

Abstract
Human bocavirus virus-1 (HBoV1), a newly discovered autonomous parvovirus with a 5,500 nt genome, efficiently infects human-polarized airway epithelia (HAE) from the apical membrane. We hypothesized that the larger genome and high airway tropism of HBoV1 would be ideal for creating a viral vector for lung gene therapy. To this end, we successfully generated recombinant HBoV1 (rHBoV1) from an open reading frames–disrupted rHBoV1 genome that efficiently transduces HAE from the apical surface. We next evaluated whether HBoV1 capsids could package oversized rAAV2 genomes. These studies created a rAAV2/HBoV1 chimeric virus (5.5 kb genome) capable of apically transducing HAE at 5.6- and 70-fold greater efficiency than rAAV1 or rAAV2 (4.7-kb genomes), respectively. Molecular studies demonstrated that viral uptake from the apical surface was significantly greater for rAAV2/HBoV1 than for rAAV2 or rAAV1, and that polarization of airway epithelial cells was required for HBoV1 capsid–mediated gene transfer. Furthermore, rAAV2/HBoV1-CFTR virus containing the full-length cystic fibrosis transmembrane conductance regulator (CFTR) gene coding sequence and the strong CBA promoter efficiently corrected CFTR-dependent chloride transport in cystic fibrosis (CF) HAE. In summary, using the combined advantages of AAV and HBoV1, we have developed a novel and promising viral vector for CF lung gene therapy and also potentially HBoV1 vaccine development.
Introduction
Cystic fibrosis (CF) is an autosomal recessive genetic disorder caused by mutations in the gene coding for the CF transmembrane conductance regulator (CFTR), for which the pulmonary disease component is the most life-threatening.1 Although major advances in pharmacologic therapies for CF are emerging, gene therapy still remains the “Holy Grail” for curing this disease.2,3 Recombinant adenoassociated viral vectors (rAAV) are currently one gene therapy agent that is being pursued for CF lung gene therapy.4
rAAV vectors for CF lung gene therapy have been under development for nearly two decades, and most serotypes appear to be effectively endocytosed from the apical surface of airway epithelia despite varying degrees of transduction (i.e., expression of an encoded transgene). Although these vectors have demonstrated good safety profiles in CF clinical trails,5,6 they have failed to achieve complementation in vivo for two significant reasons. First, post-entry barriers in virion processing following infection appear to limit nuclear translocation, and thus transgene expression, in a proteasome-dependent manner.7,8,9,10 This feature of rAAV2 is reflected in CF clinical trials where viral genomes persisted in the airway epithelia of test subjects without detection of transgene-derived CFTR mRNA or clinical improvement in lung function.5,6 Identifying an appropriate rAAV serotype that bypassed these limitations has proved challenging due to species-specific differences between animal models and humans.11,12,13 In our hands, rAAV1 proves to be the most efficient serotype for apical infection of human airway epithelial cells,11,14,15 whereas others have found success using directed capsid evolution to enhanced the tropism of rAAV for apical human-polarized airway epithelia (HAE) transduction.16 However, effective CFTR complementation in CF HAE still requires the use of proteasome inhibitors to enhance transduction.16,17
A second major barrier to efficient CFTR expression from rAAV vectors is their limited packaging capacity (~4.9 kb) that necessitates the use of small, weak promoters and/or the use of CFTR minigenes. The first generation rAAV-CFTR tested in a clinical trial used the cryptic promoter within the AAV2 ITR to drive the expression of a full-length CFTR cDNA,5 and this was later improved by the incorporation of a short 83 bp synthetic promoter.17 Other efforts to circumvent the small packing capacity of rAAV vectors have included trimming down size of the CFTR cDNA by deletion of non-critical sequences (such as partial deletion at the R-domain) to expand room for core promoter elements such as a shortened CMV promoter.16,18 Although these strategies have improved expression of CFTR, it is clear that pushing the packaging limits of rAAV can lead to inconsistent deletions at the 5′ end of rAAV genome,19 thus further jeopardizing genome stability and expression. For these reasons, there remains a great need to expand the packaging capabilities of rAAV vectors for CF lung gene therapy.
Human Bocavirus 1 (HBoV1), a newly identified autonomous human parvovirus, was discovered in patient nasopharyngeal aspirates by molecular virology screening using random DNA amplification, cloning, large-scale sequencing, and phylogenetic analyses.20 Mounting evidence indicates HBoV1 is an etiological agent of acute respiratory tract infections associated with wheezing in infants and young children.21,22 HBoV1 is often associated with other viral infections, and thus it remains unclear if HBoV1 alone can gives rise to respiratory disease.23 However, our recent studies in differentiated HAE grown at an air–liquid interface (ALI) have demonstrated that HBoV1 efficiently infects HAE from the apical membrane, resulting in replication of progeny viruses and cytopathology.24 Impressively, HBoV1 infection of HAE at extremely low multiplicity of infection (MOI) of 10−3 DNase I–resistant particles (DRP) per cell results in a productive infection.25 Recently, the full-length 5,543 nt HBoV1 complete genome (including terminal palindromic sequences at both ends) was cloned, and cell culture systems for HBoV1 production have been established.24 Given the high efficiency of HBoV1 infection from the apical surface of HAE, we sought to evaluate whether this virus might be suitable for engineering recombinant vectors for human airway gene therapy.
HBoV1 is a relative of AAV and other Parvoviridae family members. HBoV1 belongs to the genus Bocavirus, whereas AAV is in the genus Dependovirus.26 HBoV1 and AAV are both small single-stranded DNA viruses, but 90% of encapsidated HBoV1 genomes are of the minus strand, whereas for AAV, an equal ratio of plus and minus strands are encapsidated.27 These two viruses differ greatly in their lytic phase life cycle; AAV requires coinfection with a helper virus, whereas HBoV1 autonomously replicates progeny in permissive cells.24 The HBoV1 genome size is 5,543 nt, 18.5% (863 nt) larger than that of AAV2 (4,679 nt), and its structural features include asymmetrical hairpins with unique palindromic sequences at 5′ and 3′ termini and a single P5 promoter that transcribes all viral structural and non-structural proteins.24,28 This is in contrast to the inverted terminal repeats and multiple internal promoters found in AAV genomes. The HBoV1 genome encodes three major open reading frames. Two of them code for non-structural proteins, NS1/NS2 and NP1, which are essential for virus replication. The third open reading frames encodes two structural capsid proteins VP1 and VP2. By contrast, the AAV cap open reading frames encodes three capsid proteins, VP1, VP2, and VP3.27 HBoV1 capsid surface topology possesses common features with other parvoviruses (icosahedral capsid), and is most closely similar to human parvovirus B19.29 Like the cloned AAV genome, a plasmid that encodes the HBoV1 proviral genome is infectious and can be used to produce infectious particles through transfection into HEK293 cells without the need for helper virus coinfection.24
Cross-genera pseudopackaging between Parvoviridae was first established when a rAAV genome was encapsidated into a human parvovirus B19 capsid.30 This resultant cross-genera chimera was able to deliver the rAAV genome into human bone marrow cells that are resistant to rAAV infection.30 Thus, we hypothesized that pseudotyping the rAAV genome into HBoV1 capsid might create a novel chimeric vector with unique properties for gene therapy of CF and other pulmonary diseases. In this report, we demonstrate the feasibility of producing rHBoV1 vectors and chimeric rAAV2/HBoV1 vectors. These new viral vectors both efficiently transduced HAE from the apical surface and were capable of delivering genomes ranging from 4.8 to 5.5 kb in size. Using the larger packaging capacity of HBoV1, we engineered a rAAV2/HBoV1-CFTR vector harboring a strong chimeric promoter consisting of the human CMV immediate gene enhancer and the chicken β-actin promoter (CBA promoter) and demonstrated ~30% restoration of CFTR-mediate chloride currents in CF HAE. These studies lay the foundation for future applications of rHBoV1-based vectors for airway gene therapy and vaccine development.
Results
Production of a recombination HBoV1 vector and its transduction properties in HAE model systems
The plasmid clone of the full-length HBoV1 genome (pIHBoV1) can be used to produce infectious HBoV1 virions following transfection in HEK293 cells without the need for helper virus functions.24 We first attempted to generate recombination HBoV1 (rHBoV1) by testing if HBoV1 viral proteins could transcomplement and rescue replication and packaging of a rHBoV1 genome in HEK293 cells. The structure of the wild-type HBoV1 genome found in the infectious plasmid (pIHBoV1), the rHBoV1 proviral plasmid (prHBoV1-CBAluc), and the trans-helper plasmid (pHBoV1KUm630) are schematically shown in Figure 1a. The prHBoV1-CBAluc contained a 5.5 kb rHBoV1 proviral genome encoding a CBA promoter–driven luciferase gene. The ability of pHBoV1KUm630 to support rHBoV1 genome rescue and replication from prHBoV1-CBAluc was confirmed by cotransfecting these plasmids into HEK293 cells. Hirt DNA extracted from transfected cells was evaluated by Southern blot following DpnI digestion to eliminate the methylated plasmid background signal. As shown in Figure 1b, rHBoV1 replication intermediates were only observed in cells cotransfected with prHBoV1-CBALuc and pHBoV1KUm630, but not with prHBoV1-CBALuc alone. Although the detected replication intermediates from the cotransfection were lower than the level from the wild-type HBoV1 proviral plasmid pIHBoV1, we conclude the pHBoV1KUm630 plasmid does provide the necessary helper functions in trans to support rHBoV1 genome replication (Figure 1b). We next attempted to generate recombinant virus by cotransfection of prHBoV1-CBAluc and pHBoV1KUm630 in HEK293 followed by purification from cell lysates with CsCl equilibrium ultracentrifugation. Fractions from this gradient were evaluated for viral genomes by TaqMan PCR and demonstrated a peak at a density of 1.45 to 1.40 g/ml (Figure 1c), suggesting successful encapsidation of viral DNA. Viral yields from thirty 150 mm plates yielded a total of 1.25 × 1011 rHBoV1 genome copies (vc) or DRP. This is roughly 20% of the yield of wild-type HBoV1 obtained from the transfection of pIHBoV1 in HEK293 cells (~2 × 1011 DRP per ten 150 mm dishes).24
Figure 1.

rHBoV1 vector production and infection of primary polarized HAE. (a) Schematic structure of the HBoV1 genome and the proviral plasmids used in this study. pHBoV1KUm630 is the helper plasmid for trans-complementation of HBoV1 viral proteins, pIHBoV1 is the infectious clone of the HBoV1 complete genome, and prHBoV1-CBAluc is the rHBoV1 cis transfer proviral plasmid. Critical restriction enzyme cutting sites used for cloning are also indicated and small deletions within NS and VP genes are marked (Δ). (b) Replication complementation assay of the rHBoV1 proviral plasmid in HEK293 cells. pIHBoV1 (lane 1), prHBoV1-CBAluc (lane 2), or prHBoV1-CBAluc + pHBoV1KUm630 (lane 3) plasmids were transfected to HEK293 cells. Hirt DNA was extracted at 48 hours after transfection and digested by DpnI before resolving on an agarose gel. HBoV1 replication intermediates (indicated by arrows) were visualized by Southern blotting with a 32P-labeled HBoV1 probe. Hirt DNA from cells transfected with the HBoV1 infectious clone (pIHBoV1, lane 1) was used as positive control. Short and long exposures are shown on the left and right of the panel, respectively. (c) DNase I–digested cell lysates from HEK293 cotransfected with prHBoV1-CBAluc and pHBoV1KUm630 were fractionated by CsCl equilibrium ultracentrifugation. The plot shows the distribution of rHBoV1.CBAluc (solid dots) genomes against the observed density of the gradient (open dots). The genome copies in each fraction (~750 µl) were determined by TagMan PCR. (d) Transduction assay following rHBoV1.CBAluc infection of HEK293 cells, IB3 cells, undifferentiated (UD) CuFi8 cell monolayers, polarized CuFi8 cells in ALI cultures, and primary HAE ALI cultures. Data represent the mean (±SEM) relative luciferase activity per well at 2 days after infection (n = 4). (e) Transgene expression from rHBoV1.CBAluc infected HAE ALI cultures at different time points after infection. Data represent the mean (±SEM) relative luciferase activity per well (n = 3). ALI, air–liquid interface; HAE, human-polarized airway epithelia.
Twice-CsCl banded rHBoV1.CBAluc was then evaluated for its transduction properties following infection of HEK293 cells, IB3 cells (a CF human airway cell line), monolayers of CuFi8 cells (a conditional transformed CF airway cell line),31 and ALI cultures derived from CuFi cells and primary human airway epithelial cells (Figure 1d). Results demonstrated that rHBoV1.CBAluc was only capable of transducing (i.e., expressing its encoded transgene) polarized and differentiated ALI cultures derived from either primary or CuFi airway cells. These findings are similar to conditions required for productive infection with wild-type HBoV1.24 Following apical infection of polarized HAE with rHBoV1.CBAluc, maximal transgene expression occurred at 3 days after infection and gradually declined by 11 days after infection (Figure 1e).
Encapsidation of a recombinant AAV2 genome in HBoV1 virions through cross-genera parvovirus pseudotyping
Although wild-type HBoV1 and rHBoV1 virions can be assembled in HEK293 cells following plasmid(s) transfection, the viral yields are relatively low. By contrast, rAAV vector replication is very efficient in HEK293 with the proper helper plasmids. We reasoned that HEK293 cell machinery supporting HBoV1 replication could be less efficient because the HEK293 cells are not a biologically permissive cell line for HBoV1 productive infection. Therefore, we explored whether pseudotyping rAAV2 genomes with HBoV1 capsids would generate a rAAV2/HBoV1 chimeric vector with higher yields in these cells. Using this approach, the 4.85 kb rAAV genome from the cis rAAV proviral plasmid (pAV2-F5tg83luc) was successfully packaged into HBoV1 virions following cotransfection with pAD4.1 (encoding all the necessary helper function for AAV replication from adenovirus), pAV-Rep2 (encoding the AAV2 Rep genes), and pHBoV1KUm630 (encoding the HBoV1 capsids and NS genes). There were several reasons for using a helper plasmid that expressed all HBoV1 viral proteins to support encapsidation of the rAAV2 genome. First, the temporal regulation and transcriptional profiles of HBoV1 genes required to support packaging remain unknown. Second, the functional roles of HBoV1 NS and NP1 proteins in capsid assembly are unclear. Lastly, it is possible that NS proteins associate with AAV2 ITRs and this could potentially help to facilitate packaging of AAV2 genomes into HBoV1 capsids. Three days after cotransfection, substantial DNase I–resistant viral particles were recovered from crude lysates by CsCl banding (Figure 2a). Omitting pAV-Rep2 from the transfection cocktail failed to produce DNase I–resistant viral particles. Of note, the density of the chimeric rAAV2/HBoV1 virions was ~1.435 g/ml, similar to that of the rHBoV1 virions (Figure 1a). This density was slightly heavier than the 1.414 g/ml density of the rAAV2/2 virions (Figure 1a). The typical yields of the chimeric rAAV/HBoV1 vector were 10- to 20-fold less than rAAV2 vector, but two- to fourfold greater than rHBoV1 vector, when generated on a similar scale. By contrast, the pseudopackaging of a rAAV genome into human parvovirus B19 capsid is much less efficient than what we observed for rAAV2/HBoV1.30
Figure 2.

Pseudopackaging rAAV2 genomes in HBoV1 capsid. (a) DNase I–digested cell lysates from the indicated HEK293 cell plasmid transfections were fractionated by CsCl equilibrium ultracentrifugation. The number of viral genomes in each fraction was determined by TaqMan PCR. (b) HEK293 cells were transfected with the indicated combinations of plasmids (M: Molecular weight marker; lane 1: pAV2-F5tg83luc + pAV-Rep2; lane 2: pAV2-F5tg83luc + pAVRC2.3; lane 3: pAV2-CF5tg83luc + pAV-Rep2; land 4: pAV2-CF5tg83 + pAV-Rep2 + pHBoV1KUm630) together with the Ad helper pAd4.1. Low molecular weight (Hirt) DNA was extracted from transfected cells after 48 hours and digested with Dpn I, followed by Southern blotting using a 32P-labeled luciferase probe. The 4.8 kb and 5.4 kb replicative form (RF) DNA of the rAV2.F5tg83Luc and rAV2.CF5tg83Luc genomes are indicated by arrows. (c) Negatively stained transmission electron micrographs of the chimeric vector rAV2/HBc.F5tg83luc, (bar = 100 nm in the 15,000× image and 50 nm for the 50,000× image). The virus-like particle with incompletely packaged viral DNA (<1% of total virions) is marked by a white arrow in the inset. (d) A two-color western blot (red: AAV; green: HBoV1) was performed on the indicated viral preparations using an Infrared Image System. Converted single channel images are also shown with dark arrows pointing to the AAV2 and HBoV1 VP proteins in the left and right panels, respectively. Gray arrows and white arrows mark cleaved protein fragments from HBoV1 VP1 and VP2 proteins.
Examination of rAAV2 genome rescue and replication demonstrated that rAAV2 replication from DNA (RF DNA) in the rAAV2/HBoV1 production system was approximately two- to threefold less abundant than that during rAAV2 production (Figure 2b, compare lanes 2 and 4). This is consistent with previous reports describing that the parvovirus capsid plays a feedback role in virion formation.32,33 Supporting this, we observed that less rAAV2 RF DNA was present following cotransfection of the AAV2 proviral plasmids (pAV2.F5tg83luc and pAV2.CF5tg83luc) and the pAV2-Rep helper (missing intact AAV capsid genes) (Figure 2b, lane 1 and lane 3), than following cotransfection of pAV2.F5tg83luc and the AAV2 Rep-Cap helper plasmid pAVRC2.3 (Figure 2b, lane 2). Of note, the expression of the HBoV1 viral proteins appeared to not interfere with rAAV2 genome rescue and replication (Figure 2b, compare lane 4 vs. lane 1 or 3). Transmission electron microscopy demonstrated that rAAV2/HBoV1 virions had a typical parvovirus icosahedral structure that was 26 nm in diameter (Figure 2c), similar to wild-type HBoV1 virions.24 The density of the interior of the virions by electron microscopy also suggested that >99% of virions were fully packaged with DNA. Examination of the chimeric rAAV2/HBoV1 virions by western blot with anti-HBoV1 VP2 antiserum demonstrated the presence of HBoV1 VP1 and VP2 proteins, but no AAV2 VP proteins were detected in the rAAV/HBoV1 stock using an anti-AAV2 capsid monoclonal antibody B1 (Figure 2d).
Characterization of viral genome polarity and capacity of rAAV2/HBoV1 virions
One significant difference between wild-type AAV2 and HBoV1 virions is the polarity of packaged genomes—AAV packages both plus and minus DNA strands with equal efficiencies, whereas HBoV1 selectively encapsidates the minus DNA strand more than 90% of the time.27 The two terminal palindromic sequences of HBoV1 are asymmetric (differing in size, primary sequence, and predicted structure),24 whereas for AAV, terminal palindromic sequences are identical inverted repeats. The terminal sequences in parvovirus genomes are critical to the formation of concatameric duplex replication intermediates and excision of single stranded progeny genomes for packaging.34 Given that the rAAV2/HBoV1 vector genome has identical inverted repeats at the ends of its genomes, we hypothesized that rAAV2/HBoV1 would adopt unbiased packaging of both the plus and minus strands. This was indeed the case. Using sense and antisense probes against the luciferase transgene, rAAV2/HBoV1 virion DNA demonstrated approximately equal proportions of plus and minus strands, whereas rHBoV1 vector DNA demonstrated a preference (~87%) for packaging the minus strand. These differences may, in part, account for the two- to threefold higher yield in generation of rAAV2/HBoV1 over that of rHBoV1.
A second major difference between AAV and HBoV1 genomes are their size—the AAV genome is 4,679 nt in length, whereas the HBoV1 is 5,543 nt in length. The packaging capacity of rAAV vectors has been extensively studied and has limits of 4.9–5.0 kb.35,36 This is a significant hurdle for delivery of the CFTR gene by rAAV, and one that might be potentially overcome with rAAV2/HBoV1 vectors. Thus, we hypothesized that the rAAV2/HBoV1 particle might offer a significant advantage for CFTR delivery by virtue of its ability to package oversized rAAV genomes up to 5.5 kb, as observed with rHBoV1 (Figure 1d). To explore this possibility, we generated two rAAV proviral genomes with identical luciferase expression cassettes that differed in length by 600 bp (pAV2/HBc.F5tg83Luc at 4.8 kb and pAV2/HBc.CF5tg83Luc at 5.4 kb). Each of these proviral plasmids was used to generate rAAV2/2 (4.8 kb) and/or rAAV2/HBoV1 (4.8 and 5.4 kb) viruses, and the viral DNA was evaluated by alkaline-denatured agarose gel electrophoresis followed by Southern blot analysis. The viral yields of 5.4 kb rAAV2/HBoV1 were similar to 4.8 kb rAAV2/HBoV1. The Southern blot analysis of viral DNA revealed only genomes of the appropriate size with no obvious truncated forms (Figure 3b). These findings demonstrate that HBoV1 pseudotyping accurately processes and packages both short and long rAAV genomes without altering genome integrity.
Figure 3.
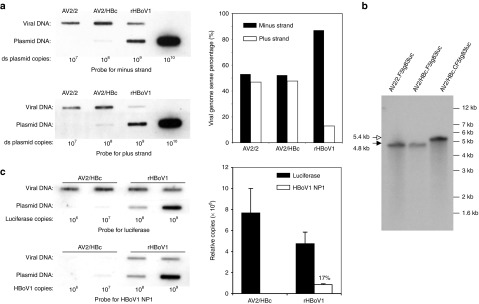
Package polarity and capacity of rHBoV1 and rAAV2/HBoV1 vectors. (a) Viruses AV2/2.F5tg83luc, AV2/HBc.F5tg83luc, and rHBoV1.CBAluc were loaded on nylon membrane by slot blotting and visualized with 32P-labeled 32-mer oligonucleotide probes against the minus and plus strand of the luciferase gene (left panels). The percentages of the minus and plus strands in each viral preparation was calculated based on the signal density quantitated with NIH ImageJ software (right panel). (b) 2 × 108 DRP of rAAV vector AV2/2.F5tg83luc, chimeric viruses AV2/HBc.F5tg83luc, and AV2/HBc.CF5tg83luc were heated in alkaline gel loading buffer at 95 °C for 10 minutes and then resolved in a 0.9% alkaline agarose gel. Following transferred to Nylon membrane, Southern blotting was performed with 32P-labeled luciferase probe. Black and white arrows mark the shorter rAV2.F5tg83luc (4.8 kb) and longer rAV2.CF5tg83luc (5.4 kb) genomes, respectively. (c) Left panels depict slot blots of AV2/HBc.F5tg83luc and rHBoV1.CBAluc viral preparations (~109 DRP based on TaqMan PCR for the luciferase transgene) probed with 32P-labeled fragments recognizing the luciferase gene (1.7 kb) or the HBoV1 genome region unique to the helper plasmid (a 2.64 kb HindIII/BglII fragment covering the NP1 coding region). Right panel depicts the relative copies of luciferase or NP1 gene fragments based on the signal intensity relative to the plasmid standards. NIH ImageJ software was used to quantify the mean (±range) signal density for rAAV2/HBoV1 and rHBoV1 viral preparations shown. DRP, DNase I–resistant particles.
Viral genome recombination plays an important role in the evolution of many viruses, and strand recombination also occurs during the replication of single-stranded viruses.37 Enteric human bocavirus infections are also associated with a high level of viral genome recombination.38 We thus sought to evaluate whether recombination products between the helper and proviral plasmids were packaged into rHBoV1 and rAAV2/HBoV1 virions. Slot blot analyses of viral genomes from purified rAAV2/HBoV1 and rHBoV1 were conducted using a luciferase transgene probe and HBoV1 helper-specific viral probe (i.e., the HindIII/BglII 2.64 kb fragment replaced by the luciferase cassette in the rHBoV1 vector). Results from this analysis demonstrated that 17% of the viral genomes in purified rHBoV1 stocks contained HBoV1 sequences found only in the helper plasmid (Figure 3c). The inclusion of 1.4 kb and 1.1 kb HBoV1 genome fragments flanking the luciferase cassette in the rHBoV1.CBAluc genome is the mostly likely cause of these recombination events. Although both the NS and VP protein coding domains were mutated in the rHBoV1.CBAluc genome (Figure 1a), if recombination occurred outside these mutations in a double cross-over event, replication-competent rHBoV1 genomes would be expected in rHBoV1 viral stocks. The presence of replication-competent virus could be one of the reasons for the time-dependent decline in transgene expression of rHBoV1 infected HAE ALI cultures (Figure 1e). In contrast to rHBoV1, HBoV1 helper genomes were not detected in purified rAAV2/HBoV1 virus, as would be expected, since there is no sequence homology between the proviral and helper plasmids. Thus, further development of packaging strategies for rHBoV1 are needed to eliminate the chance of generating replication-competent HBoV1 (i.e., expression of NS and VP genes on separate helper plasmids and minimal cis-elements for packaging in the proviral genome). However, improved knowledge of the regulation of NS/VP viral genes and HBoV1 genome packaging will be needed before similar strategies that eliminate replication-competent virus in the generation of rAAV can be applied.
Chimeric rAAV2/HBoV1 vectors mediate highly efficient transduction from the apical, but not basolateral membrane, of HAE
We next examined the transduction characteristics of the rAAV2/HBoV1 chimeric vectors. A rAAV2/HBoV1 vector encoding the luciferase transgene failed to transduce HEK293 cells at even high MOIs (50,000 DRP/cell), whereas the analogous rAAV2/2 vector efficiently expressed luciferase at much lower MOIs (2,500 DRP/cell) (Figure 4a). Experiments in primary HAE and CuFi ALI cultures confirmed that both AV2/HBc.F5tg83luc (4.8 kb genome) and AV2/HBc.CF5tg83luc (5.4 kb genome) gave rise to similar levels of luciferase expression following apical infection (data not shown), suggesting that vector size within this range did not impact transduction. We next compared the transduction of rAAV2/HBoV1 with that of rAAV vectors under the same infection conditions. This direct comparison was only possible with AV2/HBc.F5tg83luc, AV2/1.F5tg83luc and AV2/2.F5tg83luc (with identical proviral genomes derived from pAV2-F5tg83luc), because the pAV2-CF5tg83luc genome (5.4 kb) was too large to be packaged into AAV capsids. The transduction patterns for primary HAE following apical and basolateral infection with AV2/HBc.F5tg83luc and AV2/2.F5tg83luc were strikingly different (Figure 4b). As previously observed, rAAV2/2 transduced HAE with a strong basolateral preference15,39—the luciferase expression following apical infection was 210-fold lower than that following basolateral infection (Figure 4b). By contrast, rAAV2/HBoV1 demonstrated a 206-fold greater level of transduction following apical infection of primary HAE as compared with basolateral infection (Figure 4b). Importantly, the level of transgene expression achieved following apical infection with AV2/HBc.F5tg83luc was 70-fold greater than that from AV2/2.F5tg83luc, and 5.6-fold greater than AV2/1.F5tg83luc. As previously demonstrated, rAAV2/1 lacked a polarity bias for transduction in polarized HAE, and had better apical transduction efficiency than rAAV2.15
Figure 4.
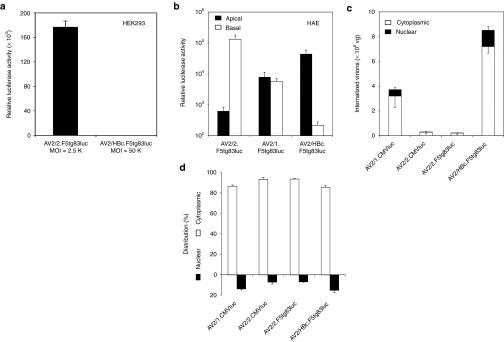
Transduction comparisons between rAAV2/HBoV1 and rAAV vectors. (a) Luciferase expression at 2 days following infection of HEK293 cells with AV2/2.F5tg83luc (MOI = 2,500 DRP/cell) or AV2/HBc.F5tg83luc (MOI = 50,000 DRP/cell). Results show the mean (±SEM, N = 4) relative luciferase activities per well of a 24-well plate. (b) Primary HAE ALI cultures were infected with AV2/2.F5tg83luc, AV2/1.F5tg83luc, or AV2/HBc.F5tg83luc from the apical or basolateral surface. The vector amount in the inoculum was 1010 DRP for each Millicell insert, roughly 5,000 to 10,000 DRP/cell. Data represent the mean (±SEM) relative luciferase activities measured at 7 days after infection (RLU/well) for N = 6 independent infections of HAE ALI cultures derived from three donors. (c,d) Virion internalization and subcellular distribution analyses were performed at 18 hours after primary HAE ALI cultures were apically infected with rAAV2/1, rAAV2/2, and rAAV2/HBoV1 vectors of 1010 DRP per Millicell insert. Viral genomes in the cytoplasmic and nuclear fractions were quantified by TaqMan PCR. The total viral genomes detected in each culture is presented in (c) with the black bars representing the nuclear fraction, whereas bars representing the cytoplasmic fraction. The percentage of viral genomes in each fraction is presented in (d). Data represent the mean (±SEM) viral genome copies (per well) for N = 3 independent infections. ALI, air–liquid interface; DRP, DNase I–resistant particles; HAE, human-polarized airway epithelia.
Given that HBoV1 is a newly discovered virus, little is known about HBoV1-cell interactions in HAE. We found that the wild-type HBoV1 can infect HAE at MOIs as low as 0.001 DRP/cell,25 suggesting that viral entry from the apical surface of HAE is quite efficient. However, with the added variable of viral replication, the efficiency of HBoV1 internalization and intracellular trafficking to the nucleus is difficult to directly evaluate. The creation of a replication defective rAAV2/HBoV1 chimeric virus provides the opportunity to directly evaluate these processes and furthermore compare the efficiency of these steps in transduction with rAAV vectors. To this end, we compared virion uptake and viral genome distribution in primary ALI cultures of HAE at 18 hours following apical infection. As controls, we included two rAAV pseudotyped vectors, rAAV2/1 (AV2/1.CMVluc) and rAAV2/2 (AV2/2.CMVluc), which are known to transduce HAE from the apical membrane with different efficiencies.15 rAAV1 has thus far been shown to be one of the most efficient AAV serotype for transduction of HAE, with greater virion uptake and faster nuclear translocation following apical infection.14,15 In addition, we evaluated both AV2/HBc.F5tg83luc and AV2/2.F5tg83luc viruses that contain the identical viral genome. Results from these comparisons demonstrated that AV2/1.CMVluc and AV2/HBc.F5tg83luc showed ~14-fold and ~32-fold more viral uptake from the apical membrane of primary HAE than the rAAV2/2 vectors (AV2/2.CMVluc and AV2/2.F5tg83luc), respectively (Figure 4c). AV2/HBc.F5tg83luc viral uptake was also 2.3-fold more efficient than AV2/1.CMVluc (P = 0.026). Furthermore, the post-entry processing of rAAV2/HBoV1 to the nucleus appeared to be more rapid than for rAAV2/2, with ~15% of internalized AV2/HBc.F5tg83luc genomes detected in the nuclear fraction by 18 hours after infection, as compared with ~7% for rAAV2/2 (Figure 4c,d). Interestingly, the cytoplasmic/nuclear distribution of viral genomes was similar for rAAV2/1 (86.4/13.6%) and rAAV2/HBoV1 (85.0/15.0%) (Figure 4c,d). In contrast to both rAAV serotypes tested, rAAV2/HBoV1 poorly transduced HAE from the basolateral membrane (Figure 4b). Overall, these findings suggest that rAAV2/HBoV1 viral uptake and nuclear translocation is highly efficient following apical infection of primary differentiated HAE.
Modulating proteasome activity during the infection period greatly enhances transduction following apical infection with rAAV2/HBoV1
Despite the fact that rAAV2/HBoV1 demonstrates a high transduction efficiency following apical infection in HAE ALI, the majority (85%) of internalized rAAV2/HBoV1 virions are retained in the cytoplasm at 18 hours after infection. This suggested that intracellular barriers limiting effective nuclear transport of the virus, as observed for rAAV2 and rAAV1 transduction of HAE,7,40 may also exist for HBoV1. Thus, the transduction efficiency of rAAV2/HBoV1 could be further improved by overcoming these barriers. Impaired endosomal processing/intracellular trafficking is one of the major barriers that limit rAAV vector transduction of polarized HAE following apical infection. This barrier can be partially overcome by the application of proteasome inhibitors at the time of infection or within a certain period after infection.7,9,39 These studies have demonstrated that both tripeptidyl aldehyde N-acetyl-l-leucyl-l-leucyl-l-norleucine (LLnL) and the anthracycline derivative doxorubicin (Dox) can enhance the rAAV2, rAAV5, and rAAV1 viral processing and translocation to the nucleus, leading to higher levels of transduction. Combined administration of these two distinct classes of proteasome activity modulating agents can induce transduction over 1,000-fold following apical rAAV2/2 infection of primary HAE.15,39 Although there is a significant divergence between the HBoV1 and AAV2 capsid proteins at primary sequence level, these two viruses retain some conserved core capsid sequences and also share a similar surface icosahedral topology with other parvovirus particles.29 Thus, we hypothesized that treatment with proteasome inhibitors might also enhance transduction of HAE by rAAV2/HBoV1, and sought to study the kinetics of transduction between rAAV2/2 and rAAV2/HBoV1 in the presence and absence of LLnL and Dox. Results comparing rAAV2/2 with rAAV2/HBoV1 (Figure 5a,b) demonstrated a similar rise in transgene expression between 3–7 days after infection, with a plateau at 7–11 days after infection. For both vectors, treatment with proteasome inhibitors at the time of infection enhanced transduction at all time points greater than 1,000-fold, and there was no decline in transgene expression at the 11-day period. These findings demonstrate that rAAV2/HBoV1 shares a similar proteasome-dependent barrier to transduction as observed with most other rAAV serotypes.
Figure 5.

Effect of proteasome inhibitors on rAAV2/HBoV1 transduction in polarized and non-polarized cultures of human airway epithelial cells. (a,b) Primary HAE ALI cultures were apically infected with 1010 DRP per Millicell insert with (a) AV2/2.F5tg83luc or (b) AV2/HBc.F5tg83luc for a period of 16 hours. When indicated, proteasome inhibitors (PI) LLnL (40 nmol/l) and doxorubicin (5 µmol/l) were applied only during the infection period. Luciferase expression was monitored over 11 days by biophotonic imaging of live cells using the Xenogen 200 IVIS (Xenogen). Data represent the mean (±SEM, n = 6) relative luciferase activity per well at three time points of 3, 7, and 11 days after infection. (c) CuFi8 cells cultured as a polarized epithelium at an ALI (CuFi ALI; a: apical infection, b: basolateral infection) or non-polarized undifferentiated monolayers on plastic (CuFi-UD), and HEK293 cells, were incubated with 1.5 × 109 DRP of AV2/HBc.F5tg83luc at 37 °C for 4 hours. All cultures contained ~5 × 105 cells at the time of infection. Following infection, unbound virus was washed off and cells were either detached from the culture supports with trypsin and lysed for TaqMan PCR quantification of viral genomes, or returned to the incubator for luciferase expression assays at 24 hours after infection using cell lysates. When indicated (+PI), CuFi8 cells were treated with proteasome inhibitors doxorubicin (1 µmol/l) and LLnL (8 nmol/l) during the 4 hours infection period. Data represent the mean (±SEM) total vector genomes (n = 4) at 4 hours after infection and relative luciferase activity (n = 3) at 24 hours after infection. ALI, air–liquid interface; DRP, DNase I–resistant particles; HAE, human-polarized airway epithelia.
Polarization of human airway epithelia is required for HBoV1 capsid–mediated gene transfer
The mechanism by which HBoV1 productively infects the apical membrane of polarized human airway epithelia remains unclear. In previous studies, we observed that infection of monolayers of CuFi cells with wild-type HBoV1 does not support viral replication and production of progeny virions,24 a finding similar to the lack of transduction of monolayer CuFi cells with luciferase expressing rHBoV1 (Figure 1d). By contrast, when polarized, CuFi cells efficiently produce progeny virus following apical, but not basolateral, infection with wild-type HBoV1.24 These findings suggest that polarization influences a cellular factor(s) required for productive infection, such as expression of a viral receptor/coreceptor or expression of factors involved in intracellular processing of the HBoV1 virion. As viral replication does not occur within the chimeric rAAV2/HBoV1 vector, this was an opportunity to define the step(s) following HBoV1 infection that are influenced by polarization. To this end, we performed experiments with rAAV2/HBoV1 to address whether the polarization of airway epithelial cells influences HBoV1 capsid–mediated transduction through steps involving receptor binding/uptake or the post-entry intracellular processing of virions. As the ubiquitin-proteasome pathway affects rAAV2/HBoV1 transduction in HAE ALI cultures, we also examined the influences of proteasome inhibitors on these two steps of infection.
Equivalent numbers of CuFi cells under monolayer (i.e., non-polarized) or polarized ALI culture conditions were incubated with equal amounts of AV2/HBc.F5tg83luc virus at 37 °C for 4 hours. After removal of the unbound vectors, the infected cells were either lysed for quantification of internalized vector genomes by TaqMan PCR or cultured for an additional 20 hours before assessing transgene expression (Figure 5c). Results from this analysis demonstrated that apical transduction of polarized CuFi ALI cultures with AV2/HBc.F5tg83luc was 72-fold more efficient than basolateral transduction, a finding consistent with AV2/HBc.F5tg83luc infection of primary HAE ALI cultures (Figure 4b). Under these conditions, ~40-fold more virus was taken up by CuFi epithelia following a 4 hours apical infection as compared with basolateral infection (Figure 5c), suggesting that polarization enhances the abundance of HBoV1 receptor/coreceptor on the apical membrane. Interestingly, AV2/HBc.F5tg83luc entered CuFi cell monolayers at an efficiency similar to that observed following apical infection of polarized CuFi epithelia (Figure 5c), despite significantly reduced transduction of non-polarized CuFi cultures (Figure 4b). In addition, analysis of viral uptake and transduction of HEK293 cells infected with AV2/HBc.F5tg83luc, which is not permissive to HBoV1 infection, revealed substantial viral uptake without transgene expression (Figure 5c). These two observations in CuFi and HEK293 monolayer cultures suggest that post-entry barriers, rather than receptor-mediated uptake, also play a key role in rAAV2/HBoV1 transduction.
To further investigate post-entry barriers to rAAV2/HBoV1 transduction, we also evaluated the influences of proteasome inhibitors. Overall, proteasome inhibitors had little effect on viral uptake following infection under all the conditions evaluated (apical or basolateral infection of polarized CuFi ALI or infection of non-polarized CuFi monolayers) (Figure 5c). By contrast, proteasome inhibitor application during the 4 hours infection period enhanced transduction 45-fold following apical infection of polarized CuFi epithelia, whereas only marginally enhancing transduction following basolateral infection of polarized CuFi epithelia (fivefold) or infection of CuFi monolayers (4.3-fold) (Figure 5c). These results suggest proteasome-dependent barriers to intracellular processing of HBoV1 virions are greater from the apical membrane of polarized CuFi epithelia. Furthermore, CuFi monolayers appear to have the greatest post-entry block to HBoV1 virion processing that is also less proteasome-dependent. Cumulatively, these results suggest that polarization/differentiation of airway epithelial cells alter both receptor-mediated uptake and intracellular processing of HBoV1 virions.
A rAAV2/HBoV1 vector harboring a 5.5 kb genome with a strong CBA-hCFTR expression cassette can correct CFTR-mediated chloride currents in CF HAE
The application of rAAV vectors for CF gene therapy has been hindered by the relatively small packaging capacity of the virus and the large size of full-length CFTR cDNA (4,443 bp of coding sequence). This has necessitated the use of very small synthetic weaker promoters and/or the deletion of CFTR domains not critical for chloride channel function.18 Both of these approaches are suboptimal for CFTR-mediated gene therapy. A rAAV2/HBoV1 vector would have enough space to accommodate strong transcription regulatory elements for human CFTR gene expression. To provide the proof-of-concept for this approach, we constructed a rAAV2 proviral plasmid harboring a 5.5 kb genome containing a 5.2 kb human CFTR expression cassette driven by the strong CBA promoter. When this proviral plasmid was packaged into HBoV1 virions, the resultant viral yield (AV2/HBc.CBAhCFTR) averaged 1.5 × 1011 DRP from ten 150 mm dishes of transfected HEK293 cells, a similar level of production for rAAV2/HBoV1 vectors with luciferase reporters. The integrity of the 5.5 kb rAAV genome within AV2/HBc.CBAhCFTR was confirmed by alkaline agarose gel analysis (data not shown).
To validate the function of AV2/HBc.CBAhCFTR virus, we infected primary differentiated CF HAE with 1010 DPR (5,000–10,000 DRP/cell) from the apical surface in the presence of proteasome inhibitors and assessed CFTR function at 10 days following infection. CFTR function was evaluated as the change in cAMP-mediated short-circuit current (Isc) following stimulation with IBMX and forskolin and inhibition with GlyH101 (a CFTR inhibitor). DIDS and amiloride were used to block non-CFTR chloride channels and ENaC-mediated sodium currents before cAMP induction. Results comparing complementation of CFTR-mediate chloride currents following apical infection with AV2/HBc.CF5tg83Luc (control vector) and AV2/HBc.CBAhCFTR are shown in Figure 6a. A significant change in cAMP-inducible Isc was observed following AV2/HBc.CBAhCFTR infection, as compared control AV2/HBc.CF5tg83Luc infected samples, and this current was blocked by the addition of the CFTR inhibitor GlyH101. Figure 6b summarizes the ΔIsc(cAMP) following cAMP agonist induction and the ΔIsc(glyH) following GlyH101 inhibition for the two infections conditions and non-CF controls. The level of correction following AV2/HBc.CBAhCFTR infection (ΔIsc(cAMP) = 2.60 ± 0.96 µA/cm2 and ΔIsc(glyH) = 2.98 ± 0.73 µA/cm2) reflects ~30% of the ΔIsc(cAMP) and ΔIsc(glyH) observed in non-CF HAE. Expression of hCFTR protein on the apical surface of AV2/HBc.CBAhCFTR infected CF HAE was also confirmed by immunofluorescent staining (Figure 6c). Little immunoreactivity was observed in the AV2/HBc.F5tg83Luc infected samples, as might be expected since ΔF508-CFTR is efficiently degraded in primary CF HAE.41
Figure 6.

Partial correction of CFTR-dependent chloride transport by primary CF HAE ALI cultures following infection with AV2/HBc.CBAhCFTR. CF HAE ALI cultures derived from two CF patient donors (genotypes: ΔF508/ΔF508 homozygous) were infected with AV2/HBc.CF5tg83luc or AV2/HBc.CBAhCFTR at 1010 DRP per Millicell insert (MOI of 5,000–10,000 DRP/cell) in the presence of proteasome inhibitors LLnL (40 nmol/l) and doxorubicin (5 µmol/l). Uninfected non-CF HAE were also cultured for electrophysiologic comparisons and experimental cultures were evaluated at 10 days after infection. (a) Representative traces of transepithelial short-circuit current (Isc) of CF HAE following the sequential addition of various inhibitors and agonists as indicated. Amiloride and DIDS were used to block ENaC-mediated sodium currents and non-CFTR chloride channels before cAMP agonists (forskolin and IBMX) induction and GlyH101 inhibition of CFTR currents. ΔIsc(cAMP) reflects the activation of CFTR-mediated chloride currents following cAMP agonist induction and ΔIsc(glyH) reflects the inhibition of CFTR-mediated chloride currents following addition of GlyH101. (b) Summary data of the ΔIsc(cAMP) and ΔIsc(glyH) (mean ± SEM, n = 6 independent transwells) for both CF infected cultures and non-CF controls. (c) Immunofluorescent detection of CFTR expression (green) in CF HAE following infection with AV2/HBc.CBAhCFTR (left panels) or AV2/HBc.CF5tg83luc (right panels). ALI, air–liquid interface; DRP, DNase I–resistant particles; HAE, human-polarized airway epithelia.
Discussion
The newly discovered and partially characterized HBoV1 provides several potential attractive advantages for the design of CF airway gene therapy vectors. First, wild-type HBoV1 efficiently infects HAE from the apical surface at extremely low MOIs, suggesting that its capsid proteins are highly adapted for airway infection. Second, wild-type HBoV1 has a genome of 5,500 nt, suggesting that larger CFTR expression cassettes could be efficiently packaged into a recombinant HBoV1 virus. For these reasons, we successfully generated both replication-defective rHBoV1 and pseudotyped rAAV2/HBoV1 vectors and studied their transduction profiles in HAE. Our findings demonstrate that rAAV2/HBoV1 vectors may indeed be an attractive alternative to rAAV vectors for gene therapy of CF. In addition, our studies evaluating these new recombinant HBoV1-based vectors have uncovered interesting biology regarding how polarization/differentiation influences HBoV1 capsid–mediated infection and transduction from the apical and basolateral membranes of HAE.
Although cross-genera parvovirus pseudopackaging has been known to be possible for some time, the efficiency appears much higher for HBoV1-based vectors. For example, the efficiency of the rAAV2 genome encapsidation in parvovirus B19 capsids yields viral titers of ~109 DRP/ml,30 whereas yields of rAAV2/HBoV1 vectors are ~2 × 1011 DPR/ml. Yields of rAAV2/HBoV1 were slightly higher (approximately two- to fourfold) than that for rHBoV1, but similar to that of wild-type HBoV1 production in HEK293 cells following transfection of the infectious clone pIHBoV1.24 However, it remains clear that improvements in viral packaging are still needed, as the yield of rAAV2/HBoV1 vectors remains ~10% of the level for rAAV2.
One unique aspect of the HBoV1 capsid is the fact it more efficiently transduces (>100-fold) polarized airway epithelia from the apical surface as compared with the basolateral surface. This membrane polarity of HAE transduction is distinct from all other rAAV serotypes studied to date. For example, rAAV2, rAAV5, and rAAV6 preferentially transduce HAE from basolateral membrane with a ~100-fold preference,14,15,40 whereas rAAV1 demonstrates an equal preference for transduction from both apical and basolateral membranes.15 In the context of HBoV1 capsid, polarization appears to be key to induce viral receptors and/or coreceptors required for efficient transduction from the apical membrane. Indeed, enhanced viral genome uptake from the apical, as compared with basolateral, membranes of polarized CuFi epithelia suggests that the expression of an HBoV1 receptor(s) is likely regulated by polarization. The ability of proteasome inhibitors to effectively enhance rAAV2/HBoV1 transduction from the apical, but not basolateral, membrane also suggests that infection from these two membranes differs with respect to capsid processing biology of internalized HBoV1 virions.
Interestingly, the process of HBoV1 infection of non-polarized CuFi cells represents a biologic process that is uniquely different from that of apical or basolateral infection of polarized cells. In this context, CuFi monolayers exhibit efficient uptake of rAAV2/HBoV1, as seen following apical infection of CuFi ALI cultures, but largely lack proteasome responsiveness as observed following basolateral infection of CuFi ALI cultures. One potential explanation for these findings might be the partitioning of certain binding receptor and coreceptor pairs at the apical membrane that route virus to be productively processed through a proteasome-interacting pathway (Figure 7). For example, following polarization, a binding receptor/coreceptor that efficiently processes internalized virus may be shuttled to the apical membrane, resulting in low viral uptake from the basolateral membrane (Figure 7a). In the case of CuFi monolayers, efficient binding receptors may remain on the surface and interact with a more abundant second coreceptor that shuttles virus to an intracellular compartment that is less efficient for transduction and non-responsive to proteasome inhibition (Figure 7b). This second inefficient coreceptor may be sequestered in the basolateral membrane of polarized cells, thus preventing interference with apical infection (Figure 7a). This is only one scenario of many, and assumes the expression of binding receptors and coreceptors do not change following polarization. Alternative explanations for the differences in transduction biology between the three CuFi models may involve uniquely expressed binding receptors and/or coreceptors that are influences by polarization and differentiation.
Figure 7.

One potential model for how polarization of human airway epithelia cells influences HBoV1 virion infection and transduction. (a) Polarized HAE may contain multiple binding and/or coreceptors for HBoV1. In this illustrated scenario, a single binding receptor exists on the apical membrane and is significantly reduced or absent on the basolateral membrane. Two different coreceptors exist including an efficient coreceptor-1 on the apical membrane and a more abundant inefficient coreceptor-2 on the basolateral membrane. Endocytosis through coreceptor-1 leads to functionally efficient (from a transduction standpoint) virion processing that is highly influences by activity of the proteasome, whereas internalization through coreceptor-2 is ineffective at processing the virion and not influenced by proteasome function. This model is consistent with significantly less viral uptake and transduction from the basolateral surface, as compared with the apical membrane. Other models not shown might include a second type of binding receptor on the basolateral surface that is inefficiently endocytosed with coreceptor-1 or coreceptor-2. (b) In non-polarized human airway cells, the primary binding receptor, coreceptor-1, and coreceptor-2 exist in the same membrane. Both coreceptors can interact with the same binding receptor, however, coreceptor-2 is in greater abundance than coreceptor-1. Thus, endocytosis of HBoV1 virions through coreceptor-2 predominates, and since this pathway inefficiently processes HBoV1 virions for productive transduction, transgene expression is low. These findings are consistent with high-level HBoV1 virion endocytosis, but poor transduction and weak proteasome inhibitor responsiveness, in non-polarized human airway cells. HAE, human-polarized airway epithelia.
Like most rAAV serotypes, rAAV2/HBoV1 transduction of primary HAE from the apical membrane was significantly enhanced (>1,000-fold) by the addition of proteasome inhibitors at the time of apical infection (Figure 5b). Proteasome inhibitors have been shown to enhance trafficking of rAAV virions to the nucleus by promoting ubiquitination of the capsid9 and our results demonstrating no change in rAAV2/HBoV1 viral uptake following proteasome inhibitor treatment are consistent with action at a post-entry point following infection. However, it remains unclear if the mechanism of the proteasome-sensitive post-entry barrier is similar for rAAV and HBoV1 virions. For example, although we observed similar patterns of cytoplasmic and nuclear distribution between the rAAV2/HBoV1 and rAAV2/1 following apical infection of primary HAE, rAAV2/HBoV1 was approximately tenfold more sensitive to enhancement of transduction by proteasome inhibitors than previous observed for rAAV1.15 Differences in the mechanism of virion processing and uncoating between rAAVs and HBoV1 may be responsible for these observations and warrants further investigation.
One of the most important differences between rAAV2/HBoV1 and rAAV vectors is the packaging capacity for a transgene cassette. rAAV2/HBoV1 vectors can carry genomes up to 5.5 kb as compared with 4.9 kb for rAAV vectors. Although the upper limit of genome packaging within HBoV1 capsids was not evaluated in this study, the 12% increase in genome size (600 bp) is very significant for delivery of CFTR expression cassettes. rAAV2/HBoV1 packaging enabled the use of a strong CBA promoter–driven CFTR expression cassette, and resulted in very reasonable correction of CFTR-dependent chloride currents in CF HAE. Based on the ability of rAAV to effectively package ~5% more DNA than the wild-type genome, it is reasonable to expect that HBoV1-based vectors may be capable of efficiently packaging genomes up to 5.8 kb in length. In addition, it may be possible to encapsidate self-complementary (sc) double-stranded forms of rAAV genomes (2.7 to 2.8 kb in length) into HBoV1 capsids.
In conclusion, we have developed two new types of recombinant HBoV1-based vectors for efficient gene therapy to the human airway. However, chimeric rAAV2/HBoV1 vectors have three clear advantages over the authentic rHBoV1 vectors for human gene therapy. First, rAAV2/HBoV1 vector yields were significantly greater in an HEK293 cell production system than that for rHBoV1. Second, the rAAV2 genome has already been used in clinical trials and avoids potential safety concerns that might accompany use of a new viral genome. Third, the application of rHBoV1 vectors could be hampered by the potential for contamination of replication-competent virus in the vector stocks, which could be generated through homologous recombination of helper plasmids. This later concern is likely theoretical as we did not observe cytopathology in HAE following infection with rHBoV1. Nonetheless, further vector development is required for the application of rHBoV1 to both minimize the potential for replication-competent virus and improve vector yields.
Despite the promise of this new rAAV2/HBoV1 vector system, several unknowns require further investigation before considering clinical applications. For example, is there pre-existing airway humoral immunity to HBoV1 caspids in most CF patients, and if so, does this impact rAAV2/HBoV1 infection? Studies have suggested that ~71% of humans ranging from birth to 41 years of age contain circulating antibodies against HBoVs;42 however, nothing is known concerning neutralizing antibodies to this virus in the airway. Given that HBoV1 infections primarily occur within the first year of life, it is assumed that such immunity is protective to secondary infections. However, the frequent detection of HBoVs in stool from both healthy children and adults, as well as seroepidemiology studies, suggests that viral shedding can occur for long periods and/or patients can have frequent recurrent infections.38,43 Secondary infections or anamnestic immune responses also appear to commonly occur, whereas primary infections are strongly associated with respiratory illness, secondary immuno-activation by HBoV1 is not.44 It remains unclear whether such humoral immunity can prevent infection from the airway surface or acts to limit replication and spread of HBoV1.45 The fact that wild-type HBoV1 shows long-term and low-level persistence in the respiratory tract following primary infection21,46 suggests that this virus may have methods of evading immune detection. The development of rHBoV1 and rAAV2/HBoV1 vectors should enable such questions to be addressed using HAE reconstitution experiments combining recombinant reporter virus with serum or bronchioalveolar lavage samples from HBoV1 infected patients. Despite these unknowns, the development of HBoV1-based recombinant vectors may have unique utilities for CF gene therapy and/or vaccination of infants to protect from wild-type HBoV1 infections.
Materials and Methods
Plasmids. pIHBoV1 is the infectious clone plasmid containing the 5,543 bp full-length HBoV1 genome.24 prHBoV1-CBAluc is a recombinant HBoV1 (rHBoV1) transfer plasmid derived from pIHBoV1 and was constructed by replacing the 2.64 kb HindIII/BglII fragment of the HBoV1 genome by a 2.74 kb fragment containing the CMV enhancer/chicken β–actin promoter–driven luciferase gene. The NP1 gene, which plays an essential role in HBoV1 DNA replication, was completely removed in the resulting prHBoV1-CBAluc plasmid. To reduce the probability of rescued wild-type virus through recombination, the 5′ remained NS gene coding region was further disrupted by elimination of a BspE1 site using blunt ligation, and the 3′ VP partial coding region was further deleted by removal of a 145 bp PstI to EcoRI fragment. The helper plasmid pHBoV1KUm630 harbors the 5,299 bp HBoV1 genome (99 to 5,395 nt) without terminal repeats,28 with the P5 promoter and 3′ polyA signal being retained for the expression of viral genes. pAV-Rep2 and pAD4.1 are the helper plasmids supporting rAAV2 genome rescue and replication from proviral plasmids in 293 cells as described previously.15,47 pAV2-F5tg83luc is a rAAV2 cis transfer plasmid, containing a 4.85 kb rAAV proviral genome with a 180 bp synthetic promoter driving firefly luciferase gene. pAV2-CF5tg83luc is a longer form of pAV2-F5tg83luc, and was derived by inserting 600 bp of stuffer sequence upstream the 180 bp synthetic promoter to generate a rAAV2 proviral genome 5.4 kb in length. pAV2-CBAhCFTR harbors an oversized 5.5 kb rAAV2 proviral genome containing a human CFTR expression cassette with a 580 bp CMV IE enhancer plus β-actin promoter (CBA promoter), a 50 bp synthetic polyA signal, and a 4,443 bp human CFTR cDNA containing 56 bp 5′UTR and 45 bp 3′UTR.
Recombinant virus production. rAAV vectors, rAAV2/2.F5tg83luc and AV2/1.F5tg83luc, were generated by triple plasmid cotransfection using an adenovirus-free system in HEK293 cells as previously described;15 this system uses the rAAV trans-helper plasmid pAVRC2.3, adenovirus helper plasmid pAD Helper 4.1, and rAAV2 proviral plasmid pAV2-F5tg83luc, transfected at a ratio of 2:3:1, respectively. The rHBoV1 vector stock (HBc.CBAluc) was generated by cotransfection of helper pHBoV1KUm630 and proviral plasmid prHBoV1-CBAluc into HEK293 cell at a ratio of 3:1, respectively. Chimeric rAAV2/HBoV1 vectors were generated by pseudotyping the rAAV genome within HBoV1 capsid following cotransfection of pAV-Rep2, pAd4.1, pHBoV1KUm630 together with the rAAV cis proviral plasmid into HEK293 cells at a ratio of 1.5:3:3:1, respectively. The rAAV proviral plasmids used for the chimeric vector production were pAV2-F5tg83luc (4.8 kb), pAV2-CF5tg83luc (5.4 kb), and pAV2-CBAhCFTR (5.5 kb). All viruses were recovered from the cell pellets at 72 hours after transfection and the cell crude lysates were treated as for rAAV vector production as previous describe.14 After DNase I digestion, all viruses were purified by two rounds of CsCl ultracentrifugation and dialyzed against phosphate-buffered saline. The titers of viral preparations as DRP were determined by TaqMan real-time quantification PCR and confirmed with slot blot assays using a 32P-labeled probe against the luciferase gene.14
Western blotting. 5 × 109 DRP of rAAV2 and chimeric rAAV2/HBoV1 were resolved by 10% SDS-PAGE. Following transfer to nitrocellulose membranes, two-color Westerns were performed with mouse anti-AAV capsid monoclonal antibody B1 (1:1,000) and rat anti-HBoV1 VP2 antiserum (recognizing both VP1 and VP2 proteins)28 (1:200). Infrared detection used 1:10,000 dilution of the secondary antibody goat anti-mouse-IRDye700 (red, for AAV) and goat anti-rat-IRDye800 (green, for HBoV1). Images were then scanned using an Odyssey Infrared Image System.
Cell culture and virus infection conditions. HEK293 and IB3 cells were cultured as monolayers in Dulbecco's modified Eagle medium, supplemented with 10% fetal bovine serum and penicillin-streptomycin, and maintained in a 37 °C incubator at 5% CO2. Undifferentiated immortalized CF human airway cells (CuFi8) were cultured as monolayers in bronchial epithelial cell growth medium (Lonza, Basel, Switzerland).31 Polarized primary human airway epithelia were generated as previously described from lung transplant airway tissue39,48 and were obtained from the Tissue and Cell Culture Core of The Center for Gene Therapy at the University of Iowa. Epithelia were grown on 12 mm Millicell membrane inserts (Millipore, Billerica, MA) and differentiated with 2% USG medium at an ALI before use. Polarization of CuFi8 cells at an ALI was performed using similar conditions to primary HAE. To apically infect the polarized airway epithelia, 1 × 1010 DRP of virus was diluted in USG medium to the final volume of 50 µl and applied to the upper chamber of the Millicell insert. For basolateral infections, 1 × 1010 DRP of virus was directly added to the culture medium in the bottom chamber. Viruses were typically exposed to epithelia for 16 hours and then removed. At this time, the Millicell inserts were briefly washed with a small amount USG medium and fed with fresh USG medium in the bottom chamber only. Approximately 1–2 × 106 cells are in each Millicell insert and thus the MOI is estimated ~5,000–10,000 DRP/cell. Transduction was assessed by luciferase reporter assays at various time points after infection using cell lysates or IVIS Biophotonic Imaging.
Measurement of luciferase reporter expression. Luciferase enzyme activity in cell lysates was determined using the Luciferase Assay System (Promega, Madison, WI) in a 20/20 luminometer equipped with an automatic injector (Turner Biosystems, Sunnyvale, CA). Quantification of luciferase activity in live cells was performed using the IVIS Biophotonic Imaging system according to the manufacturer's instructions. Images were captured 15 minutes after adding the VivoGlo Luciferin substrate (Promega) to the basolateral culture medium only, and quantification of images were processed with the Living Image 2.51 software (Xenogen, Alameda, CA).
Analysis of internalized viral genome. Fully differentiated HAE were infected with 1 × 1010 particles of rAAV or chimeric rAAV/HBoV1 vectors. After a 4 hours infection period, virus was removed, and epithelia were extensively washed with phosphate-buffered saline. The Millicell inserts then were fed with fresh medium in the bottom chamber and placed in a 37 °C incubator for 18 hours. Before harvesting cells for all viral internalization assays, Millicell inserts were washed thoroughly with 40 ml phosphate-buffered saline in a 50 ml conical tube three times. The cells were then detached from the support membrane of the Millicell inserts by trypsin digestion. The cell pellets were then washed three more times with 1 ml phosphate-buffered saline before subcellular fractionation and viral genome quantification. Control experiments using virus bound for 1 hours at 4 °C demonstrated >98% removal of virus from the cell surface using this washing and trypsin digestion method (data not shown). Nuclei were isolated with the Nuclei EZ pre kit (Sigma, St Louis, MO) as previous described.49 The cytoplasmic fractions were pooled during the nuclei preparation and 1/10 was dried in a SpeedVac. The nuclei pellet and dried cytoplasmic fraction were dissolved in 50 µl digestion buffer (50 mmol/l KCl, 2.5 mmol/l MgCl2, 10 mmol/l Tris pH 8.0, 0.5% NP40, 0.5% Tween-20 and 400 µg/ml proteinase K). After digestion at 56 °C for 45 minutes and heat-inactivation at 95 °C for 15 minutes, 0.1 µl of the nuclear and 1µl of the cytoplasmic digestion were used for TaqMan PCR to quantify viral genomes. When total viral internalization assays were performed in CuFi ALI cultures, non-polarized CuFi and HEK293 cell monolayers, the same washing and trypsinization procedure was used to remove cell-surface bound virions. However, the washed cell pellets were directly lysed with the above digestion buffer and used for viral genome quantification.
Quantitative analysis of rAAV genomes by TaqMan PCR. TaqMan real-time PCR was used to quantify the physical titer of the viral stocks and copies of viral genomes in cell lysates from AAV infected cells as previously described.15 The PCR primers used were 5′-TTTTTGAAGCGAAGGTTGTGG-3′ (forward) and 5′-CACACACAGTTCGCCTCTTTG-3′ (reverse) and amplify a 73 bp fragment of the rAAV2.Luc genome. The Taqman probe (5′-ATCTGGATACCGGGAAAACGCTGGGCGTTAAT-3′) was synthesized by IDT (Coralville, IA). This probe was tagged with 6-carboxy fluorescein (FAM) at the 5′-end as the reporter and Dark Hole Quencher 1 (BHQ1) at the 3′-end as the quencher. The PCR reaction was performed and analyzed using Bio-Rad MyIQ Real-time PCR detection system and software.
Short-circuit current measurements. Transepithelial short-circuit currents (Isc) were measured using an epithelial voltage clamp (Model EC-825) and a self-contained Ussing chamber system (both purchased from Warner Instruments, Hamden, CT) as previously described.50 Throughout the experiment the chamber was kept at 37 °C, and the chamber solution was aerated. The basolateral side of the chamber was filled with buffered Ringer's solution containing 135 mmol/l NaCl, 1.2 mmol/l CaCl2, 1.2 mmol/l MgCl2, 2.4 mmol/l KH2PO4, 0.2 mmol/l K2HPO4, and 5 mmol/l Hepes, pH 7.4. The apical side of the chamber was filled with a low chloride Ringer's solution in which 135 mmol/l Na-gluconate was substituted for NaCl. Transepithelial voltage was clamped at zero with current pulses every 5 seconds to record the short-circuit current using a VCC MC8 multichannel voltage/current clamp (Physiologic Instruments, San Diego, CA) with Quick DataAcq software. The following chemicals were sequentially added into the apical chamber: (i) amiloride (100 µmol/l) for inhibition of epithelial sodium conductance by ENaC; (ii) 4,4′-diisothiocyanato-stilbene-2,2′-disulfonic acid (DIDS) (100 µmol/l) to inhibit non-CFTR chloride channels; (iii) cAMP agonists forskolin (10 µmol/l)/3-isobutyl-l-methylxanthine (IBMX) (100 µmol/l) to activate CFTR chloride channels; and (iv) 10 µmol/l CFTRinh-GlyH-101 (N-(2-naphthalenyl)-[(3,5-dibromo-2,4-dihydroxyphenyl) methylene] glycine hydrazide) to block Cl− secretion through CFTR. ΔIsc calculations were made by taking the difference of the plateau measurements average over 45 seconds before and after each experiment conditions (chemical stimulus).
CFTR immunostaining. Following short-circuit current measurements, the HAE on the support membrane was cut out from the Millicell insert and embedded in OCT medium. 10µm cryosections were fixed in 4% paraformaldehyde followed by blocking and immunofluorescence staining using an anti-CFTR antibody cocktail consisting of equal amount (at a 1:200 dilution) of three mouse anti-CFTR antibodies (clone MM13-4 (Millipore), clone M3A7 (Millipore), and clone 13-1 (R&D System, Minneapolis, MN)) and finally incubation with donkey anti-mouse FITC-labeled secondary antibody.
Acknowledgments
This work was supported by NIH grants HL108902 (to J.F.E.), the Roy J Carver Chair in Molecular Medicine (to J.F.E.), the University of Iowa Center for Gene Therapy (DK54759; to J.F.E), and AI085236 (to J.Q.). The authors declare no conflict of interest.
References
- Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352:1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- Mueller C, Flotte TR. Gene therapy for cystic fibrosis. Clin Rev Allergy Immunol. 2008;35:164–178. doi: 10.1007/s12016-008-8080-3. [DOI] [PubMed] [Google Scholar]
- Driskell RA, Engelhardt JF. Current status of gene therapy for inherited lung diseases. Annu Rev Physiol. 2003;65:585–612. doi: 10.1146/annurev.physiol.65.092101.142426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flotte TR. Recombinant adeno-associated virus vectors for cystic fibrosis gene therapy. Curr Opin Mol Ther. 2001;3:497–502. [PubMed] [Google Scholar]
- Aitken ML, Moss RB, Waltz DA, Dovey ME, Tonelli MR, McNamara SC, et al. A phase I study of aerosolized administration of tgAAVCF to cystic fibrosis subjects with mild lung disease. Hum Gene Ther. 2001;12:1907–1916. doi: 10.1089/104303401753153956. [DOI] [PubMed] [Google Scholar]
- Moss RB, Milla C, Colombo J, Accurso F, Zeitlin PL, Clancy JP, et al. Repeated aerosolized AAV-CFTR for treatment of cystic fibrosis: a randomized placebo-controlled phase 2B trial. Hum Gene Ther. 2007;18:726–732. doi: 10.1089/hum.2007.022. [DOI] [PubMed] [Google Scholar]
- Duan D, Yue Y, Yan Z, Yang J, Engelhardt JF. Endosomal processing limits gene transfer to polarized airway epithelia by adeno-associated virus. J Clin Invest. 2000;105:1573–1587. doi: 10.1172/JCI8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding W, Zhang L, Yan Z, Engelhardt JF. Intracellular trafficking of adeno-associated viral vectors. Gene Ther. 2005;12:873–880. doi: 10.1038/sj.gt.3302527. [DOI] [PubMed] [Google Scholar]
- Yan Z, Zak R, Luxton GW, Ritchie TC, Bantel-Schaal U, Engelhardt JF. Ubiquitination of both adeno-associated virus type 2 and 5 capsid proteins affects the transduction efficiency of recombinant vectors. J Virol. 2002;76:2043–2053. doi: 10.1128/jvi.76.5.2043-2053.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, Li B, Jayandharan G, Mah CS, Govindasamy L, Agbandje-McKenna M, et al. Tyrosine-phosphorylation of AAV2 vectors and its consequences on viral intracellular trafficking and transgene expression. Virology. 2008;381:194–202. doi: 10.1016/j.virol.2008.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flotte TR, Fischer AC, Goetzmann J, Mueller C, Cebotaru L, Yan Z, et al. Dual reporter comparative indexing of rAAV pseudotyped vectors in chimpanzee airway. Mol Ther. 2010;18:594–600. doi: 10.1038/mt.2009.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Luo M, Guo C, Yan Z, Wang Y, Engelhardt JF. Comparative biology of rAAV transduction in ferret, pig and human airway epithelia. Gene Ther. 2007;14:1543–1548. doi: 10.1038/sj.gt.3303014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Luo M, Trygg C, Yan Z, Lei-Butters DC, Smith CI, et al. Biological Differences in rAAV Transduction of Airway Epithelia in Humans and in Old World Non-human Primates. Mol Ther. 2007;15:2114–2123. doi: 10.1038/sj.mt.6300277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Lei-Butters DC, Keiser NW, Engelhardt JF. Distinct transduction difference between adeno-associated virus type 1 and type 6 vectors in human polarized airway epithelia. Gene Ther. 2013;20:328–337. doi: 10.1038/gt.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Lei-Butters DC, Liu X, Zhang Y, Zhang L, Luo M, et al. Unique biologic properties of recombinant AAV1 transduction in polarized human airway epithelia. J Biol Chem. 2006;281:29684–29692. doi: 10.1074/jbc.M604099200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Zhang L, Johnson JS, Zhijian W, Grieger JC, Ping-Jie X, et al. Generation of novel AAV variants by directed evolution for improved CFTR delivery to human ciliated airway epithelium. Mol Ther. 2009;17:2067–2077. doi: 10.1038/mt.2009.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LN, Karp P, Gerard CJ, Pastor E, Laux D, Munson K, et al. Dual therapeutic utility of proteasome modulating agents for pharmaco-gene therapy of the cystic fibrosis airway. Mol Ther. 2004;10:990–1002. doi: 10.1016/j.ymthe.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Zhang L, Wang D, Fischer H, Fan PD, Widdicombe JH, Kan YW, et al. Efficient expression of CFTR function with adeno-associated virus vectors that carry shortened CFTR genes. Proc Natl Acad Sci USA. 1998;95:10158–10163. doi: 10.1073/pnas.95.17.10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapranov P, Chen L, Dederich D, Dong B, He J, Steinmann KE, et al. Native molecular state of adeno-associated viral vectors revealed by single-molecule sequencing. Hum Gene Ther. 2012;23:46–55. doi: 10.1089/hum.2011.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allander T, Tammi MT, Eriksson M, Bjerkner A, Tiveljung-Lindell A, Andersson B. Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proc Natl Acad Sci USA. 2005;102:12891–12896. doi: 10.1073/pnas.0504666102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allander T, Jartti T, Gupta S, Niesters HG, Lehtinen P, Osterback R, et al. Human bocavirus and acute wheezing in children. Clin Infect Dis. 2007;44:904–910. doi: 10.1086/512196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Gu X, Zhao X, Luo J, Luo Z, Wang L, et al. High viral load of human bocavirus correlates with duration of wheezing in children with severe lower respiratory tract infection. PLoS ONE. 2012;7:e34353. doi: 10.1371/journal.pone.0034353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streiter M, Malecki M, Prokop A, Schildgen V, Lüsebrink J, Guggemos A, et al. Does human bocavirus infection depend on helper viruses? A challenging case report. Virol J. 2011;8:417. doi: 10.1186/1743-422X-8-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Deng X, Yan Z, Cheng F, Luo Y, Shen W, et al. Establishment of a reverse genetics system for studying human bocavirus in human airway epithelia. PLoS Pathog. 2012;8:e1002899. doi: 10.1371/journal.ppat.1002899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Yan Z, Luo Y, Xu J, Cheng F, Li Y, et al. In vitro modeling of human bocavirus 1 infection of polarized primary human airway epithelia. J Virol. 2013;87:4097–4102. doi: 10.1128/JVI.03132-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tijssen P, Agbandje-McKenna M, Almendral JM, Bergoin M, Flegel TW, Hedman K, et al. 2011Virus taxonomy: classification and nomenclature of viruses: Ninth Report of the International Committee on Taxonomy of Viruses. King M, Adams MJ, Carstens E, Lefkowitz EJ.eds). Parvoviridae Elsevier: San Diego, CA; 405–425. [Google Scholar]
- Schildgen O, Qiu J, Söderlund-Venermo M. Genomic features of the human bocaviruses. Future Virol. 2012;7:31–39. doi: 10.2217/fvl.11.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AY, Cheng F, Lou S, Luo Y, Liu Z, Delwart E, et al. Characterization of the gene expression profile of human bocavirus. Virology. 2010;403:145–154. doi: 10.1016/j.virol.2010.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurda BL, Parent KN, Bladek H, Sinkovits RS, DiMattia MA, Rence C, et al. Human bocavirus capsid structure: insights into the structural repertoire of the parvoviridae. J Virol. 2010;84:5880–5889. doi: 10.1128/JVI.02719-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnazhagan S, Weigel KA, Raikwar SP, Mukherjee P, Yoder MC, Srivastava A. Recombinant human parvovirus B19 vectors: erythroid cell-specific delivery and expression of transduced genes. J Virol. 1998;72:5224–5230. doi: 10.1128/jvi.72.6.5224-5230.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabner J, Karp P, Seiler M, Phillips SL, Mitchell CJ, Saavedra M, et al. Development of cystic fibrosis and noncystic fibrosis airway cell lines. Am J Physiol Lung Cell Mol Physiol. 2003;284:L844–L854. doi: 10.1152/ajplung.00355.2002. [DOI] [PubMed] [Google Scholar]
- Cheng F, Chen AY, Best SM, Bloom ME, Pintel D, Qiu J. The capsid proteins of Aleutian mink disease virus activate caspases and are specifically cleaved during infection. J Virol. 2010;84:2687–2696. doi: 10.1128/JVI.01917-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Deng X, Best SM, Bloom ME, Li Y, Qiu J. Internal polyadenylation of parvoviral precursor mRNA limits progeny virus production. Virology. 2012;426:167–177. doi: 10.1016/j.virol.2012.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotmore S, Tattersall P.1996Parvovirus DNA replication. DePamphilis M.ed). DNA replication in eukaryotic cells. Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY; 799–813. [Google Scholar]
- Dong JY, Fan PD, Frizzell RA. Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Hum Gene Ther. 1996;7:2101–2112. doi: 10.1089/hum.1996.7.17-2101. [DOI] [PubMed] [Google Scholar]
- Wu Z, Yang H, Colosi P. Effect of genome size on AAV vector packaging. Mol Ther. 2010;18:80–86. doi: 10.1038/mt.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DP, Biagini P, Lefeuvre P, Golden M, Roumagnac P, Varsani A. Recombination in eukaryotic single stranded DNA viruses. Viruses. 2011;3:1699–1738. doi: 10.3390/v3091699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A, Simmonds P, Slikas E, Li L, Bodhidatta L, Sethabutr O, et al. Human bocaviruses are highly diverse, dispersed, recombination prone, and prevalent in enteric infections. J Infect Dis. 2010;201:1633–1643. doi: 10.1086/652416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Zak R, Zhang Y, Ding W, Godwin S, Munson K, et al. Distinct classes of proteasome-modulating agents cooperatively augment recombinant adeno-associated virus type 2 and type 5-mediated transduction from the apical surfaces of human airway epithelia. J Virol. 2004;78:2863–2874. doi: 10.1128/JVI.78.6.2863-2874.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan D, Yue Y, Yan Z, McCray PB, Jr, Engelhardt JF. Polarity influences the efficiency of recombinant adenoassociated virus infection in differentiated airway epithelia. Hum Gene Ther. 1998;9:2761–2776. doi: 10.1089/hum.1998.9.18-2761. [DOI] [PubMed] [Google Scholar]
- Fisher JT, Liu X, Yan Z, Luo M, Zhang Y, Zhou W, et al. Comparative processing and function of human and ferret cystic fibrosis transmembrane conductance regulator. J Biol Chem. 2012;287:21673–21685. doi: 10.1074/jbc.M111.336537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schildgen O, Müller A, Allander T, Mackay IM, Völz S, Kupfer B, et al. Human bocavirus: passenger or pathogen in acute respiratory tract infections. Clin Microbiol Rev. 2008;21:291–304, table of contents. doi: 10.1128/CMR.00030-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantola K, Hedman L, Arthur J, Alibeto A, Delwart E, Jartti T, et al. Seroepidemiology of human bocaviruses 1-4. J Infect Dis. 2011;204:1403–1412. doi: 10.1093/infdis/jir525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meriluoto M, Hedman L, Tanner L, Simell V, Mäkinen M, Simell S, et al. Association of human bocavirus 1 infection with respiratory disease in childhood follow-up study, Finland. Emerging Infect Dis. 2012;18:264–271. doi: 10.3201/eid1802.111293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Körner RW, Söderlund-Venermo M, van Koningsbruggen-Rietschel S, Kaiser R, Malecki M, Schildgen O. Severe human bocavirus infection, Germany. Emerging Infect Dis. 2011;17:2303–2305. doi: 10.3201/eid1712.110574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin ET, Fairchok MP, Kuypers J, Magaret A, Zerr DM, Wald A, et al. Frequent and prolonged shedding of bocavirus in young children attending daycare. J Infect Dis. 2010;201:1625–1632. doi: 10.1086/652405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan D, Yan Z, Yue Y, Ding W, Engelhardt JF. Enhancement of muscle gene delivery with pseudotyped adeno-associated virus type 5 correlates with myoblast differentiation. J Virol. 2001;75:7662–7671. doi: 10.1128/JVI.75.16.7662-7671.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp PH, Moninger TO, Weber SP, Nesselhauf TS, Launspach JL, Zabner J, et al. An in vitro model of differentiated human airway epithelia. Methods for establishing primary cultures. Methods Mol Biol. 2002;188:115–137. doi: 10.1385/1-59259-185-X:115. [DOI] [PubMed] [Google Scholar]
- Chen AY, Kleiboeker S, Qiu J. Productive parvovirus B19 infection of primary human erythroid progenitor cells at hypoxia is regulated by STAT5A and MEK signaling but not HIFa. PLoS Pathog. 2011;7:e1002088. doi: 10.1371/journal.ppat.1002088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Luo M, Zhang L, Ding W, Yan Z, Engelhardt JF. Bioelectric properties of chloride channels in human, pig, ferret, and mouse airway epithelia. Am J Respir Cell Mol Biol. 2007;36:313–323. doi: 10.1165/rcmb.2006-0286OC. [DOI] [PMC free article] [PubMed] [Google Scholar]