show numbers of cells counted. Bars = 100 µm. AAV, adeno-associated virus; GFP, green fluorescent protein; ChAT, choline acetyltransferase; GFAP, glial fibrillary acidic protein; P1, postnatal day 1; P21, postnatal day 21; P85, postnatal day 85.
Although we have previously reported that transduced astrocytes in wild-type spinal cords persist with continued GFP accumulation for at least 7 weeks postinjection,18 longevity of mutant SOD1 astrocytes (and their continued synthesis of genes encoded by the AAV9 episome) during active ALS-like disease was untested. Therefore, SOD1G93A mice were injected at P1 and P21 with AAV9-CB-GFP and followed to end stage (~P130; n = 3 per group) (Figure 1c,d,h,i,m,n,r,s). Immunofluorescent examination of the end-stage SOD1G93A spinal cords from animals injected at P1 and P21 showed a comparable number of GFP-expressing astrocytes as were found 21 days after AAV9 injection (P1: 42 ± 2%, P21: 61 ± 2%). These data are consistent with the survival of transduced astrocytes for the duration of disease (~110 days postinjection at P21) in SOD1G93A mice and that AAV9-encoded gene expression is maintained.
Furthermore, recognizing SOD1-mutant–mediated damage, including astrocytic and microglial activation and early changes in the blood–brain barrier develop during disease in SOD1-mutant mice,20 we tested if this damage affected AAV9 transduction. SOD1G93A mice were injected at P85 with AAV9-CB-GFP and killed at end stage (n = 3; Figure 1e,j,o,t). Analysis of the spinal cords revealed that the transduction pattern seen in P85 animals was similar to P21-treated animals with astrocytes as the predominant cell type transduced at all levels (51 ± 6% GFP+/GFAP+ cells in lumbar gray matter).
Development of an shRNA sequence specific for human SOD1
To specifically target the human SOD1 mRNA, we generated four shRNA constructs that had a minimum of four base mismatches compared with the mouse mRNA sequence (Figure 2a). The base numbers for the human sequences shown correspond to record number CCDS33536.1 in the NCBI CCDS database. Each shRNA sequence was inserted into an expression cassette to place it under control of the H1 promoter. Human 293 cells were transfected with each cassette, lysates were harvested 72 hours posttransfection, and SOD1 levels were analyzed by immunoblotting. All four sequences reduced SOD1 protein levels by >50% (Figure 2b,c). shRNA130 was selected because it produced the most consistent knockdown across three separate transfection experiments. It was cloned into a self-complementary AAV9 vector that also contained a GFP gene whose expression would identify transduced cells (referred to as AAV9-SOD1-shRNA). To confirm that the shRNA could suppress accumulation of human SOD1, SOD1G93A mice (n = 3) were injected intravenously with AAV9-SOD1-shRNA at either P1 or P21. Animals were killed 3 weeks postinjection and the spinal cords were harvested and analyzed by immunoblotting for both human (mutant) and murine (wild-type) SOD1 protein. P1- and P21-injected spinal cords showed 60 and 45% reductions in mutant SOD1 protein, respectively (Figure 2d,e). Murine SOD1 levels remained unchanged in response to human SOD1 knockdown.
Figure 2.

shRNA constructs show efficient reduction of human SOD1 protein in vitro and in vivo. (a) Sequence alignments between human and mouse SOD1 for the regions targeted by the four different shRNA constructs tested. (b) shRNA sequences were cloned into an H1 expression construct and transiently transfected into 293 cells. Lysates were collected 72 hours posttransfection and analyzed by western blot. (c) Quantification of in vitro suppression of human SOD1 from three separate transient transfections showed >50% reduction in SOD1. (d) shRNA 130 was packaged into AAV9 and injected into SOD1G93A mice at either P1 or P21. Spinal cords (n = 3 per time point) were harvested 3 weeks postinjection and analyzed by western blot for human SOD1 protein levels. (e) Quantification of in vivo suppression of human SOD1 within the spinal cord of ALS mice. P1- and P21-injected spinal cords showed 60 and 45% reductions in mutant SOD1 protein. GAPDH, glyceraldehyde 3 phosphate dehydrogenase; hSOD1, human superoxide dismutase 1; mSOD1, mouse superoxide dismutase 1; P1, postnatal day 1; P21, postnatal day 21.
AAV9-SOD1-shRNA is safe and well tolerated in wild-type mice
To determine whether high-dose AAV9-SOD1-shRNA would be safe, normal mice of both sexes were intravenously injected at P1 or P21 (P1: 5 males, 5 females at 3.6 × 1014 vg/kg; P21: 5 males, 5 females at 1.7 × 1014 vg/kg (vector genomes per kg body weight)) and then monitored up to 6 months of age. Both P1- and P21-injected mice showed a steady increase in body mass similar to untreated mice (Supplementary Figure S1). Weekly behavioral tests observed no significant differences between injected and control groups in motor skills (measured by rotarod) as well as in hindlimb grip strength. At 150 and 180 days of age, blood samples were collected. Complete and differential blood counts of both treated and untreated groups showed similar blood chemistry parameters (Supplementary Figure S2). Serum samples from both groups showed no significant differences in the levels of alkaline phosphatase, creatinine, blood urea nitrogen, potassium, sodium, and chloride. Finally, all the animals were killed at the age of 180 days. Histopathological analyses by a pathologist blinded to treatment group revealed no significant alterations in the AAV9-SOD1-shRNA treated animals compared with the uninjected controls (data not shown). We conclude that both administration of AAV9 and sustained shRNA expression were apparently safe and well tolerated.
Extended survival of SOD1G93A mice from AAV9-mediated reduction in mutant SOD1 even when initiated mid-disease
To test the efficacy of AAV9-mediated SOD1 reduction, we treated cohorts of SOD1G93A mice with a single intravenous injection of AAV9-SOD1-shRNA before (P1: 3.6 × 1014 vg/kg, n = 6 and P21: 1.7 × 1014 vg/kg, n = 9) or after (P85: 1.6 × 1014 vg/kg, n = 5) onset, recognizing that many astrocytes, but few motor neurons, would be transduced at the two later time points. Onset of disease (measured by weight loss from denervation-induced muscle atrophy) was significantly delayed by a median of 39.5 days (Figure 3a,c; uninjected, 103 days; P1: 142.5 days; P < 0.05, Wilcoxon signed-rank test) in the P1-injected cohort but was not affected by either of the later injections (P21: 110 days; P85: 105 days). P1- and P21-treated animals maintained their weights, had better rotarod performance and hindlimb grip strength when compared with the age-matched controls, indicating treated animals maintained muscle tone and motor function during their prolonged survival (Figure 3f–h). Survival was significantly extended by AAV9 injection at all three ages, yielding survival times 30–51.5 days beyond that of uninjected SOD1G93A mice (uninjected: 132 days; P1: 183.5 days; P21: 171 days; P85: 162 days; log-rank test, P ≤ 0.0001, P ≤ 0.0003, and P ≤ 0.001, respectively) (Figure 3b,e). Defining disease duration as the time from onset to end stage revealed that the P21 treatment group had significantly increased duration, indicative of slowed disease progression, compared with the uninjected controls (uninjected: 29.5 days; P21: 49 days; Wilcoxon signed-rank test, P = 0.01), with trends toward slowed disease progression in animals injected at the other two ages (P1: 41 days; P85: 40 days; P = 0.06 and P = 0.12, respectively) (Figure 3d). The lower percentage of targeted non-neuronal cells at P1 versus those targeted at P21 (Figure 1u) suggests that a minimum percentage of non-neuronal cells must be targeted to slow disease progression in the fast-progressing SOD1G93A model (Figure 1u).
Figure 3.

Intravenous delivery of AAV9-SOD1-shRNA improves survival and motor performance in SOD1G93A mice. SOD1G93A mice received a single intravenous injection of AAV9-SOD1-shRNA at P1 (n = 6, green), P21 (n = 9, red), or P85 (n = 5, blue). Treated mice were monitored up to end stage and compared with noninjected control SOD1G93A mice (n = 15, gray). (a,c) AAV9-SOD1-shRNA injection into P1 SOD1G93A mice significantly delayed median disease onset 39.5 days compared with control animals (uninjected: 103 days; P1: 142.5 days; P < 0.05). Injection in P21 (red) or P85 (blue) ALS animals had no effect on disease onset (P21: 110 days; P85: 105 days). However, AAV9-SOD1-shRNA administered at P1, P21, or P85 all significantly extended median survival (b,e) (uninjected: 132 days; P1: 183.5 days; P21: 171 days; P85: 162 days; all comparisons to control P < 0.001). The P21 group had a significant extension in median disease duration (d) indicating a slowing of disease (uninjected: 29.5 days; P1: 41 days; P21: 49 days; P85: 40 days; Wilcoxon signed-rank test, P = 0.06, P = 0.01, and P = 0.12, respectively). (f-h) P1- and P21-treated animals maintained their weights, had better hindlimb grip strength and rotarod performance as compared with age-matched controls, indicating treated animals retained muscle tone and motor function during their prolonged survival. Lines between bars in (c–e) indicate statistically significant differences. *P < 0.05. P1, postnatal day 1; P21, postnatal day 21; P85, postnatal day 85.
Reduction of mutant SOD1 in AAV9-infected cells in treated SOD1G93A mice
Indirect immunofluorescence with an antibody that recognizes human, but not mouse SOD1, was used to determine accumulated mutant SOD1 levels in end-stage spinal cords of treated and control mice. Human SOD1 levels in end-stage spinal cord sections inversely correlated with increased survival (Figure 4a–d). At end stage, P1 (Figure 4b), P21 (Figure 4c), and P85 (Figure 4d), AAV9-SOD1-shRNA-injected animals had lower levels of mutant SOD1 when compared with the uninjected SOD1G93A animals (Figure 4a). SOD1 expression within transduced motor neurons (identified by GFP- and choline acetyltransferase (ChAT)-expressing cells) was reduced compared with the surrounding neurons that had not been transduced to express viral-encoded GFP (Figure 4h,l,p,t; arrows versus arrowheads). Moreover, immunofluorescence imaging of end-stage spinal cords revealed corresponding reduction in astrogliosis but no difference was observed in microgliosis in AAV9-SOD1-shRNA-treated animals versus controls (Supplementary Figure S3).
Figure 4.

Intravenous injection of AAV9-SOD1-shRNA reduces mutant protein in spinal cords of SOD1G93A mice. (a–d) Images of lumbar spinal cord sections from (a) uninjected, (b) P1-injected, (c) P21-injected, and (d) P85-injected mice were captured with identical microscope settings to qualitatively show SOD1 levels at end stage. SOD1 levels inversely correlate with survival. (e–t) Colabeling for GFP, ChAT, and SOD1 shows that AAV9-transduced motor neurons had reduced SOD1 expression (arrows) while cells that lacked GFP maintained high levels of mutant protein (arrowheads). As described in Figure 1u, higher motor neuron transduction and corresponding SOD1 reduction was observed in (i–l) P1-injected mice as compared with (m–p) P21-injected and (q–t) P85-injected mice. Bar = 100 µm. ChAT, choline acetyltransferase; GFP, green fluorescent protein; P1, postnatal day 1; P21, postnatal day 21; P85, postnatal day 85; SOD1, superoxide dismutase 1.
Therapeutic slowing of disease progression with peripheral injection of AAV9 after onset
To determine if AAV9-mediated mutant SOD1 reduction would slow disease progression, a cohort of SOD1G37R mice6 were injected intravenously with AAV9-SOD1-shRNA after disease onset (average age at treatment = 215 days versus median onset of 197 days in treated animals; log-rank test, P = 0.46; Figure 5a). A combination of AAV9-CB-GFP (n = 9) and uninjected (n = 12) littermates were used as controls. Post hoc analysis showed no differences between GFP and uninjected animals; therefore, the groups were compiled as “control” and are shown in Figure 5. Animals were evaluated weekly for body weight and hindlimb grip strength and monitored until end stage. AAV9-SOD1-shRNA treatment after disease onset significantly extended median survival by 86.5 days over control animals (control, n = 21, 392 days; SOD1 shRNA, n = 25, 478.5 days; log-rank test, P < 0.0001). Early disease duration, defined by the time from peak weight to 10% weight loss, was significantly slowed (control: 89 days; SOD1 shRNA-treated mice: 162 days; Wilcoxon signed-rank test, P < 0.01; Figure 5c). A continuing trend toward slowing of later disease (10% weight loss to end stage) was also seen (control: 63 days; SOD1 shRNA-treated mice: 81 days; Wilcoxon signed-rank test P = 0.1389; Figure 5d). Overall disease duration following AAV9-SOD1-shRNA therapy rose to 239 days after disease onset versus 173 days in control mice (Wilcoxon signed-rank test, P < 0.0001; Figure 5e). Consistent with the slowed disease progression, AAV9 therapy maintained grip strength relative to control SOD1-mutant animals (Figure 5g). The 86.5-day extension in survival surpassed the 62-day extension seen in transgenic studies that used astrocyte-specific Cre expression to inactivate the mutant SOD1G37R transgene,8 presumably reflecting efficient AAV9 transduction of astrocytes after peripheral delivery and the possible transduction of other cell types (especially microglia)6 whose synthesis of mutant SOD1 accelerates disease progression.
Figure 5.
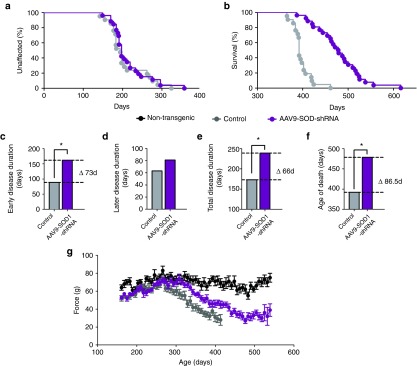
AAV9-SOD1-shRNA improves survival and motor performance in SOD1G37R mice treated after disease onset. (a) There was no difference in median disease onset between AAV9-SOD1-shRNA– and control-treated mice (average age at treatment = 215 days versus median onset of 194 days control and 197 days treated; log-rank test, P = 0.46). (b,f) Median survival of AAV9-SOD1-shRNA–treated SOD1G37R mice (n = 25) was significantly extended versus control mice (n = 21) (control, n = 21, 392 days; SOD1 shRNA, n = 25, 478.5 days; log-rank test, P < 0.0001). (c–e) The early phase of disease was significantly slowed by 73 days in treated mice as compared with control mice (control: 89 days; SOD1 shRNA: 162 days; P < 0.0001; Wilcoxon signed-rank test) while the late phase of disease showed a nonsignificant slowing (control: 63 days; SOD1 shRNA: 81 days; P = 0.14, Wilcoxon signed-rank test). Together this amounted to a 66-day increase in median disease duration (control: 173 days; SOD1 shRNA: 239 days; P < 0.0001; Wilcoxon signed-rank test). (g) A trend to improved hindlimb grip strength appeared in AAV9-SOD1-shRNA–treated mice compared with control mice.
Histological examination of end-stage SOD1G37R-treated animals revealed similar levels of intraspinal cell transduction in animals treated with AAV9-SOD1-shRNA or AAV9-GFP (Figure 6). GFP expression was predominantly observed within motor neurons and astrocytes of both groups, and SOD1 expression was detectably decreased only in animals those received AAV9-SOD1-shRNA (Figure 6k,o). Immunoblotting of whole spinal cord extracts from end-stage SOD1G37R mice revealed an 80% reduction in hSOD1 protein levels in AAV9-SOD1-shRNA-treated animals compared with controls (Supplementary Figure S4).
Figure 6.

Intravenous injection of AAV9 in adult SOD1G37R mice targets astrocytes and motor neurons within the spinal cord. (a–h) Immunofluorescence analysis revealed neuronal as well as glial transduction in both (a–d) AAV9-CB-GFP– and (e-h) AAV9-SOD1-shRNA–treated mice. (i–p) Human SOD1 levels appeared reduced in (o) AAV9-SOD1-shRNA treated mice compared with (k) AAV9-GFP treated mice. Bar = 100 µm. ChAT, choline acetyltransferase; GFP, green fluorescent protein; GFAP, glial fibrillary acidic protein; SOD1, superoxide dismutase 1.
AAV9-mediated suppression of SOD1 in nonhuman primates
To test whether SOD1 levels could be efficiently lowered using AAV9 in the nonhuman primate spinal cord, AAV9 was injected intrathecally through lumbar puncture. This method was chosen over systemic delivery to decrease the amount of virus required and to minimize any effects from reduction of SOD1 in peripheral tissues. Sequencing of cDNA copied from mRNA isolated from African Green Monkey (COS cells) and the Cynomolgus macaque verified that the 130 shRNA had a single base mismatch to either sequence (Supplementary Figure S5). The 130 shRNA expression cassette was inserted into a lentiviral vector which was then used to transduce COS cells. Quantitative RT-PCR of total RNA harvested 72 hours postinfection revealed that the monkey SOD1 mRNA was reduced by ~75% in 130 shRNA-transduced cells compared with the mock-transduced control cells (Supplementary Figure S5).
The AAV9-SOD1-shRNA virus (1 × 1013 vg/kg) was infused along with contrast agent through lumbar puncture into the subarachnoid space of the three male Cynomolgus macaques and one control subject was injected with AAV9-CB-GFP (1 × 1013 vg/kg) (Figure 7a). No side effects from the treatments were identified. Two weeks postinjection, spinal cords were harvested for the analysis of GFP expression and SOD1 RNA levels. GFP expression was seen broadly in neuronal and astrocytic cells throughout the gray and white matter of the lumbar spinal cord, the area closest to the site of injection (Figure 7b–e). Immunoblotting of extracts of lumbar spinal cord revealed 87% reduction in monkey SOD1 protein levels (Figure 7f,g). Laser capture microdissection was then used to isolate total RNA from motor neurons as well as from glia in the nearby neuropil. Analysis by quantitative RT-PCR using primers specific for monkey SOD1 (and normalized to actin) confirmed a 95 ± 3% knockdown in the motor neuron pool and a 66 ± 9% knockdown in the neuropil pool as compared with the samples from a control animal (Figure 7h).
Figure 7.

Intrathecal infusion of AAV9-SOD1-shRNA in nonhuman primates leads to efficient reduction in SOD1 levels. (a) A myelogram shortly after intrathecal infusion of AAV9-SOD1-shRNA mixed with contrast shows proper delivery into the subarachnoid space of a Cynomolgus macaque. Arrows show diffusion of the contrast agent along the entire spinal cord. (b) Lumbar spinal cord sections from treated monkeys (n = 3) were harvested 2 weeks postinjection and stained for GFP using 3,3'-diaminobenzidine staining. Sections had widespread GFP expression throughout the gray and white matter. (c–e) Immunofluorescence analysis of the lumbar spinal cord sections showed (c) robust GFP expression within (d) ChAT-positive cells indicating (e, merge) motor neuron transduction. (f) Western blot analysis of the lumbar spinal cords showed significant reduction in SOD1 levels in AAV9-SOD1-shRNA–injected animals as compared with controls. (g) In vivo quantification of SOD1 knockdown in monkey lumbar spinal cord homogenate (n = 3) showed an 87% reduction in animals that received AAV9-SOD1-shRNA compared with uninjected controls. (h) Laser capture microdissection was used to collect motor neurons or surrounding neuropil from injected and control lumbar monkey sections. Collected cells were analyzed for SOD1 levels by quantitative reverse transcriptase polymerase chain reaction. Motor neurons collected from AAV9-SOD1-shRNA animals (n = 3) had a 95 ± 3% reduction in SOD1 RNA. Non-neurons had a 66 ± 9% reduction in SOD1 RNA in AAV9-SOD1-shRNA treated animals. Scale bars: b = 100 µm; e = 50 µm. SOD1, superoxide dismutase 1.
Next, we examined the level of cell transduction throughout the spinal cord including cervical, thoracic, and lumbar segments. GFP was found to be expressed broadly within all sections analyzed (Figure 8a–c). Motor neuron counts revealed a caudal to rostral gradient in cell transduction, with the cervical region showing more than 50% of GFP/Chat+ motor neurons, increasing to 65% in the thoracic region and reaching 80% in the lumbar region (Figure 8d). In order to determine the overall level of SOD1 knockdown achieved with this transduction pattern, quantitative RT-PCR for SOD1 was performed on whole-section homogenates from cervical, thoracic, and lumbar cord segments. The results confirmed robust SOD1 reduction at all three spinal cord levels, ranging from a 60% decrease in the cervical segment, a 70% decrease in the thoracic region, and an 88% decrease in the lumbar region (Figure 8e), consistent with the proportion of cells transduced in each region.
Figure 8.

Lumbar intrathecal infusion of AAV9-SOD1-shRNA leads to efficient transduction of motor neurons and non-neuronal cells in the cervical, thoracic, and lumbar cord resulting in reduction of SOD1. (a–c) Immunofluorescence analysis of the three segments of the spinal cord; (a) cervical, (b) thoracic, (c) and lumbar, showed robust GFP (green) expression within ChAT (red)-positive cells indicating motor neuron transduction. (d) GFP+/Chat+ cell counts show a caudal to rostral gradient of motor neuron transduction ranging from 85% of transduced cells in the lumbar region to more than 50%in the cervical region. (e) SOD1 mRNA levels in cervical, thoracic, and lumbar cord section homogenates analyzed by quantitative reverse transcriptase polymerase chain reaction show significant reduction in SOD1 transcript, consistently with motor neuron transduction.SOD1 levels were normalized to β-actin– and AAV9-SOD1-shRNA–injected animals were compared with an AAV9-CB-GFP–injected control. (a–c) Scale bars: = 50 µm; (d–e) error bars: = SD.
Discussion
Only one drug is currently approved by Food and Drug Administration as a therapy for ALS, providing a modest survival benefit.21 For the 20% of familial cases caused by mutation in SOD1, attempts at improving therapy by reducing synthesis of SOD1 have been the focus of multiple therapeutic development approaches. Antisense oligonucleotides and virus-delivered RNA interference were tested in rat22 and mouse models23,24,25 that develop fatal paralysis from overexpressing human SOD1G93A. Antisense oligonucleotides infused at disease onset produced SOD1 reduction and a modest slowing of disease progression.22 Direct cerebrospinal fluid (CSF) infusion of antisense oligonucleotides has been tested clinically,26 leading to encouraging results in terms of tolerability and safety but without significant reduction in SOD1 levels at the low dosages used. In each of the prior viral studies,23,24,25 SOD1 knockdown was achieved before disease onset by direct injection into the nervous system or taking advantage of axonal retrograde transport when a virus was injected intramuscularly.23,24 These studies led to varying degrees of success in extending survival or improving motor performance, depending on the time of treatment as well as level of SOD1 knockdown achieved in the spinal cord. Although these studies provided important proof of principle, the approaches were far from being readily translated into clinical strategies. Indeed, there have been controversial reports surrounding these initial virus-mediated SOD1 suppression studies.23,24,27,28,29
Here, we have tested the therapeutic potential of the groundbreaking finding that AAV9 crosses the blood–brain barrier and successfully transduces motor neurons and astrocytes.18,19 Our results also show that intravenous administration of AAV9-SOD1-shRNA is safe and well tolerated in wild-type mice, with the absence of adverse effects after long-term assessment. Remarkably, our efforts with this approach have achieved one of the longest extensions in survival ever reported in the rapidly progressive SOD1G93A mouse model of ALS (increasing survival by 39% when treatment is initiated at birth). Even more encouraging, markedly slowed disease progression is seen even when AAV9 therapy to reduce mutant SOD1 synthesis is applied after disease onset in SOD1G37R mice, thereby significantly extending survival. Therefore, the vascular delivery paradigm in mice represents a proof of concept that mutant SOD1 knockdown after disease onset can be beneficial in both rapid and more slowly progressive models of ALS at clinically relevant points in disease. Together, these data show that robust targeting and suppression of SOD1 levels through AAV9-mediated delivery of shRNA is effective in slowing disease progression in mouse models of ALS, critically even when treatment is initiated after onset.
The significant reduction in SOD1 protein levels at end stage in both SOD1G93A and SOD1G37R mouse models (Figure 4b and Supplementary Figure S4) exceeded expectations, given the range in transduction efficiency of the virus seen at 3 weeks postinfection (Figure 1u), and the level of reduction in SOD1 levels in both cell culture (Figure 2c) and in mice at 3 weeks postinfection (Figure 2d). This raises the possibility that in the central nervous system, cells with reduced SOD1 levels might exhibit a survival advantage over time, consistent with data from Ralph et al.24 using lentiviral-mediated knockdown of SOD1 and from Reaume et al.30 in SOD1 deficient mice. Although contrasting with previous evidence that SOD1 knockdown leads to senescence in fibroblasts or cancer cells,31 together these findings suggest that sensitivity to SOD1 reduction may be cell type specific. Although not yet directly tested elsewhere in a mammalian system in vivo, the sensitivity of certain cells to SOD1 reduction may depend on metabolic status or proliferative potential, as reduction of respiration in yeast prevented the rapid viability loss in SOD1-mutant strains.32
A metabolic link underlying cell type-specific sensitivity to SOD1 reduction would also be consistent with another approach that has shown significant lifespan extension in the SOD1G93A mouse model33,34 and in the SOD1G37R mouse model.6 ALS-associated mutations in SOD1 have been shown to the increase activity of NADPH-oxidase 2 by binding to Rac1, resulting in an overproduction of extracellular reactive oxygen species released from microglia.33,34 Genetic deletion of NADPH-oxidase 2 or inhibition with apocynin treatment slowed both the onset and progression of disease.34 However, the significant neuroprotection conferred by apocynin treatment starting at P14 has not been reproduced when initiated at P21 and therefore, controversy remains.35 Most recently, Nox activation in microglial cells has also been linked to an induction of protein disulphide isomerase expression by the unfolded protein response to mutant SOD1 at an early symptomatic phase in the SOD1G93A mouse model.36 In vivo studies testing pharmacological inhibition of protein disulphide isomerase or genetic depletion in any available SOD1 mouse models would clarify the potential of these alternate therapeutic approaches targeting downstream consequences of misfolded SOD1.
Whether SOD1 is involved in sporadic disease remains controversial. Although some reports have argued against any such involvement,37,38,39 multiple recent studies have brought forward the hypothesis that wild-type SOD1 may contribute through misfolding to the pathogenic mechanism(s) that underlie sporadic ALS through a pathway similar to that triggered by mutant SOD1.12,14,40,41 Included in this body of evidence is our own demonstration that astrocytes produced from sporadic ALS patients are toxic to cocultured motor neurons and that toxicity is alleviated by siRNA-mediated reduction in wild-type SOD1.30 Several additional reports have provided data supporting SOD1 misfolding in sporadic disease.12,13,15,41,42,43,44,45 Although no consensus has yet emerged, the evidence creates the potential that a proportion of sporadic ALS patients could also benefit from an AAV9-mediated SOD1 reduction approach that we have demonstrated to be effective in slowing disease progression in mice that develop fatal, ALS-like disease from expressing ALS-causing mutations in SOD1.
Finally, for translation of an AAV9-mediated suppression of SOD1 synthesis to the human setting, we have determined that infusion directly into the CSF at the lumbar level in a nonhuman primate produces substantial SOD1 reduction by targeting both motor neurons and non-neuronal cells. This outcome provides strong support for extending these efforts to an adult human by direct injection into CSF, as previously proposed46,47 so as to limit the cost of viral production, to reduce the possibility that chronic suppression of SOD1 in the periphery may have deleterious consequences, and to reduce viral exposure to the peripheral immune system.46 These data strongly suggest that AAV9-SOD1-shRNA is a valid candidate for clinical trials in ALS and also opens the opportunity for delivering genes to non-neuronal cells in other disorders, given the recently established contribution of astrocytes in Rett syndrome and age-related decline in neurogenesis.48,49
Materials and Methods
Vectors. shRNA constructs targeting human SOD1 were generated and obtained from the Life Technologies design tool. These constructs were cloned in pSilencer 3.1 (Genscript, Piscataway, NJ) under the human H1 promoter and tested in vitro. shRNA 130 along with H1 promoter was further cloned into an AAV vector along with a reporter GFP under chicken β-actin promoter to identify the transduced cells. Self-complementary AAV9-SOD1-shRNA was produced by transient transfection procedures using a double-stranded AAV2-ITR–based CB-GFP vector, with a plasmid encoding Rep2Cap9 sequence as previously described along with an adenoviral helper plasmid pHelper (Stratagene, Santa Clara, CA) in 293 cells.18
Cells. HEK-293 cells were maintained in Iscove's modified Dulbecco's media containing 10% FBS, 1% L-glutamine, and 1% penicillin/streptomycin. Upon reaching ~60% confluence, cells were transfected with pSilencer 3.1 containing the shRNAs being tested. Protein lysates were prepared 72 hours posttransfection and analyzed for SOD1 levels by western blot.
Cos-7 cells were maintained in Dulbecco's Modified Eagle Medium with 10% FBS and 1% penicillin/streptomycin. Cells were infected with a lentiviral vector expressing SOD1 shRNA 130 under the H1 promoter and RFP under cytomegalovirus promoter. RNA was extracted from infected and noninfected cells 72 hours postinfection using an RNAeasy Kit (Qiagen, Valencia, CA). cDNA was prepared using RT2 First strand synthesis kit (SABiosciences, Valencia, CA). SOD1 transcript levels were analyzed by quantitative reverse transcriptase polymerase chain reaction (RT-PCR).
Animals. All procedures performed were in accordance with the NIH Guidelines and approved by the Research Institute at Nationwide Children's Hospital (Columbus, OH), University of California (San Diego, CA), or Mannheimer Foundation (Homestead, FL) Institutional Animal Care and Use Committees.
High-copy SOD1G93A mice were obtained from Jackson Laboratories (Bar Harbor, ME) and bred in the Kaspar laboratory. Animals were genotyped before the treatment to obtain SOD1G93A-expressing mice and their wild-type littermates. Only female mice were included in the SOD1G93A experiments. loxSOD1G37R ALS mice, carrying a human-mutant SOD1G37R transgene flanked by lox p sites under its endogenous promoter were maintained in the Cleveland laboratory, as previously described.8
One-year-old Cynomolgus macaques (Macacafascicularis) with average body weight of 2 kg were used for this study at the Mannheimer Foundation. Regular monitoring of overall health and body weight was performed before and after the injections to assess the welfare of the animals.
Injections. For neonatal mouse injections, P1–P2 SOD1G93A pups were used. Total volume of 50 μl containing 5 × 1011 (3.6 × 1014 vg/kg) DNase-resistant viral particles of AAV9-SOD1-shRNA (Virapur LLC, San Diego, CA) was injected through temporal vein as previously described.18 A correct injection was verified by noting blanching of the vein. After the injection, pups were returned to their cage. For adult tail vein injections, animals were placed in a restraint that positioned the mouse tail in a lighted, heated groove. The tail was swabbed with alcohol and then injected intravenously with AAV9-SOD1-shRNA. SOD1G93A mice were injected ~21 days and ~85 days of age with 200 μl or 300 μl viral solution containing 2 × 1012 or 3 × 1012 DNase-resistant viral particles, for an average dose of 1.7 × 1014 vg/kg at P21 or 1.6 × 1014 vg/kg at P85. Mutant lox SOD1G37R mice were injected at ~215 days of age with 300 μl containing 3 × 1012 viral particles of AAV9-SOD1-shRNA (average dose of 1.1 × 1014 vg/kg) or AAV9-GFP or empty capsid (average dose of 1.0 × 1014 vg/kg).
For nonhuman primate injections, three anesthetized cynomolgus monkeys received intrathecal injections of 1 × 1013 vg/kg AAV9-SOD1-shRNA and one received 1 × 1013 vg/kg AAV9-CB-GFP. The injection was performed by lumbar puncture into the subarachnoid space of the lumbar thecal sac. AAV9 was resuspended with omnipaque (GE, Fairfield, CT) (iohexol), an iodinated compound routinely used in the clinical setting. Iohexol is used to validate successful subarachnoid space cannulation and was administered at a dose of 100 mg/kg. The subject was placed in the lateral decubitus position and the posterior midline injection site at ~L4/5 level identified (below the conus of the spinal cord). Under sterile conditions, a spinal needle with stylet was inserted and subarachnoid cannulation was confirmed with the flow of clear CSF from the needle. In order to decrease the pressure in the subarachnoid space, 0.8 ml of CSF was drained, immediately followed by injection with a mixture containing 0.7 ml iohexol (300 mg/ml formulation) mixed with 2.1 ml of virus (2.8 ml total).
Perfusion and tissue processing. Control and treated SOD1G93A mice were killed at either 21 days postinjection or at end stage for immunohistochemical analysis. Animals were anesthetized with xylazene/ketamine cocktail, transcardially perfused with 0.9% saline, followed by 4% paraformaldehyde. Spinal cords were harvested, cut into blocks of tissue 5–6 mm in length, and then cut into 40-µm thick transverse sections on a vibratome (Leica, Bannockburn, IL). Serial sections were kept in a 96-well plate that contained 4% paraformaldehyde and were stored at 4 °C. End-stage loxSOD1G37R mice were anesthetized using isoflurane and perfused with 4% paraformaldehyde. Spinal cord segments, including cervical, thoracic, and lumbar segments were dissected. Following cryoprotection with 20% sucrose or 4% paraformaldehyde overnight, spinal cords were frozen in isopentane at −65 °C, and serial 30-µm coronal sections were collected free floating using a sliding microtome.
For safety studies, P1- and P21-treated mice and control wild-type mice were killed at 180 days of age. Animals were anesthetized using xylazene/ketamine cocktail and perfused with 0.9% saline. Different tissues were removed and stored in 10% buffered formalin. These tissues were further processed, blocked, and mounted for hematoxilin & eosin staining by the Nationwide Children's Hospital Morphology Core.
Cynomolgus monkeys injected with virus were killed 2 weeks postinjection. Animals were anesthetized with sodium pentobarbital at the dose of 80–100 mg/kg intravenously and perfused with saline solution. Brain and spinal cord dissection were performed immediately and tissues were processed either for nucleic acid isolation (snap frozen) or postfixed in 4% paraformaldehyde and subsequently cryoprotected with 30% sucrose and frozen in isopentane at −65 °C. Coronal sections of 12 µm were collected from lumbar cord using a cryostat for free floating immunostaining.
Immunohistochemistry. Mouse spinal cords were stained as floating sections. Tissues were washed three times for 10 minutes each in Tris-buffered saline (TBS), then blocked in a solution containing 10% donkey serum, 1% Triton X-100, and 1% penicillin/streptomycin for 2 hours at room temperature (RT). All the antibodies were diluted with the blocking solution. Primary antibodies used were as follows: rabbit anti-GFP (1:400; Invitrogen, Carlsbad, CA), rabbit anti-SOD1 (1:200; Cell signaling, Danvers, MA), goat anti-ChAT (1:50; Millipore, Billerica, MA), mouse anti-GFAP (1:200; Millipore), chicken anti-GFAP (1:400; Abcam, Cambridge, MA), and rabbit anti-Iba1 (1:400; Wako, Richmond VA). Tissues were incubated in primary antibody at 4 °C for 48–72 hours and then washed three times with TBS. After washing, tissues were incubated for 2 hours at RT in the appropriate fluorescein isothiocyanate-, Cy3-, or Cy5-conjugated secondary antibodies (1:200; Jackson Immunoresearch, Westgrove, PA) and DAPI (1:1,000; Invitrogen). Tissues were then washed three times with TBS, mounted onto slides, and then coverslipped with PVA-DABCO. All images were captured on a Zeiss laser-scanning confocal microscope.
For 3,3'-diaminobenzidine staining, monkey spinal cord sections were washed three times in TBS, blocked for 2 hours at RT in 10% donkey serum and in 1% Triton X-100. Sections were then incubated overnight at 4 °C with rabbit anti-GFP primary antibody (1:1,000; Invitrogen) diluted in blocking buffer. The following day, tissues were washed with TBS three times, incubated with biotinylated secondary antibody antirabbit (1:200; Jackson Immunoresearch) in blocking buffer for 30 minutes at RT, washed three times in TBS, and incubated for 30 minutes at RT with ABC (Vector, Burlingame, CA). Sections were then washed for three times in TBS and incubated for 2 minutes with 3,3'-diaminobenzidine solution at RT and washed with distilled water. These were then mounted onto slides and covered with coverslips in mounting medium. All images were captured with the Zeiss Axioscope.
Motor neuron and astrocyte quantification. For motor neuron quantification, serial 40-μm thick lumbar spinal cord sections, each separated by 480 μm, were labeled as described for GFP and ChAT expression. Stained sections were serially mounted on slides from rostral to caudal, then coverslipped. Sections were evaluated using confocal microscopy (Zeiss, Munich, Germany) with a ×40 objective and simultaneous fluorescein isothiocyanate and Cy3 filters. The total number of ChAT-positive cells found in the ventral horns with defined soma was tallied by careful examination through the entire z-extent of the section. GFP-labeled cells were quantified in the same manner, while checking for colocalization with ChAT. For astrocyte quantification, as with motor neurons, serial sections were stained for GFP, GFAP and then mounted. Using confocal microscopy with a ×63 objective and simultaneous fluorescein isothiocyanate and Cy5 filters, random fields in the ventral horns of lumbar spinal cord sections from tail vein-injected animals were selected. The total numbers of GFP- and GFAP-positive cells were counted from a minimum of at least 24 fields per animal while focusing through the entire z extent of the section. Spinal cord sections of three animals per group were examined for motor neuron and astrocyte quantification.
Immunoblot analysis. Spinal cords were harvested from P1- and P21-injected mice and control SOD1G93A mice 21 days postinjection and from treated and control monkeys 2 weeks postinjection of AAV9-SOD1-shRNA. Spinal cords were homogenized and protein lysates were prepared using T-Per (Pierce, Rockford, IL) with protease inhibitor cocktail. Samples were resolved on SDS-PAGE according to the manufacturer's instructions. Primary antibodies used were rabbit anti-SOD1 (1:750; Cell signaling) mouse anti-SOD1 (1:750; Millipore), rabbit anti-SOD1 (1:1,000; Abcam), rabbit anti-Actin (1:1,000; Abcam) and mouse anti-GAPDH (1:1,000, Millipore). Secondary antibodies used were anti-rabbit HRP (1:10,000–1:50,000) and anti-mouse HRP (1:10,000). Densitometric analysis was performed using Image J software.
Laser capture microdissection. Lumbar spinal cord frozen sections of 12 µm were collected onto PEN membrane slides (Zeiss) and stained with 1% Cresyl violet (Sigma, St Louis, MO) in methanol. Sections were air dried and stored at −80 °C. After thawing, motor neurons were collected within 30 minutes from staining using the laser capture microdissector PALM Robo3 Zeiss) using the following settings: cut energy: 48, laser pressure catapulting energy: 20, cut focus: 80/81, laser pressure catapulting focus: 1, position speed: 100, cut speed: 50. Approximately 500 motor neurons were collected per animal. Non-neuronal cells from the ventral horn were collected from the same sections after collecting the motor neurons.
Quantitative RT-PCR. RNA from laser-captured cells or whole spinal cord sections from the cervical, thoracic, and lumbar segments was isolated using the RNaqueous Micro Kit (Ambion, Grand Island, NY) according to the manufacturer's instructions. RNA was then reverse transcribed into cDNA using the RT2 HT First Strand Kit (SABiosciences). RNA of 12.5 ng was used in each Q-PCR reaction using SyBR Green (Invitrogen) to establish the relative quantity of endogenous monkey SOD1 transcript in animals who had received the AAV9-SOD1-shRNA compared with the animals who had received only AAV9-GFP. Each sample was run in triplicate and relative concentration calculated using the ddCt values normalized to endogenous actin transcript.
Behavior and survival analysis. Treated and control SOD1G93A mice were monitored for changes in body mass twice a week. loxSOD1G37R mice were weighed on a weekly basis. Motor coordination was recorded using a rotarod instrument (Columbus Instruments, Columbus, OH). Each weekly session consisted of three trials on the accelerating rotarod beginning at 5 rpm/minute. The time each mouse remained on the rod was registered. Both SOD1G93A and loxSOD1G37R mice were subjected to weekly assessment of hindlimb grip strength using a grip strength meter (Columbus Instruments). Each weekly session consisted of three (SOD1G93A mice) or five (loxSOD1G37R mice) tests per animal. Survival analysis was performed using Kaplan–Meier survival analysis. End stage was defined as an artificial death point when animals could no longer “right” themselves within 30 seconds after being placed on its back. Onset and disease progression were determined from retrospective analysis of the data. Disease onset is defined as the age at which the animal reached its peak weight. Disease duration is defined as the time period between disease onset and end stage. Early disease duration is the period between peak weight and loss of 10% of body weight while late disease duration is defined as the period between 10% loss of body weight until disease end stage. Due to shorter life span of SOD1G93A animals, we did not assess the distinction between the early and late progression.
For toxicity analysis following injection at P1- or P21-treated mice and control WT mice were subjected to behavioral analysis starting at ~30 days of age and monitored up to 6 months. Body mass was recorded weekly while rotarod performance and hindlimb grip strength were recorded biweekly.
Hematology and serum studies. Blood samples were collected in (dipotassium ethylene diamine tetraacetic acid) EDTA microtainer tubes (Becton Dickinson, San Jose, CA) from treated and control wild-type mice at 150 days of age by mandibular vein puncture. The same animals were bled at 180 days of age, and blood was collected in serum separator microtainer tubes. The blood was allowed to clot for an hour and was then centrifuged at 10,000 rpm for 5 minutes. The clear upper phase (serum) was collected and frozen at −80 °C. Hematological and serum analysis were conducted by AniLytics, Gaithersburg, MD.
Statistical analysis. All statistical tests were performed using the GraphPad Prism (San Diego, CA) software package. Kaplan–Meier survival analyses were analyzed by the log-rank test. Comparisons of median disease durations and survival times were analyzed by the Wilcoxon signed-rank test.
SUPPLEMENTARY MATERIAL Figure S1. AAV9-shRNA-SOD1 administration is well tolerated in WT mice. Figure S2. Hematology and serum chemistry of AAV9-SOD1-shRNA-treated WT animals. Figure S3. AAV9-SOD1-shRNA treatment in SOD1G93A mice reduces astrogliosis. Figure S4. Intravenous injection of AAV9-SOD1-shRNA efficiently reduces levels of mutant SOD1 protein in spinal cords of SOD1G37R mice. Figure S5. shRNA 130 efficiently reduces the levels of monkey SOD1 in vitro.
Acknowledgments
We thank Pablo Morales, Mannheimer Foundation, Homestead, FL for expert nonhuman primate studies. We thank UCSD undergraduate students Christie Duong and Yiyang Zou for their contributions in processing tissue samples. This work was funded by US National Institutes of Health (NIH) R21-NS067238, NS027036, RC2 NS69476-01, Project A.L.S. and Packard Center for ALS Research (P2ALS) and Helping Link Foundation. D.L.S. was a San Diego IRACDA Postdoctoral Fellow supported by NIH Grant K12 GM068524. D.D. is supported by a Ruth Kirschstein National Research Service Award for Individual Postdoctoral Fellows from the NINDS, NIH F32 NS073269 02. K.M. is supported by a fellowship from the Swiss National Science Foundation. L.F. is supported by a Marie Curie Fellowship. D.W.C. receives salary support from the Ludwig Institute for Cancer Research.
Supplementary Material
References
- Da Cruz S, Cleveland DW. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr Opin Neurobiol. 2011;21:904–919. doi: 10.1016/j.conb.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay M, Valentine JS. Aggregation of copper-zinc superoxide dismutase in familial and sporadic ALS. Antioxid Redox Signal. 2009;11:1603–1614. doi: 10.1089/ars.2009.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudencio M, Hart PJ, Borchelt DR, Andersen PM. Variation in aggregation propensities among ALS-associated variants of SOD1: correlation to human disease. Hum Mol Genet. 2009;18:3217–3226. doi: 10.1093/hmg/ddp260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Kang SH, Li Y, Fukaya M, Lorenzini I, Cleveland DW, Ostrow LW, et al. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci. 2013;16:571–579. doi: 10.1038/nn.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11:251–253. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giorgio FP, Boulting GL, Bobrowicz S, Eggan KC. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell. 2008;3:637–648. doi: 10.1016/j.stem.2008.09.017. [DOI] [PubMed] [Google Scholar]
- Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci. 2007;10:608–614. doi: 10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetto MC, Muotri AR, Mu Y, Smith AM, Cezar GG, Gage FH. Non-cell-autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell. 2008;3:649–657. doi: 10.1016/j.stem.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011;29:824–828. doi: 10.1038/nbt.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13:1396–1403. doi: 10.1038/nn.2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokrishevsky E, Grad LI, Yousefi M, Wang J, Mackenzie IR, Cashman NR. Aberrant localization of FUS and TDP43 is associated with misfolding of SOD1 in amyotrophic lateral sclerosis. PLoS ONE. 2012;7:e35050. doi: 10.1371/journal.pone.0035050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg K, Jonsson PA, Andersen PM, Bergemalm D, Graffmo KS, Hultdin M, et al. Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS ONE. 2010;5:e11552. doi: 10.1371/journal.pone.0011552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal S, Cudkowicz M. ALS drug development: reflections from the past and a way forward. Neurotherapeutics. 2008;5:516–527. doi: 10.1016/j.nurt.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Cutting FB, Zhai P, Andrus PK, Hall ED. Pathogenic mechanisms in familial amyotrophic lateral sclerosis due to mutation of Cu, Zn superoxide dismutase. Pathol Biol. 1996;44:51–56. [PubMed] [Google Scholar]
- Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duque S, Joussemet B, Riviere C, Marais T, Dubreil L, Douar AM, et al. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol Ther. 2009;17:1187–1196. doi: 10.1038/mt.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Deane R, Ali Z, Parisi M, Shapovalov Y, O'Banion MK, et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci. 2008;11:420–422. doi: 10.1038/nn2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2012;3:CD001447. doi: 10.1002/14651858.CD001447. [DOI] [PubMed] [Google Scholar]
- Smith RA, Miller TM, Yamanaka K, Monia BP, Condon TP, Hung G, et al. Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Invest. 2006;116:2290–2296. doi: 10.1172/JCI25424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoul C, Abbas-Terki T, Bensadoun JC, Guillot S, Haase G, Szulc J, et al. Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat Med. 2005;11:423–428. doi: 10.1038/nm1207. [DOI] [PubMed] [Google Scholar]
- Ralph GS, Radcliffe PA, Day DM, Carthy JM, Leroux MA, Lee DC, et al. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat Med. 2005;11:429–433. doi: 10.1038/nm1205. [DOI] [PubMed] [Google Scholar]
- Miller TM, Kaspar BK, Kops GJ, Yamanaka K, Christian LJ, Gage FH, et al. Virus-delivered small RNA silencing sustains strength in amyotrophic lateral sclerosis. Ann Neurol. 2005;57:773–776. doi: 10.1002/ana.20453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TM, Pestronk A, David W, Rothstein J, Simpson E, Appel SH, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 2013;12:435–442. doi: 10.1016/S1474-4422(13)70061-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towne C, Raoul C, Schneider BL, Aebischer P. Systemic AAV6 delivery mediating RNA interference against SOD1: neuromuscular transduction does not alter disease progression in fALS mice. Mol Ther. 2008;16:1018–1025. doi: 10.1038/mt.2008.73. [DOI] [PubMed] [Google Scholar]
- Towne C, Setola V, Schneider BL, Aebischer P. Neuroprotection by gene therapy targeting mutant SOD1 in individual pools of motor neurons does not translate into therapeutic benefit in fALS mice. Mol Ther. 2011;19:274–283. doi: 10.1038/mt.2010.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel RJ, Lowenstein PR, Byrne BJ. AAV6-mediated gene silencing fALS short. Mol Ther. 2011;19:231–233. doi: 10.1038/mt.2010.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- Blander G, de Oliveira RM, Conboy CM, Haigis M, Guarente L. Superoxide dismutase 1 knock-down induces senescence in human fibroblasts. J Biol Chem. 2003;278:38966–38969. doi: 10.1074/jbc.M307146200. [DOI] [PubMed] [Google Scholar]
- Longo VD, Gralla EB, Valentine JS. Superoxide dismutase activity is essential for stationary phase survival in Saccharomyces cerevisiae. Mitochondrial production of toxic oxygen species in vivo. J Biol Chem. 1996;271:12275–12280. doi: 10.1074/jbc.271.21.12275. [DOI] [PubMed] [Google Scholar]
- Marden JJ, Harraz MM, Williams AJ, Nelson K, Luo M, Paulson H, et al. Redox modifier genes in amyotrophic lateral sclerosis in mice. J Clin Invest. 2007;117:2913–2919. doi: 10.1172/JCI31265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harraz MM, Marden JJ, Zhou W, Zhang Y, Williams A, Sharov VS, et al. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Invest. 2008;118:659–670. doi: 10.1172/JCI34060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trumbull KA, McAllister D, Gandelman MM, Fung WY, Lew T, Brennan L, et al. Diapocynin and apocynin administration fails to significantly extend survival in G93A SOD1 ALS mice. Neurobiol Dis. 2012;45:137–144. doi: 10.1016/j.nbd.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaronen M, Vehviläinen P, Malm T, Keksa-Goldsteine V, Pollari E, Valonen P, et al. Protein disulfide isomerase in ALS mouse glia links protein misfolding with NADPH oxidase-catalyzed superoxide production. Hum Mol Genet. 2013;22:646–655. doi: 10.1093/hmg/dds472. [DOI] [PubMed] [Google Scholar]
- Liu HN, Sanelli T, Horne P, Pioro EP, Strong MJ, Rogaeva E, et al. Lack of evidence of monomer/misfolded superoxide dismutase-1 in sporadic amyotrophic lateral sclerosis. Ann Neurol. 2009;66:75–80. doi: 10.1002/ana.21704. [DOI] [PubMed] [Google Scholar]
- Kerman A, Liu HN, Croul S, Bilbao J, Rogaeva E, Zinman L, et al. Amyotrophic lateral sclerosis is a non-amyloid disease in which extensive misfolding of SOD1 is unique to the familial form. Acta Neuropathol. 2010;119:335–344. doi: 10.1007/s00401-010-0646-5. [DOI] [PubMed] [Google Scholar]
- Brotherton TE, Li Y, Cooper D, Gearing M, Julien JP, Rothstein JD, et al. Localization of a toxic form of superoxide dismutase 1 protein to pathologically affected tissues in familial ALS. Proc Natl Acad Sci USA. 2012;109:5505–5510. doi: 10.1073/pnas.1115009109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synofzik M, Ronchi D, Keskin I, Basak AN, Wilhelm C, Gobbi C, et al. Mutant superoxide dismutase-1 indistinguishable from wild-type causes ALS. Hum Mol Genet. 2012;21:3568–3574. doi: 10.1093/hmg/dds188. [DOI] [PubMed] [Google Scholar]
- Guareschi S, Cova E, Cereda C, Ceroni M, Donetti E, Bosco DA, et al. An over-oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc Natl Acad Sci USA. 2012;109:5074–5079. doi: 10.1073/pnas.1115402109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Valdmanis PN, Dion P, Rouleau GA. Oxidized/misfolded superoxide dismutase-1: the cause of all amyotrophic lateral sclerosis. Ann Neurol. 2007;62:553–559. doi: 10.1002/ana.21319. [DOI] [PubMed] [Google Scholar]
- Gruzman A, Wood WL, Alpert E, Prasad MD, Miller RG, Rothstein JD, et al. Common molecular signature in SOD1 for both sporadic and familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2007;104:12524–12529. doi: 10.1073/pnas.0705044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Blitterswijk M, Gulati S, Smoot E, Jaffa M, Maher N, Hyman BT, et al. Anti-superoxide dismutase antibodies are associated with survival in patients with sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2011;12:430–438. doi: 10.3109/17482968.2011.585163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterström P, Andersen PM, Brännström T, Marklund SL. Misfolded superoxide dismutase-1 in CSF from amyotrophic lateral sclerosis patients. J Neurochem. 2011;117:91–99. doi: 10.1111/j.1471-4159.2011.07177.x. [DOI] [PubMed] [Google Scholar]
- Bevan AK, Duque S, Foust KD, Morales PR, Braun L, Schmelzer L, et al. Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol Ther. 2011;19:1971–1980. doi: 10.1038/mt.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray SJ, Matagne V, Bachaboina L, Yadav S, Ojeda SR, Samulski RJ. Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol Ther. 2011;19:1058–1069. doi: 10.1038/mt.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioy DT, Garg SK, Monaghan CE, Raber J, Foust KD, Kaspar BK, et al. A role for glia in the progression of Rett's syndrome. Nature. 2011;475:497–500. doi: 10.1038/nature10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda CJ, Braun L, Jiang Y, Hester ME, Zhang L, Riolo M, et al. Aging brain microenvironment decreases hippocampal neurogenesis through Wnt-mediated survivin signaling. Aging Cell. 2012;11:542–552. doi: 10.1111/j.1474-9726.2012.00816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.