

Abstract
Gene therapy provides a significant opportunity to treat a variety of inherited and acquired diseases. However, adverse immune responses toward the adeno-associated virus (AAV) antigens may limit its success. The mechanisms responsible for immunity or tolerance toward AAV-encoded transgene products remain poorly defined. Studies in mice demonstrate that AAV2/8 gene transfer to liver is associated with immunological hyporesponsiveness toward both AAV vector and antigenic transgene product. To evaluate the role of activation-induced cell death (AICD) and cytokine withdrawal (intrinsic cell death) in the deletion of mature T lymphocytes, we compared immunological responses in hepatic AAV2/8 transfer in murine recipients lacking the Fas receptor, and recipients overexpressing Bcl-xL, to WT murine counterparts. Prolonged transgene expression was dependent on both Fas signaling and Bcl-xL–regulated apoptosis in T cells. Abrogation of intrinsic cell death enhanced Th1 responses, whereas AICD functioned to limit neutralizing antibody production toward AAV2/8. In addition, immune hyporesponsiveness and stable transgene expression was dependent on upregulation of FasL expression on transduced hepatocytes and a corresponding apoptosis of infiltrating Fas (+) cells. These data provide evidence that both AICD and apoptosis due to cytokine withdrawal of lymphocytes are essential for immune hyporesponsiveness toward hepatic AAV2/8-encoded transgene product in the setting of liver gene transfer.
Introduction
Gene therapy holds great promise for the treatment of genetic diseases. Viral gene delivery vehicles are among the most effective systems at transferring and achieving therapeutic levels of the transgene products in gene therapy applications.1 However, an immune response toward the viral gene delivery vehicle or the therapeutic transgene product can compromise the success of long-term gene replacement therapy and potentially precipitate a robust inflammatory response that can be pathogenic for the recipient.2 Viral gene delivery vehicles derived from adeno-associated viruses (AAVs) are leading candidates for clinical gene therapy applications because they fulfill much of the criteria listed above, are non-pathogenic, non-toxic, and replication deficient.1,3
Numerous studies in mice demonstrate that gene transfer to the liver using the AAV serotype, AAV2/8, is associated with immunological hyporesponsiveness toward both the AAV vector and antigenic transgene products.4,5 A variety of mechanisms have been proposed to explain host nonresponsiveness, including antigen-specific regulatory T-cell induction,4,6,7,8 tolerogenic Kupffer cells with immunosuppressive properties,7 impaired T-cell activation and anergy,9 failure of AAV2/8 to transduce dendritic cells,10 and reduced sensitivity of the liver toward T-cell–mediated effects.11 However, apoptosis of mature T lymphocytes is integral for regulating the induction of immune hyporesponsiveness following stimulation with self and foreign antigens12,13 and investigation of this mechanism as a mediator of immune regulation following hepatic AAV2/8 gene transfer remains to be studied.
T-cell apoptosis occurs through two primary pathways: apoptosis due to cytokine withdrawal (intrinsic cell death) and antigen driven death (activation-induced cell death, AICD).14 Intrinsic cell death occurs through growth factor (i.e., IL-2) withdrawal and is regulated by the Bcl-2 protein family, which consists of both pro-apoptotic members (BIM, Bax, Bak, Bid, Bad, Noxa, and Puma) and anti-apoptotic members (Bcl-2, Bcl-xL, Mcl-1, and A1). BIM has been studied recently as the counterpart of BcL-xL.15,16,17 Following a death signal, such as cytokine withdrawal, BIM activates the pro-apoptotic factors, Bax and Bak, which destablize the mitochondrial outer membrane to induce cell death.15 BcL-xL, in turn, binds BIM to block association of the protein with the pro-apoptotic factors. Bcl-xL overexpression protects T cells from apoptosis following cytokine withdrawal compared with wild-type (WT) cells. Expression of Fas on T cells and ligation with FasL on a target cell induces AICD.18 T-cell receptor activation and IL-2 stimulation induces resistance to Fas ligand–mediated apoptosis. However, upon reactivation and in the presence of the death cytokine, T cells can undergo apoptosis in a Fas and FasL-dependent manner.12
This study evaluated the role of AICD and intrinsic cell death in the deletion of mature T lymphocytes that could respond to vector and antigenic transgene product. We compared immunological responses in hepatic AAV2/8 gene transfer recipients lacking the functional death cytokine receptor, Fas,19 and recipients overexpressing the anti-apoptotic factor, Bcl-xL under a T-cell–specific promoter,20 with WT counterparts. We hypothesized that the blockade of these two pathways that regulate lymphocyte apoptosis would result in the accumulation of transgene-reactive cytotoxic T cells corresponding with an elimination of AAV2/8-transduced hepatocytes. We demonstrate that both AICD and intrinsic cell death of lymphocytes are critical for immune hyporesponsiveness toward hepatic AAV2/8-vectored transgene product and abrogation of these apoptotic pathways results in a non-cytolytic mechanism of transgene extinguishment.
Results
Effect of AICD and intrinsic cell death blockade on stability of transgene expression in mouse liver tissue following AAV2/8 transduction
Numerous studies in mice demonstrate that AAV2/8 gene transfer to the livers of WT C57BL/6 mice results in long-term transgene expression with minimal immune responses toward both the vector and transgene antigen.4,5,7,11,21 To evaluate the role of AICD and intrinsic cell death in deletion of mature T lymphocytes that could potentially respond to antigenic transgene product and reduce transgene expression, mice lacking functional Fas receptor19 and mice that express Bcl-xL constitutively20 were intravenously (i.v.) injected with 1E11 viral particles of AAV2/8 expressing a nuclear β-gal (nLacZ) reporter gene from the chicken β-actin (β-actin) promoter. Livers were recovered from WT and the congenic Faslpr/lpr and Bcl-xL+ mice on days 7, 14, 21, 28, and 35 after transduction and transgene expression in mouse liver tissue was assessed by X-gal histochemical stain and quantified by morphometric analysis (Figure 1). Bcl-xL–mediated death repression and the absence of the death cytokine receptor, Fas, resulted in a significant reduction in transgene expression compared with WT controls (P ≤ 0.05) by day 35 after transduction (Figure 1a,b). These data suggest a connection between increased T-cell apoptosis through both AICD and intrinsic cell death and long-term AAV2/8-vectored transgene product expression following hepatic gene transfer.
Figure 1.

Stability of transgene expression in mouse liver tissue. (a) X-gal histochemical stain of liver sections from WT, Faslpr/lpr, and Bcl-xL+ mice injected i.v. with 1E11 GC of AAV2/8CBnLacZ. Representative sections from three mice per group. Scale bar = 200 μm. (b) Quantification of nLacZ staining in the liver was performed using Image J software for day 35 after vector administration for WT, Faslpr/lpr, and Bcl-xL+ mice. Results represent the percent area fraction ± SEM of X-gal (+) stain for n = 3 WT, Faslpr/lpr, and Bcl-xL+ mice.
Progressive transgene extinguishment in the livers of mutant mice is due to a non-cytolytic effect
Prior studies examining the mechanism by which transgene levels decline following AAV hepatic gene transfer has revealed that transgene extinguishment may be due to either a cytotoxic T lymphocyte (CTL) response that results in the killing of transduced hepatocytes22,23 or to a non-cytolytic mechanism currently unidentified that acts to silence the vector genome.24 To investigate whether the reduction of transgene expression in the livers of Fas-deficient and Bcl-xL-transgenic mice may be due a CTL-mediated killing of hepatocytes, AAV2/8-nLacZ vector genomes were quantified by real-time TaqMan PCR from WT, Faslpr/lpr, and Bcl-xL+ mice on days 7, 14, 21, 28, and 35 after AAV2/8 transduction (Figure 2). No reduction in AAV2/8-nLacZ vector genome levels or persistence was observed in mutant mice compared with WT controls at any time points. These results suggest that loss of transgene expression observed following AICD and intrinsic cell death blockade is attributable to a non-cytolytic effect and not associated with diminished viral genome copy number due to CTL-mediated hepatocyte destruction.
Figure 2.

Vector genome analysis in mouse liver tissue. Vector GC in the liver were quantified by real-time TaqMan PCR from WT, Faslpr/lpr, and Bcl-xL+ mice injected i.v. with 1E11 GC of AAV2/8CBnLacZ. Results represent the mean GC/µg of DNA ± SEM from at least n = 3 recipients per group.
Effect of Fas deficiency and Bcl-xL–mediated death repression on Th1 responses toward AAV2/8-vectored transgene antigen
AAV2/8 gene transfer to the liver in WT C57BL/6 mice is associated with immune hyporesponsiveness toward both the AAV vector and the antigenic transgene products.4,11,21 To determine whether AICD or intrinsic cell death blockade affects T-cell cytokine phenotype in response to AAV2/8-vectored transgene antigen, we used ELISPOT to quantify primed nLacZ responsive IFNγ-producing cells at previously described time points (Figure 3). Splenocytes were stimulated ex vivo with either the nLacZ-specific CD8+ immunodominant peptide (Figure 3a) or the nLacZ-specific peptide library (Figure 3b). As observed previously,7 AAV2/8-nLacZ induces minimal Th1 responses toward the nLacZ-specific CD8+ immunodominant peptide in WT mice following gene transfer to the liver, although a slightly enhanced response is observed following stimulation with the entire nLacZ-specific peptide pool (Figure 3b). Fas-deficient mice exhibit similar immunoreactivity toward both the immunodominant nLacZ epitope (Figure 3a) and peptide library (Figure 3b) as WT controls. In contrast, Bcl-xL transgenic mice demonstrate enhanced Th1 responses toward the nLacZ-specific CD8+ immunodominant peptide and nLacZ-specific peptide library peaking at days 21 and 28 after AAV2/8 gene transfer. Thus, abrogation of intrinsic cell death enhances Th1 responses toward transgene antigen.
Figure 3.
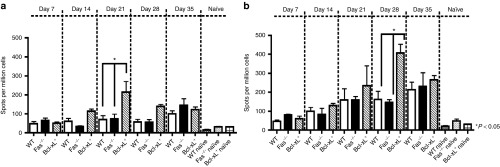
Kinetics of Th1 responses toward nLacZ antigen in WT, Faslpr/lpr, and Bcl-xL+ mice following transduction with AAV2/8CBnLacZ. Splenocytes were harvested and processed for ELISPOT assays to quantify primed (a) CD8+ nLacZ T-cell immunodominant and (b) nLacZ library peptide responsive IFNγ-producing cells. Results represent the mean ± SEM of cytokine-producing cells from at least n = 3 recipients per group.
Elevated AAV2/8 neutralizing antibody levels in Fas-deficient mice
A major limitation to AAV vector readministration is the occurrence of neutralizing antibodies following primary AAV injection.25 To determine the effect that Fas-mediated cell death and Bcl-xL+–regulated apoptosis has on the prevalence of neutralizing antibody toward AAV2/8, a neutralizing antibody (Nab) assay–based in vitro transduction inhibition was performed (Figure 4). The frequency of NAb levels toward AAV2/8 was highest for Fas-deficient mice beginning on day 21 after injection, whereas NAb levels were low and comparable between WT and Bcl-xL transgenic mice. These data suggest that AICD, but not intrinsic cell death, functions to limit neutralizing Ab production toward AAV2/8 following i.v. injection.
Figure 4.

Characterization of B-cell responses toward AAV2/8 in WT, Faslpr/lpr, and Bcl-xL+ mice. Neutralization to AAV2/8 in Huh7 cells. Values are serum dilution at which relative luminescence units was reduced 50% compared with virus control well (no test sample). Limit of detection 1/5 dilution. Results represent the mean serum dilution ± SEM from at least n = 3 recipients per group.
Diminished transgene expression corresponds with a heavy CD4+ and CD8+ T-cell infiltrate within the liver
To examine the degree and character of cellular infiltrate within the livers of mice containing dysregulated Bcl-xL or Fas pathways, liver tissue was recovered from WT, Faslpr/lpr, and Bcl-xL+ mice following AAV2/8 i.v. injection at time points, costained with CD4 and CD8 Abs, and examined by fluorescent microscopy (Figure 5). WT, Faslpr/lpr, and Bcl-xL+ mice exhibited similar levels of CD4+ T-cell infiltrate at day 21 after transduction (Figure 5a). At day 35 after AAV gene transfer, however, a significant CD4+ and CD8+ cellular infiltrate resembling neolymphoid aggregates was observed in the mutant mice but not in the WT counterparts (Figure 5a–d). The livers of naïve mutant age-matched control mice were also examined by H&E stain to insure that cellular infiltration observed in mutant mice was dependent on AAV gene transfer (data not shown). No infiltrating cells were detected in naïve controls ruling out non-specific cellular infiltration due to Fas deficiency and overexpression of Bcl-xL+. In addition, we performed flow cytometric analysis of splenocytes from BcL-xL+ and WT mice on day 35 after vector administration. A twofold increase in the percentage of CD4+ and CD8+ T cells in the spleens of Bcl-xL+ mice compared with WT were observed indicating that intrinsic apoptosis regulates T-cell populations in the peripheral lymphoid organ following AAV8 gene transfer (data not shown). These data collectively suggest that at least two mechanisms of T-cell deletion—AICD and intrinsic cell death—limit the accumulation of transgene and/or vector responsive T cells in the liver following AAV gene transfer. These T cells are probably specific to AAV capsid or transgene product because they accumulated subsequent to vector administration and were not observed in naïve age-matched congenic mice.
Figure 5.
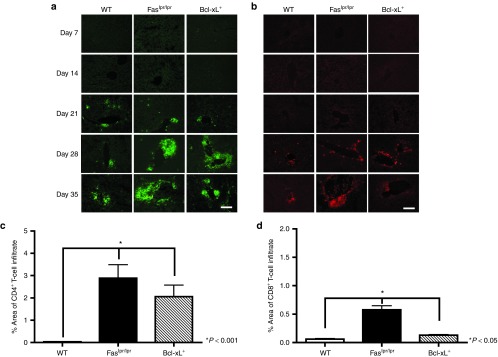
Characterization of cellular infiltrate at kinetic time points following i.v. injection of 1 × 1011 GC AAV2/8-expressing nLacZ. Liver tissue was harvested from WT, Faslpr/lpr, and Bcl-xL+ mice. Liver sections were stained with (a) anti-CD4 (green) and (b) anti-CD8 (red) Ab and examined by fluorescent microscopy. Representative sections from three mice per group are shown. Scale bar = 200 μm. Quantification of (c) CD4+ T-cell or (d) CD8+ T-cell infiltrate in the liver was performed using Image J software for day 35 after vector administration for WT, Faslpr/lpr, and Bcl-xL+ mice. Results represent the percent area fraction ± SEM of CD4+ and CD8+ stain for n = 3 WT, Faslpr/lpr, and Bcl-xL+ mice.
Enhancement of Fas/FasL expression in the mouse liver following AAV2/8 transduction
Fas-FasL interactions may play a pivotal role in peripheral T-cell deletion under certain immunological conditions.26 Although FasL is commonly detected on functional activated T cells, it can also be induced in several non-lymphoid tissues, such as the liver. To further characterize the role of Fas signaling in controlling the accumulation of CD4+ and CD8+ T cells in the liver following AAV2/8 gene transfer, liver tissue recovered from WT mice or WT age-matched naïve controls was costained with Fas-FITC and FasL-TRITC Ab at day 35 after injection and examined by fluorescent microscopy (Figure 6). A significant upregulation of FasL on hepatocytes was observed in the livers of WT mice transduced with AAV2/8-nLacZ but was not observed in naïve controls. Furthermore, a large population of infiltrating lymphocytes expressing Fas were detected in WT livers challenged with AAV2/8 compared with naïve controls. These data suggest that in WT mice receiving AAV2/8 hepatic gene transfer, vector and/or transgene-reactive lymphocytes expressing Fas infiltrate the liver, interact with FasL-expressing hepatocytes, and undergo AICD.
Figure 6.

Fas/FasL expression in the liver following AAV2/8CBnLacZ transduction. Liver tissue was harvested from WT naïve or WT mice injected i.v. with 1E11 GC of AAV2/8CBnLacZ. Liver sections were stained with Fas-FITC (green) and FasL-TRITC (red) Ab and examined by fluorescent microscopy. Representative sections from three mice per group imaged at 10X at day 35 after vector administration mice. Scale bar = 200 μm.
Apoptosis regulates the accumulation of T cells following AAV2/8 gene transfer and is dependent on AICD and intrinsic apoptosis
To investigate the relationship between AICD and intrinsic apoptosis on T-cell apoptosis, liver sections recovered from WT, Faslpr/lpr, and Bcl-xL+ mice at time points following AAV2/8-nLacZ injection were TUNEL stained and percent area of apoptotic cells were quantified (Figure 7a). Apoptotic lymphocytes were detected in the livers of WT mice challenged with AAV2/8 as early as day 7 after injection. On day 35 after AAV gene transfer, a significant enhancement of apoptotic cells were observed in the WT livers compared with the livers recovered from the mutant mice (Figure 7b). Collectively, these findings indicate that Fas-mediated apoptosis and intrinsic cell death regulates the accumulation of responding T cells within the liver following AAV2/8 gene transfer.27,28
Figure 7.

Detection of apoptotic lymphocytes in AAV2/8CBnLacZ-transduced liver. Liver tissue was harvested from WT, Faslpr/lpr, and Bcl-xL+ AAV8 gene–transferred mice at kinetic time points following AAV2/8CBnLacZ injection. (a) Sections were TUNEL stained and examined by fluorescent microscopy. Positive control shows Dnase-treated section, negative control Dnase-treated section without terminal deoxynucleotidyl transferase. Representative sections from three mice per group are shown. Scale bar = 200 μm. (b) Quantification of TUNEL stain in the liver was performed using Image J software for day 35 after vector administration for WT, Faslpr/lpr, and Bcl-xL+ mice. Results represent the percent area fraction ± SEM of TUNEL stain for n = 3 WT, Faslpr/lpr, and Bcl-xL+ mice.
Discussion
Apoptosis of lymphocytes serves to modulate T- and B-cell survival and inhibit lymphocyte immune reactivity during the course of an immune response.12 Here, we address the role of AICD and intrinsic cell death in the induction of in vivo immune hyporesponsiveness toward both vector and transgene antigen following AAV2/8 hepatic gene transfer. Our data suggest that both intrinsic and extrinsic (AICD) mechanisms of apoptosis work together to maintain long-term gene replacement following AAV2/8 hepatic gene transfer, and that repression of either of these pathways abrogates immune hyporesponsiveness toward both vector and transgene antigen and results in a significant reduction in transgene expression.
The role of Fas and Bcl-xL proteins in shaping the repertoire of lymphocytes and regulating the size and effector function of the mature lymphocyte pool has been investigated in a variety of systems.13,27,28,29 Both extrinsic and intrinsic mechanisms of apoptosis serve to control T-cell survival and dysregulation of these pathways can precipitate the induction of systemic autoimmunity,13,30 transplant rejection,31 lymphoma,30 and defective immune responses toward a number of pathogens.32 In our murine model of AAV2/8 hepatic transfer, regulated cell death through both AICD and intrinsic cell death was found to be indispensable for the maintenance of long-term gene replacement following AAV2/8 challenge. In the absence of Fas signaling and Bcl-xL–regulated apoptosis, T cells differentiate into effector cells, secrete cytokines and, in the case of Fas deficiency, provide help to B cells following AAV2/8 hepatic gene transfer. Although splenocytes isolated from WT and Faslpr/lpr mice challenged with AAV2/8 do exhibit some IFNγ secretion in response to stimulation with β-gal antigen, only Bcl-xL transgenic mice demonstrate enhanced Th1 responses. Thus, in the absence of intrinsic cell death, an increase in the survival of transgene-reactive T cells results in enhanced capacity to produce inflammatory cytokines. Similar findings have been reported in the role of intrinsic cell death in an experimental model of chronic autoimmune encephalomyelitis.27 This suggests that although both intrinsic cell death and AICD serve to limit proliferation and survival of responding T cells, only the repression of intrinsic cell death enhances differentiation of the responding T cells into fully functional effector cells. These data demonstrate that hepatic AAV2/8 gene transfer can generate activated effector T cells capable of cytokine secretion, but that T-cell apoptotic mechanisms are in place to control the accumulation and pathogenesis of these cells in the periphery leading to immune hyporesponsiveness in WT mice.
Crispe and colleagues have amassed an impressive amount of data that suggests the liver acts as a disposal site for T cells activated in peripheral lymphoid tissues.26,33 Data accumulated over the past decade supports a paradigm in which the liver eliminates the infiltrating cells through a Fas-based mechanism of apoptosis.26,33,34,35,3636 This mechanism of peripheral deletion occurs as a consequence of an immune response and under certain immunological conditions permits the liver to monitor the degree of local immune responses and express FasL to kill Fas-expressing infiltrating T cells when a critical threshold is achieved.34 In support of this mechanism, the absence of AICD results in the loss of transgene expression following AAV2/8 challenge and corresponds with a progressive accumulation of CD4+ and CD8+ cellular infiltrate within the liver resembling neolymphoid aggregates. We reasoned that Fas-mediated apoptosis of transgene and/or vector responsive T cells control the accumulation of T cells in the liver following AAV2/8 challenge and sought to further characterize the role of Fas signaling by staining for Fas/FasL expression in the livers of WT mice transduced with AAV2/8CBnLacZ. A robust upregulation of FasL was observed in the livers of WT mice transduced with AAV2/8 and corresponded with equally robust Fas expression on infiltrating T cells. The detection of apoptotic lymphocytes in the livers of WT mice challenged with AAV2/8 suggest that Fas-mediated apoptosis regulates the accumulation of transgene-reactive T cells within the periphery following AAV2/8 gene transfer.
On the basis of these observations, we propose a model representing the mechanisms by which apoptosis regulates the accumulation of effector T cells following AAV2/8 hepatic gene transfer (Figure 8). Transgene-reactive T cells in the secondary lymphoid organs normally undergo intrinsic cell death in response to growth factor deprivation in WT mice. On the basis of our findings, we hypothesize that in the absence of this form of regulated cell death, transgene-reactive T cell amplify in the lymph nodes and spleen and migrate to the liver. Accumulation of these effector T cells induces a non-cytolytic suppression of transgene expression. In parallel, during a normal immune response toward AAV2/8-vectored transgene product, T cells that escape intrinsic cell death, upregulate the death receptor, Fas, in response to T-cell stimulation with transgene antigen and subsequently migrate to the liver, where they undergo AICD in response to antigen presenting FasL (+) hepatocytes. In the absence of Fas signaling or in the case of overexpression of BcL-xL, transgene reactive T cells infiltrate and accumulate in the liver and extinguish vector transgene expression through a non-cytolytic mechanism.
Figure 8.

Proposed model for two mechanisms of T-cell deletion—intrinsic cell death and AICD—in limiting the accumulation of transgene responsive T cells in the liver following AAV2/8 gene transfer. In the secondary lymphoid organs, AAV2/8 transgene-reactive T cells undergo intrinsic cell death in response to IL-2 deprivation. T cells that escape this pathway upregulate Fas and infiltrate the liver. (a) In response to FasL-expressing AAV2/8-transduced hepatocytes and reactivation with transgene antigen, the T cells undergo AICD. (b) In the absence of intrinsic cell death and AICD, T cells are amplified, accumulate in the liver, and induce a non-cytolytic suppression of vector transgene expression.
These pathways likely cooperate to maintain immune hyporesponsiveness toward AAV associated antigens. Evidence for this is in data acquired from BIM/Fas double knockout mice.13,16,17 BcL-xL sequesters BIM to prevent binding with BAX and BAK which destablize the mitochondrial outer membrane, thereby inhibiting apoptosis. BIM deletion equally prevents death of activated T cells. Deficiency of both Fas and BIM results in an accumulation of enhanced memory T-cells populations in the periphery following viral infection, as well as an increase in the acceleration of autoimmunity.13,16,17 Deletion of both factors has a synergistic effect as the T-cell population for the double knockout mice was four- to fivefold bigger compared with the Fas-deficient mice.17 Through a cooperative effort it is possible that intrinsic and extrinsic cell death following AAV2/8 administration are important at different phases of gene transfer. For example, intrinsic cell death may predominate shortly after gene transfer, whereas both intrinsic and extrinsic apoptosis may be at play during the chronic phase of the immune response toward the vector. Lymphocytic choriomeningitis virus infection in double knockout mice supports this hypothesis as T-cell contraction following acute lymphocytic choriomeningitis virus infection required activation of both BIM and Fas and the early contraction phase required only BIM.16
Classic immunological experiments have provided strong evidence that cells that harbor intracellular viruses are destroyed and cleared by major histocompatibility complex class I–restricted CD8+ cytotoxic T cells (CTL).37 Adenovirus vectors, for instance, induce an innate inflammatory response and an adaptive immune response that results in the generation of adenovirus-specific cytotoxic CD8+ T cells and destruction of transduced cells.38,39 However, substantial data also suggests an alternative mechanism by which certain viruses are subject to control by the non-cytolytic action of inflammatory cytokines such as IFNγ, TNFα, and IL-12.40,41 This mechanism was best demonstrated in a transgenic mouse model of hepatitis B virus infection and found to be dependent on both transcriptional and posttranslational regulation.42 Consistent with a non-cytolytic mechanism of vector expression shutdown, loss of nLacZ transgene expression in mice with dysregulated Bcl-xL or Fas pathways following AAV2/8 gene transfer is not associated with diminished viral genome copy number.
The suppression of viral gene expression through the cytokine-mediated activation of intracellular pathways that block the viral life cycle but spare the infected cells is hypothesized to be the principal mechanism of viral clearance following a massive virus infection of the liver.40,41 In this scenario, the virally infected hepatocytes greatly outnumber the responding CTL, leading to an insurmountable challenge for cytolytic clearance. The control of viral replication and expression by non-cytolytic cytokine-mediated intracellular mechanisms also spares the extermination of massive numbers of hepatocytes, the death of which could lead to a severe, fatal hepatitis.41 AAV2/8 hepatic gene transfer yields remarkably efficient transduction in mice with >96% of hepatocytes exhibiting transgene expression by day 28 after vector administration (data not shown).43,44 Reduction in hepatic transgene expression in the absence of diminished genome copies following AAV2/8 gene transfer in Fas-deficient and Bcl-xL–overexpressing mice strongly suggests a non-cytolytic mechanism of control that may be dependent on the efficiency of AAV2/8 hepatocyte transduction. Additional investigation into this phenomenon is necessary for the development of AAV vectors that are capable of efficient transgene expression and resistant to non-cytolytic host immune responses that threaten to undermine gene therapy success.
In summary, our results demonstrate that two pathways of apoptosis—AICD and intrinsic cell death—eliminate transgene-reactive T cells and are critical for the induction of immune hyporesponsiveness toward AAV2/8-vectored transgene product in the liver. Furthermore, our findings reveal the requirement for apoptotic cell death of functional T cells within the target organ mediated by the Fas-FasL pathway following hepatic gene transfer of AAV2/8. Thus, a hierarchy of immunomodulatory mechanisms are at work to establish immune hyporesponsiveness toward AAV2/8-vectored transgene product following hepatic gene transfer, including antigen-specific T regulatory cell induction,4,6,7,8 tolerogenic Kupffer cells,7 and impaired T-cell activation and anergy,9 with effector T-cell deletion through AICD and intrinsic cell death at the nexus. These findings suggest that both AICD and intrinsic cell death are essential for immune hyporesponsiveness and provide insight into the ability of target organs to modulate a local immunosuppressive response through T-cell deletion rather than passively provoking an immune response. The impact of T-cell apoptosis in response to AAV2/8 hepatic gene transfer may provide a therapeutic target for enhancing immune hyporesponsiveness following AAV gene transfer. In addition, these data identify the requirement for modified AAV vectors that are resistant to non-cytolytic mechanisms of transgene suppression and, consequently, have significant implications in the clinical application of AAV.
Materials and Methods
Mice. C57BL/6 WT and B6.MRL-Faslpr/J (Faslpr/lpr)19 were ordered from the Jackson ImmunoResearch Laboratory (West Grove, PA). Mice that overexpress human Bcl-xL under the T-cell–specific lck promoter (lckpr-Bcl-xL+) backcrossed to C57BL/6 mice and referred to as Bcl-xL+ mice20 were a kind gift from Craig B Thompson and Tullia Lindstein (Memorial Sloan-Kettering Cancer Center, New York, NY). The genotyping of transgene expressing Bcl-xL mice was conducted using a HuBcl2l1 Tg probe and performed by Transnetyx (www.transnetyx.com). Both strains of mice were backcrossed to C57BL/6 mice for 12 generations to eliminate artifactual effects due to strain differences. To avoid potential complications of age-related autoimmunity, mice used in the experiments were 6 weeks of age and experiments were terminated no later than 35 days after transduction. The livers of naïve mutant age-matched control mice were examined by H&E stain to insure that cellular infiltration observed in mutant mice was dependent on AAV gene transfer: No infiltrating cells were detected in naïve controls. To confirm at 35 days after vector administration that an increased population of CD4+ and CD8+ T cells was not present in Faslpr/lpr mice (indicating lymphoproliferation unrelated to AAV vector transfer), flow cytometric analysis was performed and Faslpr/lpr mice had similar populations of T cells to WT mice (data not shown). All mice were housed under specific pathogen-free conditions in the TRL Animal Facility at the University of Pennsylvania. All animal procedure protocols were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
AAV vectors–mediated transduction of liver. An AAV8-pseudotyped vector flanked with AAV2 ITRs encoded a nuclear targeted form of β-gal (nLacZ) under the transcriptional control of a CMV-enhanced chicken β-actin promoter. AAV vectors were produced by a scaled-down version of a previously described method45 by triple transfection of vector genome, AAV helper and adenovirus helper plasmids. Purification of vectors involved a single iodixanol step gradient and subsequent DNAse treatment. Real-time PCR using a primer/probe set corresponding to the polyA region of the vector and linearized plasmid standards determined genome titer (genome copy/ml) of AAV vectors. All vectors used in this study passed the endotoxin assay using QCL-1000 chromogenic LAL test kit (Cambrex Bio Science, East Rutherford, NJ). Vectors were produced by Penn Vector Core at the University of Pennsylvania. Mice were injected via the tail vein with 1011 vector genomes of AAV.
Vector genome analysis. At kinetic time points, mice were euthanized and total cellular DNA was extracted from the liver using a QiAamp DNA Mini Kit according to manufacturer's instructions (Qiagen, Venlo, Netherlands). Extraction of target nucleic acid from livers was followed by nucleic acid amplification in duplicate TaqMan PCR reactions using a primer/probe set corresponding to the polyA region of the vector. The mean quantity of target detected for each sample is indicated as genome copies/µg (GC/µg).
Immunohistochemistry. To examine expression of nuclear β-gal, X-gal staining of snap-frozen liver cryosections was performed according to standard protocols.46 Representative sections from three mice per group were imaged by brightfield microscopy with a 10X objective. To analyze the infiltrating cell types within the liver, immunostaining and fluorescent microsopy were performed on acetone-fixed cryosections stained with rat antibodies from BD Pharmingen (Franklin Lakes, NJ) acetone-fixed cryosections stained with rat antibodies from BD Pharmingen.
For Fas/FasL stain, frozen sections were fixed in 4% paraformaldehyde permeabilized with Triton and incubated with a rabbit polyclonal Ab against Fas (1/50; Santa-Cruz Biotechnology, Santa-Cruz, CA) or a goat polyconal Ab against FasL (1/50; Santa-Cruz). Following incubation with anti-rabbit/anti-goat secondary Abs (1/100; Jackson ImmunoResearch Laboratories), sections were mounted in Vectashield with DAPI.
TUNEL stain to detect apoptotic lymphocytes in the liver was performed according to manufacturer's instructions (DeadEnd Colorimetric TUNEL System; Promega Kit, Promega, Madison, WI). Frozen sections were fixed and permeabilized as described above, incubated briefly in Equilibration Buffer and subsequently in TdT reaction mix. Sections were washed two times in saline sodium citrate and one time in phosphate-buffered saline and mounted in Vectashield with DAPI.
Images were acquired with a fluorescent microscope. Representative sections from three mice per group imaged using a 10X objective.
Morphometric analysis of AAV-mediated transgene expression, T-cell infiltrate, and TUNEL stain in the liver. To quantify X-gal expression, area fraction of X-gal stain was calculated from 10 to 12 areas per liver section using ImageJ software (W Rasband, National Institutes of Health, http://rsb.info.nih.gov/ij/). Similarly, to quantify T-cell infiltrate and TUNEL stain, area fraction of CD4+/CD8+ or TUNEL stain was calculated from 10 to 12 areas per liver section using ImageJ software. All sections were analyzed with a 10X objective under identical settings. A minimum of three individual livers were analyzed per group.
Culture medium. T-cell medium consisted of the following: DMEM (Cellgro; Mediatech, Manassas, VA) supplemented with 10% heat-inactivated fetal bovine serum (Hyclone, Thermo Scientific, Waltham, MA), 1% penicillin/streptomycin (Cellgro; Mediatech), 1% L-glutamine (Cellgro; Mediatech), 10 mmol/l HEPES buffer (Cellgro; Mediatech), 0.1 mmol/l nonessential amino acids (Invitrogen, Carlsbad, CA), 2 mmol/l sodium pyruvate (Cellgro; Mediatech) and 10–6 M 2-ME (Cellgro; Mediatech).
ELISPOT assays for cytokine-producing cells. Splenocytes were isolated by mechanical dissociation followed by RBC lysis via hypotonic shock and resuspended at a concentration of 5 × 106 cells/ml and plated at 5 × 105 cells/well in triplicate on 96-well round-bottom plates. ELISPOT assays were performed according to the manufacturer's instructions (BD Biosciences, Franklin Lakes, NJ). T-cell medium supplemented with 2 μg/ml of H2-kb–restricted β-gal CD8 T-cell epitope (ICPMYARV) (Mimotopes, Victoria, Australia) or 2 μg/ml of β-gal peptide libraries (Mimotopes) was used to stimulate splenocytes. Splenocytes were incubated at 37 °C, 5% CO2 for 18 hours. Spots were visualized by addition of 3-amino-9-ethylcarbazole (AEC) substrate set (BD Biosciences) and counted using the AID ELISPOT reader system (Cell Technology, Danvers, MA).
AAV Nab assay. Serum samples were heat inactivated at 56 °C for 30 minutes. NAb assays were performed on Huh7 cells as previously described.47 The NAb titer was reported as the highest serum dilution that inhibited AAV.CMV.LacZ transduction (β-gal expression) by ≥50%, compared with the mouse serum control (Sigma, St Louis, MO).
Statistical analysis. Data were analyzed with GraphPad Prism 4.0c software (GraphPad Software, La Jolla, CA) using unpaired Student's t-tests. P values of ≤0.05 were considered statistically significant.
Acknowledgments
We thank Martin Lock at the Penn Vector Facility (University of Pennsylvania) for supplying the recombinant AAV vectors and Deirdre McMenamin for animal study assistance. We thank Craig B Thompson and Tullia Lindstein for the kind gift of the Bcl-xL+ mice. We also thank Douglas McCarty (Ohio State University) and Joseph Rabinowitz (Thomas Jefferson University) for their critical review of the manuscript. J.M.W. is a consultant to ReGenX Holdings, and is a founder of, holds equity in, and receives a grant from affiliates of ReGenX Holdings; in addition, he is an inventor on patents licensed to various biopharmaceutical companies, including affiliates of ReGenX Holdings. This work was supported by Grants ReGenXSRA (J.M.W.) and T32-AI007324 (S.M.F.) from the National Institutes of Health. The other authors declare no conflict of interest.
References
- Wang J, Faust SM, Rabinowitz JE. The next step in gene delivery: molecular engineering of adeno-associated virus serotypes. J Mol Cell Cardiol. 2011;50:793–802. doi: 10.1016/j.yjmcc.2010.10.017. [DOI] [PubMed] [Google Scholar]
- Nayak S, Herzog RW. Progress and prospects: immune responses to viral vectors. Gene Ther. 2010;17:295–304. doi: 10.1038/gt.2009.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daya S, Berns KI. Gene therapy using adeno-associated virus vectors. Clin Microbiol Rev. 2008;21:583–593. doi: 10.1128/CMR.00008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao O, Dobrzynski E, Wang L, Nayak S, Mingle B, Terhorst C, et al. Induction and role of regulatory CD4+CD25+ T cells in tolerance to the transgene product following hepatic in vivo gene transfer. Blood. 2007;110:1132–1140. doi: 10.1182/blood-2007-02-073304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoDuca PA, Hoffman BE, Herzog RW. Hepatic gene transfer as a means of tolerance induction to transgene products. Curr Gene Ther. 2009;9:104–114. doi: 10.2174/156652309787909490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi F, Liu YL, Dobrzynski E, Kaufhold A, Liu JH, Wang Y, et al. Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J Clin Invest. 2003;111:1347–1356. doi: 10.1172/JCI16887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breous E, Somanathan S, Vandenberghe LH, Wilson JM. Hepatic regulatory T cells and Kupffer cells are crucial mediators of systemic T cell tolerance to antigens targeting murine liver. Hepatology. 2009;50:612–621. doi: 10.1002/hep.23043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman BE, Martino AT, Sack BK, Cao O, Liao G, Terhorst C, et al. Nonredundant roles of IL-10 and TGF-ß in suppression of immune responses to hepatic AAV-factor IX gene transfer. Mol Ther. 2011;19:1263–1272. doi: 10.1038/mt.2011.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrzynski E, Mingozzi F, Liu YL, Bendo E, Cao O, Wang L, et al. Induction of antigen-specific CD4+ T-cell anergy and deletion by in vivo viral gene transfer. Blood. 2004;104:969–977. doi: 10.1182/blood-2004-03-0847. [DOI] [PubMed] [Google Scholar]
- Lu Y, Song S. Distinct immune responses to transgene products from rAAV1 and rAAV8 vectors. Proc Natl Acad Sci USA. 2009;106:17158–17162. doi: 10.1073/pnas.0909520106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somanathan S, Breous E, Bell P, Wilson JM. AAV vectors avoid inflammatory signals necessary to render transduced hepatocyte targets for destructive T cells. Mol Ther. 2010;18:977–982. doi: 10.1038/mt.2010.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenardo M, Chan KM, Hornung F, McFarland H, Siegel R, Wang J, et al. Mature T lymphocyte apoptosis–immune regulation in a dynamic and unpredictable antigenic environment. Annu Rev Immunol. 1999;17:221–253. doi: 10.1146/annurev.immunol.17.1.221. [DOI] [PubMed] [Google Scholar]
- Hutcheson J, Scatizzi JC, Siddiqui AM, Haines GK, 3rd, Wu T, Li QZ, et al. Combined deficiency of proapoptotic regulators Bim and Fas results in the early onset of systemic autoimmunity. Immunity. 2008;28:206–217. doi: 10.1016/j.immuni.2007.12.015. [DOI] [PubMed] [Google Scholar]
- Van Parijs L, Biuckians A, Abbas AK. Functional roles of Fas and Bcl-2-regulated apoptosis of T lymphocytes. J Immunol. 1998;160:2065–2071. [PubMed] [Google Scholar]
- Kim H, Tu HC, Ren D, Takeuchi O, Jeffers JR, Zambetti GP, et al. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell. 2009;36:487–499. doi: 10.1016/j.molcel.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weant AE, Michalek RD, Khan IU, Holbrook BC, Willingham MC, Grayson JM. Apoptosis regulators Bim and Fas function concurrently to control autoimmunity and CD8+ T cell contraction. Immunity. 2008;28:218–230. doi: 10.1016/j.immuni.2007.12.014. [DOI] [PubMed] [Google Scholar]
- Hughes PD, Belz GT, Fortner KA, Budd RC, Strasser A, Bouillet P. Apoptosis regulators Fas and Bim cooperate in shutdown of chronic immune responses and prevention of autoimmunity. Immunity. 2008;28:197–205. doi: 10.1016/j.immuni.2007.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallach D, Kovalenko AV, Varfolomeev EE, Boldin MP. Death-inducing functions of ligands of the tumor necrosis factor family: a Sanhedrin verdict. Curr Opin Immunol. 1998;10:279–288. doi: 10.1016/s0952-7915(98)80166-0. [DOI] [PubMed] [Google Scholar]
- Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–317. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- Chao DT, Linette GP, Boise LH, White LS, Thompson CB, Korsmeyer SJ. Bcl-XL and Bcl-2 repress a common pathway of cell death. J Exp Med. 1995;182:821–828. doi: 10.1084/jem.182.3.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino AT, Nayak S, Hoffman BE, Cooper M, Liao G, Markusic DM, et al. Tolerance induction to cytoplasmic beta-galactosidase by hepatic AAV gene transfer: implications for antigen presentation and immunotoxicity. PLoS ONE. 2009;4:e6376. doi: 10.1371/journal.pone.0006376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi F, Maus MV, Hui DJ, Sabatino DE, Murphy SL, Rasko JE, et al. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat Med. 2007;13:419–422. doi: 10.1038/nm1549. [DOI] [PubMed] [Google Scholar]
- Herzog RW. Hepatic AAV gene transfer and the immune system: friends or foes. Mol Ther. 2010;18:1063–1066. doi: 10.1038/mt.2010.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Léger A, Le Guiner C, Nickerson ML, McGee Im K, Ferry N, Moullier P, et al. Adeno-associated viral vector-mediated transgene expression is independent of DNA methylation in primate liver and skeletal muscle. PLoS ONE. 2011;6:e20881. doi: 10.1371/journal.pone.0020881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Calcedo R, Vandenberghe LH, Figueredo JM, Wilson JM. Impact of preexisting vector immunity on the efficacy of adeno-associated virus-based HIV-1 Gag vaccines. Hum Gene Ther. 2008;19:663–669. doi: 10.1089/hum.2008.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crispe IN, Dao T, Klugewitz K, Mehal WZ, Metz DP. The liver as a site of T-cell apoptosis: graveyard, or killing field. Immunol Rev. 2000;174:47–62. doi: 10.1034/j.1600-0528.2002.017412.x. [DOI] [PubMed] [Google Scholar]
- Issazadeh S, Abdallah K, Chitnis T, Chandraker A, Wells AD, Turka LA, et al. Role of passive T-cell death in chronic experimental autoimmune encephalomyelitis. J Clin Invest. 2000;105:1109–1116. doi: 10.1172/JCI8607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda T, Nagata S. Why do defects in the Fas-Fas ligand system cause autoimmunity. J Allergy Clin Immunol. 1997;100 6 Pt 2:S97–101. doi: 10.1016/s0091-6749(97)70013-7. [DOI] [PubMed] [Google Scholar]
- Welsh RM, McNally JM. Immune deficiency, immune silencing, and clonal exhaustion of T cell responses during viral infections. Curr Opin Microbiol. 1999;2:382–387. doi: 10.1016/s1369-5274(99)80067-8. [DOI] [PubMed] [Google Scholar]
- Do Y, Rafi-Janajreh AQ, McKallip RJ, Nagarkatti PS, Nagarkatti M. Combined deficiency in CD44 and Fas leads to exacerbation of lymphoproliferative and autoimmune disease. Int Immunol. 2003;15:1327–1340. doi: 10.1093/intimm/dxg132. [DOI] [PubMed] [Google Scholar]
- Wells AD, Li XC, Li Y, Walsh MC, Zheng XX, Wu Z, et al. Requirement for T-cell apoptosis in the induction of peripheral transplantation tolerance. Nat Med. 1999;5:1303–1307. doi: 10.1038/15260. [DOI] [PubMed] [Google Scholar]
- Wood KL, Twigg HL, 3rd, Doseff AI. Dysregulation of CD8+ lymphocyte apoptosis, chronic disease, and immune regulation. Front Biosci. 2009;14:3771–3781. doi: 10.2741/3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol. 2009;27:147–163. doi: 10.1146/annurev.immunol.021908.132629. [DOI] [PubMed] [Google Scholar]
- Bonfoco E, Stuart PM, Brunner T, Lin T, Griffith TS, Gao Y, et al. Inducible nonlymphoid expression of Fas ligand is responsible for superantigen-induced peripheral deletion of T cells. Immunity. 1998;9:711–720. doi: 10.1016/s1074-7613(00)80668-8. [DOI] [PubMed] [Google Scholar]
- Liu ZX, Govindarajan S, Okamoto S, Dennert G. Fas-mediated apoptosis causes elimination of virus-specific cytotoxic T cells in the virus-infected liver. J Immunol. 2001;166:3035–3041. doi: 10.4049/jimmunol.166.5.3035. [DOI] [PubMed] [Google Scholar]
- Pinkoski MJ, Brunner T, Green DR, Lin T. Fas and Fas ligand in gut and liver. Am J Physiol Gastrointest Liver Physiol. 2000;278:G354–G366. doi: 10.1152/ajpgi.2000.278.3.G354. [DOI] [PubMed] [Google Scholar]
- Shresta S, Pham CT, Thomas DA, Graubert TA, Ley TJ. How do cytotoxic lymphocytes kill their targets. Curr Opin Immunol. 1998;10:581–587. doi: 10.1016/s0952-7915(98)80227-6. [DOI] [PubMed] [Google Scholar]
- Muruve DA. The innate immune response to adenovirus vectors. Hum Gene Ther. 2004;15:1157–1166. doi: 10.1089/hum.2004.15.1157. [DOI] [PubMed] [Google Scholar]
- Jooss K, Chirmule N. Immunity to adenovirus and adeno-associated viral vectors: implications for gene therapy. Gene Ther. 2003;10:955–963. doi: 10.1038/sj.gt.3302037. [DOI] [PubMed] [Google Scholar]
- Guidotti LG, Chisari FV. To kill or to cure: options in host defense against viral infection. Curr Opin Immunol. 1996;8:478–483. doi: 10.1016/s0952-7915(96)80034-3. [DOI] [PubMed] [Google Scholar]
- Guidotti LG, Chisari FV. Cytokine-induced viral purging–role in viral pathogenesis. Curr Opin Microbiol. 1999;2:388–391. doi: 10.1016/s1369-5274(99)80068-x. [DOI] [PubMed] [Google Scholar]
- Guidotti LG, Matzke B, Schaller H, Chisari FV. High-level hepatitis B virus replication in transgenic mice. J Virol. 1995;69:6158–6169. doi: 10.1128/jvi.69.10.6158-6169.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang H, Bell P, McCarter RJ, He J, Calcedo R, et al. Systematic evaluation of AAV vectors for liver directed gene transfer in murine models. Mol Ther. 2010;18:118–125. doi: 10.1038/mt.2009.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai H, Fuess S, Storm TA, Muramatsu S, Nara Y, Kay MA. Unrestricted hepatocyte transduction with adeno-associated virus serotype 8 vectors in mice. J Virol. 2005;79:214–224. doi: 10.1128/JVI.79.1.214-224.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock M, Alvira M, Vandenberghe LH, Samanta A, Toelen J, Debyser Z, et al. Rapid, simple, and versatile manufacturing of recombinant adeno-associated viral vectors at scale. Hum Gene Ther. 2010;21:1259–1271. doi: 10.1089/hum.2010.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell P, Limberis M, Gao G, Wu D, Bove MS, Sanmiguel JC, et al. An optimized protocol for detection of E. coli beta-galactosidase in lung tissue following gene transfer. Histochem Cell Biol. 2005;124:77–85. doi: 10.1007/s00418-005-0793-2. [DOI] [PubMed] [Google Scholar]
- Calcedo R, Vandenberghe LH, Gao G, Lin J, Wilson JM. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J Infect Dis. 2009;199:381–390. doi: 10.1086/595830. [DOI] [PMC free article] [PubMed] [Google Scholar]