

Abstract
Epalrestat (EPS), approved in Japan, is the only aldose reductase inhibitor that is currently available for the treatment of diabetic neuropathy. Here we report that EPS at near-plasma concentration increases the intracellular levels of glutathione (GSH), which is important for protection against oxidative injury, through transcription regulation. Treatment of Schwann cells with EPS caused a dramatic increase in intracellular GSH levels. EPS increased the mRNA levels of γ-glutamylcysteine synthetase (γ-GCS), the enzyme catalyzing the first and rate-limiting step in de novo GSH synthesis. Nuclear factor erythroid 2-related factor 2 (Nrf2) is a key transcription factor that plays a central role in regulating the expression of γ-GCS. ELISA revealed that EPS increased nuclear Nrf2 levels. Knockdown of Nrf2 by siRNA suppressed the EPS-induced GSH biosynthesis. Furthermore, pretreatment with EPS reduced the cytotoxicity induced by H2O2, tert-butylhydroperoxide, 2,2'-azobis (2-amidinopropane) dihydrochloride, and menadione, indicating that EPS plays a role in protecting against oxidative stress. This is the first study to show that EPS induces GSH biosynthesis via the activation of Nrf2. We suggest that EPS has new beneficial properties that may prevent the development and progression of disorders caused by oxidative stress.
Keywords: Epalrestat, Glutathione, γ-glutamylcysteine synthetase, Nuclear factor erythroid 2-related factor 2, Oxidative stress
Graphical abstract

Highlights
-
•
Epalrestat is available for the treatment of diabetic neuropathy.
-
•
Epalrestat stimulated GSH biosynthesis by up-regulating γ-GCS via Nrf2 activation.
-
•
Epalrestat reduced the cytotoxicity induced by oxidants.
Introduction
Epalrestat (5-[(1Z,2E)-2-methyl-3-phenyl propenylidene]-4-oxo-2-thioxo-3-thiazolidine acetic acid; EPS2), a carboxylic acid derivative (Fig. 1), is an inhibitor of aldose reductase, a rate-limiting enzyme in the polyol pathway. Under hyperglycemic conditions, EPS reduces intracellular sorbitol accumulation, which is implicated in the pathogenesis of diabetic complications [1]. Diabetic neuropathy is one of the most common long-term complications in patients with diabetes mellitus. EPS is approved in Japan for the alleviation of subjective neuropathy symptoms, abnormality of vibration sense, and abnormal changes in heartbeat associated with diabetic peripheral neuropathy. Data from experimental studies indicate that EPS reduces sorbitol accumulation in the sciatic nerve, erythrocytes, and ocular tissues in animals, and in erythrocytes in humans. An evaluation of data from six clinical trials showed that 50 mg of EPS administered three times a day might improve motor and sensory nerve conduction velocity and subjective neuropathy symptoms as compared with baseline and placebo [2]. However, the basic effects of EPS on cells and tissues, other than aldose reductase inhibition, have not been reported so far.
Fig. 1.

Chemical structure of epalrestat.
Cell stress can greatly increase reactive oxygen species (ROS) levels [3]. Because of their highly reactive nature, ROS can modify other oxygen species, proteins, or lipids, a situation often termed oxidative stress. In this regard, maintaining normal cellular ROS levels is vital to the proper physiologic function of numerous cell types in the body. The excessive production or the decreased scavenging of ROS has been implicated in the pathogenesis of such diseases as neurodegeneration [4,5], diabetes [6], and atherosclerosis [7,8]. Reduced glutathione (GSH), the most abundant non-protein thiol antioxidant in cells, is important for protection against oxidative injury [9]. γ-Glutamylcysteine synthetase (γ-GCS) is the enzyme catalyzing the first and rate-limiting step in the de novo GSH synthesis [10]. Thus, the regulation of γ-GCS expression and activity is critical for GSH homeostasis. Nuclear factor erythroid 2-related factor 2 (Nrf2) is a key transcription factor that plays a central role in regulating the expression of antioxidant genes, including γ-GCS [11–13]. Nrf2 is retained in the cytoplasm as an inactive complex with its cytosolic repressor, Kelch-like ECH associated protein-1 (Keap1). The dissociation of Nrf2 from Keap1 is crucial for its nuclear translocation, followed by binding to DNA and activation of cytoprotective genes. Therefore, Nrf2 is important for the maintenance of intracellular GSH levels and redox homeostasis.
In this study, we examined the effect of EPS on GSH biosynthesis in rat Schwann cells (SCs). We found that EPS increases GSH levels through transcription regulation and induces the nuclear translocation of Nrf2.
Materials and methods
Schwann cell culture and treatment with EPS
Rat SCs were purchased from Dainippon Sumitomo Pharma Co., Ltd. (Osaka, Japan). Cells were grown to 80–90% confluence in DMEM containing 10% fetal bovine serum (FBS), L-glutamine (4 mM), penicillin (100 U/ml), and streptomycin (100 μg/ml) at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. Then, the cells were passaged by trypsinization.
Before treating the cells with EPS (Wako Pure Chemical Industries, Ltd., Osaka, Japan), the culture medium was replaced with DMEM containing 2% FBS because serum may include antioxidants, chelates of transition metal ions, and high-density lipoproteins [14]. EPS (10–200 μM) was subsequently added to the medium.
Cell viability
Cell viability was assessed by using CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS assay) from Promega (Madison, WI, USA). Briefly, SCs on 96-well plates were treated with EPS for 24 h. After the treatment with EPS, the medium containing detached cells was removed. Cells remaining on the 96-well plates were washed with DMEM (FBS-free) and incubated with fresh DMEM (100 μl) and MTS assay solution (10 µl) at 37 ˚C for 60 min. The produced MTS formazan was measured at 490 nm with a Bio-Rad Model 680 microplate reader (Hercules, CA, USA).
Flow cytometric detection of activated caspase-3
After treating SCs with EPS, the cells were fixed with 4% p-formaldehyde. Activated caspase-3 was detected by reacting with an antibody specific for the cleaved form of caspase-3, anti-mouse caspase-3 phycoerythrin (PE)-conjugated monoclonal antibody (sc-7272 PE; Santa Cruz Biotechnology, Inc., CA, USA). Following incubation with the antibody, the cells were washed with DPBS, collected with a cell scraper, resuspended in DPBS, and analyzed with a flow cytometer (Beckman Colter, Fullerton CA, USA). Fluorescence was detected with fluorescence channel 2 (FL2).
Measurement of γ-GCS and Nrf2 mRNA levels
Quantitative RT-PCR analysis was used to measure γ-GCS mRNA and Nrf2 mRNA levels. Total RNA from treated cells was extracted with RNAspin Mini (GE Healthcare, Buckinghamshire, UK) according to the manufacturer's protocol. mRNAs were reverse-transcribed into cDNA with a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Quantitative RT-PCR was performed with a 7500 Fast Real-Time PCR System (Applied Biosystems). Primers for rat γ-GCS (Rn00689046_g1) and rat Nrf2 (Rn00477784_m1) were purchased from Applied Biosystems. mRNA levels were acquired from the value of the threshold cycle (Ct) of γ-GCS or Nrf2, normalized to that of GAPDH. Relative mRNA levels were compared and expressed as percentage of control levels.
Determination of nuclear Nrf2 translocation
Nuclear extracts of SCs were prepared using the Active Motif Nuclear Extract Kit (Tokyo, Japan) according to the manufacturer's protocol. The amount of active Nrf2 in the nuclear extracts was determined by subjecting samples of 20 μg protein to assay with a TransAM Nrf2 DNA Binding ELISA Kit (Active Motif). The assay was performed according to the manufacturer's protocol.
Knockdown of Nrf2 with small interfering RNA (siRNA)
Oligonucleotides directed against rat Nrf2 and control siRNA (Ambion, Austin, TX, USA) were transfected into SCs using Lipofectamine RNAiMAX (Invitrogen, Eugene, OR, USA), according to the manufacturer's protocol. Briefly, both Nrf2 siRNA and control siRNA were diluted with Opti-MEM medium and then, diluted Lipofectamine RNAiMax was added. The transfection mixture was incubated at room temperature for 20 min. When SCs reached 30–50% confluence, the culture medium was replaced with DMEM (without FBS) and the transfection mixture was added to each well. The final concentration of siRNA was 50 nM. After SCs transfected with Nrf2 siRNA and control siRNA were treated with EPS, the effect of Nrf2 knockdown on intracellular GSH levels was assessed.
Other procedures
Intracellular GSH levels were measured by spectrophotometric methods, as described previously [15]. Aldose reductase activity was measured according to the method described by Kawasaki et al. [16] with DL-glyceraldehyde as the substrate. Protein concentrations were determined using the Bradford method with bovine serum albumin as the standard.
Statistical analysis
All experiments were performed independently at least three times. Data were combined and expressed as means±S.D. Statistical significance between two groups was evaluated using the Student's t-test after analysis of variance or the Scheffe test after the Kruskal–Wallis test. A P value of <0.05 was considered to be significant.
Results
Effect of EPS on viability of SCs
SCs were treated with EPS at the indicated concentrations for 24 h. As shown in Fig. 2A, aldose reductase activity was decreased by 30–50% after treatment with 10–100 µM EPS. In this case, however, dose-dependent differences were not observed. Fig. 2B shows cell viability estimated by the MTS assay. EPS at 10 and 50 µM had no effect on cell viability. A significant reduction in cell viability could be observed at EPS concentrations of 100 and 200 µM. At those EPS concentrations, loss of viability was 4% and 18%, respectively, compared to control. Caspase-3 activation is involved in the pathways for apoptosis. Fig. 2C shows the results of flow cytometric analyses with phycoerythrin-conjugated anti-caspase-3 antibody. The fluorescence intensities of SCs treated with 100 and 200 µM EPS were shifted to the right side of the panel compared to control. The increases in fluorescence intensity were approximately 1.7- and 2.6-fold (Fig. 2D). These results suggest that increasing EPS concentration induces apoptosis in SCs.
Fig. 2.
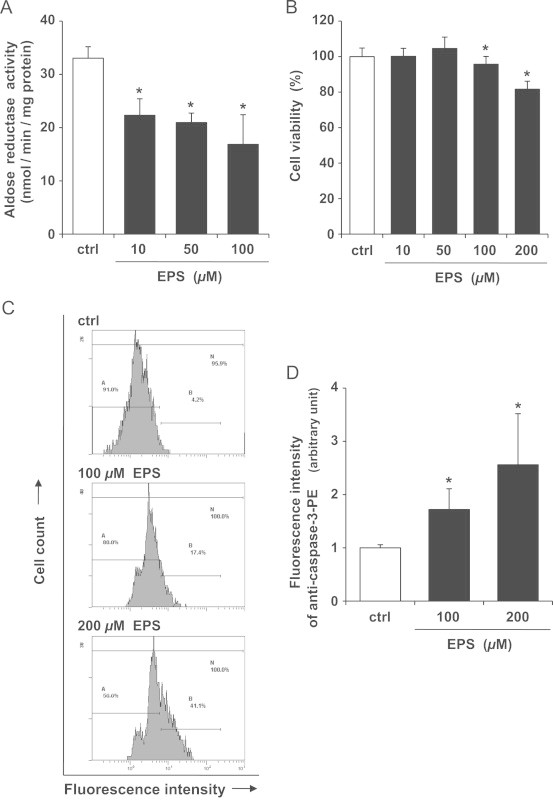
Effect of EPS on aldose reductase activity and viability of SCs. SCs were treated with EPS at the indicated concentrations for 24 h. (A) Aldose reductase activity. Values are means±SD of three experiments. (B) Cell viability estimated by MTS assay. Values are means±SD of six experiments. (C) Caspase-3 activity measured by flow cytometry using phycoerythrin-conjugated anti-caspase-3 antibody. Each panel shows the typical fluorescence intensity from three independent experiments. (D) Fluorometric analysis of data shown in C. Values are means±SD of three experiments. *Significant difference from the value of control (P<0.05).
Effect of EPS on GSH in SCs
Next, we examined the effect of EPS on intracellular GSH levels. As shown in Fig. 3A, treatment of SCs with EPS at 10 and 50 µM caused a dramatic increase in intracellular GSH levels: the increases were approximately 3.2- and 5.6-fold, respectively, compared to control. A further increase in GSH levels could be observed at the EPS concentration of 100 µM (data not shown). However, this EPS concentration caused a loss of cell viability (Fig. 2), which was probably concomitant with cell membrane damage. On the other hand, sorbinil, another aldose reductase inhibitor [17], failed to increase GSH levels (Fig. 3B). Fig. 3C demonstrates that EPS increased the mRNA levels of γ-GCS, the enzyme catalyzing the first and rate-limiting step in de novo GSH synthesis. The γ-GCS mRNA levels were increased by 1.2- and 2.6-fold by 10 and 50 µM EPS, respectively. These results indicate that EPS increases intracellular GSH levels in SCs through transcription regulation.
Fig. 3.
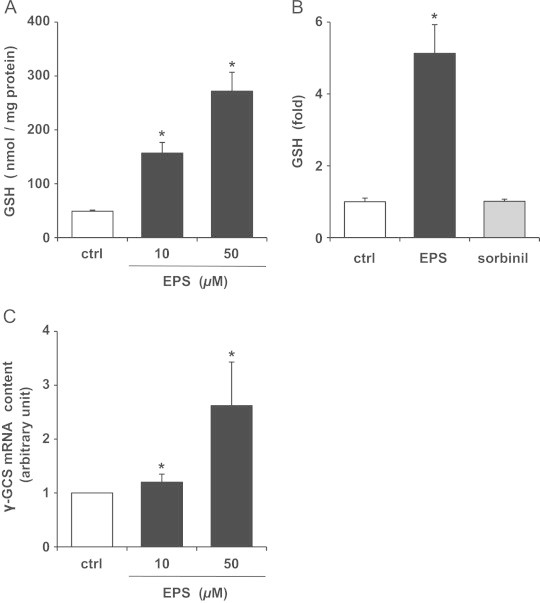
Effect of EPS on GSH and γ-GCS mRNA levels in SCs. (A) Intracellular GSH levels. SCs were treated with 10 or 50 µM EPS for 24 h. (B) GSH levels were measured after treatment with 50 µM EPS or 50 µM sorbinil for 24 h. (C) γ-GCS mRNA levels. SCs were treated with 10 or 50 µM EPS for 4 h. Values are means±SD of three experiments. *Significant difference from the value of control (P<0.05).
Effect of EPS on Nrf2 in SCs
Then, we further studied how EPS increased the levels of γ-GCS. Recent studies have reported that Nrf2 plays a pivotal role in inducing the expression of genes encoding detoxifying/defensive proteins, including γ-GCS, by binding to the antioxidant response element (ARE) [11–13]. The effect of EPS on Nrf2 activation/nuclear translocation in SCs was tested by binding EPS to an ARE-specific double-stranded oligonucleotide. Fig. 4A demonstrates that EPS caused an increase in the nuclear level of active Nrf2. The nuclear levels of active Nrf2 were increased by 1.8- and 3.8-fold by treatment with 10 and 50 µM EPS, respectively, and the increase was similar to that of γ-GCS mRNA levels. In contrast, EPS failed to increase Nrf2 mRNA levels (Fig. 4B).
Fig. 4.
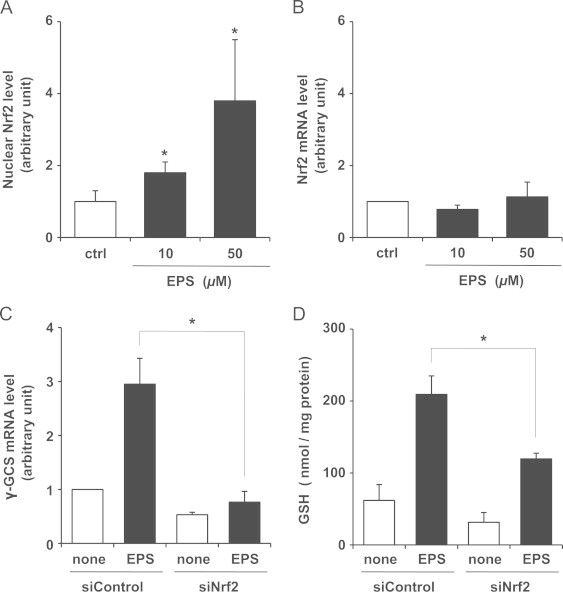
Effect of EPS on Nrf2 in SCs. Nuclear levels of active Nrf2 (A) and Nrf2 mRNA levels (B) were measured after SCs were treated with 10 or 50 µM EPS for 4 h. Values are means±SD of three experiments. *Significant difference from the value of control (P<0.05). In C and D, SCs were transfected with control siRNA (siControl) or Nrf2 siRNA (siNrf2) and were treated or not treated with 50 µM EPS for 24 h. Subsequently, γ-GCS mRNA levels (C) and GSH levels (D) were measured. Values are means±SD of three experiments. *Significant difference from the value of siControl treated with EPS (P<0.05).
We examined whether Nrf2 levels could alter the increases in γ-GCS mRNA and GSH levels of cells treated with 10 and 50 µM EPS, by means of Nrf2 knockdown in SCs. SCs were transfected with control siRNA (siControl) or Nrf2 siRNA (siNrf2). Nrf2 mRNA expression levels in cells transfected with Nrf2 siRNA were reduced by 61% relative to those in control siRNA transfected cells (data not shown). As shown in Fig. 4C and D, the increase in γ-GCS mRNA and GSH levels after EPS treatment was inhibited by the knockdown of Nrf2 expression using siRNA. These results suggest that EPS induces GSH biosynthesis by up-regulating γ-GCS via the activation of Nrf2 in SCs.
Effect of EPS on oxidative stress in SCs
Finally, we examined whether EPS could protect SCs from oxidative stress. SCs were pretreated with EPS (50 µM) for 16 h and then exposed to oxidizing agents. After exposure for 24 h, cell viability was assessed by the MTS assay. The exposures to H2O2 and tert-butylhydroperoxide resulted in approximately 70% and 80% cytotoxicity, respectively (Fig. 5A and B). EPS significantly reduced the cytotoxicity induced by the exposure to the peroxides. In addition, EPS reduced the cytotoxicity induced by 2,2'-azobis (2-amidinopropane) dihydrochloride, a free-radical-generating azo compound [18] (Fig. 5C). Furthermore, EPS reduced the cytotoxicity induced by menadione, a redox-cycling quinone known to cause oxidative stress [19], in SCs (Fig. 5D). N-Acetyl-L-cysteine (a precursor of GSH) or GSH monoethyl ester (a cell-permeable derivative of GSH) also reduced the cytotoxicity induced by these oxidants, while DL- buthionine-(S,R)-sulfoximine (an inhibitor of γ-GCS) completely abolished the protective effect of EPS against H2O2-induced cytotoxicity (data not shown).
Fig. 5.
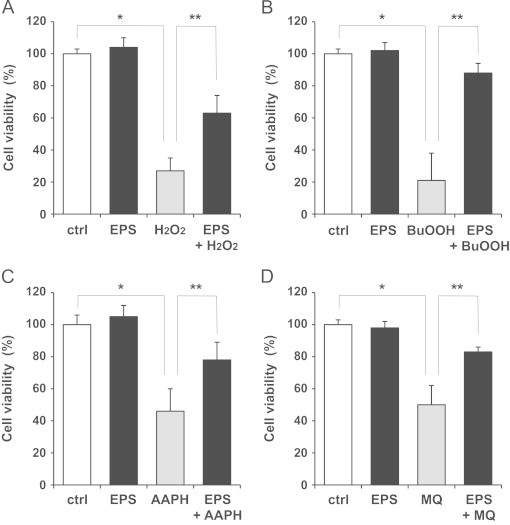
Effect of EPS on oxidative stress in SCs. SCs were pretreated with EPS (50 µM) for 16 h. Subsequently, the untreated or EPS-treated cells were exposed to 100 µM H2O2 (A), 100 µM tert-butylhydroperoxide (BuOOH) (B), 25 mM 2,2'-azobis (2-amidinopropane) dihydrochloride (AAPH) (C), and 100 µM menadione sodium bisulfide (MQ) for 24 h. Values are means±SD of six experiments. *Significant difference from the value of control (P<0.05). **Significant difference from the value of oxidizing agent alone (P<0.05).
Discussion
EPS (Ono Pharmaceuticals, Osaka, Japan), approved in Japan in 1992, is the only aldose reductase inhibitor currently available for the treatment of diabetic neuropathy. EPS is easily absorbed by neural tissue and inhibits aldose reductase with minimum adverse effects [20]. A recent study showed that treatment with EPS at an early stage delayed the progression of diabetic neuropathy and prevented the onset/progression of retinopathy and nephropathy [21]. The usual dosage of EPS for oral use is 50 mg three times a day. The plasma EPS concentration of 3.9 µg/ml (12 µM) was observed 1 h after a single oral dose of 50 mg [22]. In this study, we demonstrated for the first time that EPS at near-plasma concentration exerted biological effects other than aldose reductase inhibition on cells.
GSH, the most abundant antioxidant, plays an essential role in maintaining the cellular redox state [9]. The present study demonstrated that EPS increased intracellular GSH and γ-GCS mRNA levels in SCs (Fig. 3A and C). Increasing the concentration of EPS resulted in an increase in GSH level. In contrast, no significant dose-dependent effect of EPS on aldose reductase activity was observed (Fig. 2A), implying that the inhibition of aldose reductase does not contribute to the ability of EPS to increase GSH levels. Indeed, sorbinil, another aldose reductase inhibitor, [17] failed to increase GSH levels (Fig. 3B). On the other hand, the knockdown of Nrf2, which regulates GSH levels by controlling the expression of γ-GCS gene [11–13], suppressed the increase in GSH and γ-GCS mRNA levels after EPS treatment (Fig. 4C and D). Moreover, the activation/nuclear translocation of Nrf2 was observed after treatment with EPS, although EPS did not affect the mRNA level of Nrf2 (Fig. 4A and B). Taken together, these results provide strong evidence that EPS stimulates GSH biosynthesis by up-regulating γ-GCS via Nrf2. Under unstimulated conditions, Nrf2 is sequestered in the cytoplasm where it associates with Keap1 [11–13]. Nrf2 can be translocated into the nucleus to bind to ARE when the Keap1–Nrf2 complex is dissociated by some form of cellular stimuli. Therefore, the EPS-stimulated Nrf2 activation may be mediated by the dissociation of the Keap1–Nrf2 complex, rather than by Nrf2 transcription. Meanwhile, treatment with siNrf2 did not completely decrease intracellular GSH to baseline levels despite complete inhibition of γ-GCS mRNA (Fig. 4C and D). A very recent study has demonstrated that activation of Nrf2 results in increased release of GSH [23]. It is possible that treatment with siNrf2 decreases GSH released from cells, thereby leading to the intracellular GSH accumulation.
Oxidative stress is associated with the development and progression of numerous pathological conditions, such as atherosclerosis [7,8], diabetes [6], and neurodegeneration [4,5]. It is important to find ways to increase the intracellular GSH level in order to prevent and/or minimize ROS-induced cellular damage. We assumed that EPS might play a role in protecting against oxidative stress. To determine whether EPS protects SCs from oxidative stress, we performed experiments by using H2O2, tert-butylhydroperoxide, 2,2'-azobis (2-amidinopropane) dihydrochloride or menadione [18,19] as the source of oxidative stress. Pretreatment with EPS at 50 µM clearly protected SCs from toxicity induced by these oxidizing agents (Fig. 5), indicating that EPS acts to suppress oxidative damage in cells and tissues. It was shown that EPS improved impaired superoxide generation in streptozotocin-induced diabetic rats [24]. In addition, EPS reduced plasma thiobarbituric acid reactive substances [25] or lipid peroxide level [26], an index of oxidative stress, in type 2 diabetic patients. These findings suggest that EPS has new beneficial properties that may prevent the development and progression of disorders caused by oxidative stress.
Hypoxia, hyperglycemia, and oxidative stress contribute directly and indirectly to SC dysfunction [27]. SCs respond to oxidative stress via the activation of Nrf2 [28]. Nrf2 is a key transcriptional activator of the antioxidant response pathway. Our present data showed that EPS protected SCs from oxidative stress, presumably by increasing GSH levels through the up-regulation of γ-GCS via the activation of Nrf2. However, Nrf2 controls not only γ-GCS gene but also the genes of many cytoprotective enzymes, such as superoxide dismutase, catalase, glutathione peroxidase, and heme oxygenase-1 [28–30]. Therefore, we speculate that the EPS-induced resistance to oxidative stress in SCs is associated with the increased expression of some other cytoprotective enzymes, including γ-GCS. EPS contains an α,β-unsaturated ketone moiety within its structure. α,β-unsaturated ketones can act as inducers of ARE genes through the oxidation of sulfhydryl groups in Keap1 leading to dissociation and nuclear translocation of Nrf2 [31]. EPS-stimulated Nrf2 activation is most likely to be achieved by alterations in the structure of Keap1. On the other hand, the main metabolite of EPS is known to be the sulfate conjugate of a mono-OH or a di-OH compound [22]. However, at present, the contribution of EPS metabolites to the increase in GSH levels is unclear. Further extensive investigations will be required to clarify the protective mechanism of EPS against oxidative stress.
In the present study, a significant reduction in cell viability was observed at EPS concentrations of 100 and 200 µM, concomitantly causing caspase-3 activation (Fig. 2). This result indicates that increasing EPS concentration may induce the apoptosis of SCs. One report described that exposing human colon adenocarcinoma cells to 50 or 100 µM EPS enhanced lipid peroxide levels and cell survival, indicating that the inhibition of aldose reductase leads to carbonyl-induced cell death [32]. A very recent study has shown that fidarestat, a potent inhibitor of aldose reductase [33], potentiates the apoptosis of human colon cancer cells through the down-regulation of cell survival proteins and the up-regulation of death receptors [34]. Consequently, one possibility is that the strong inhibition of aldose reductase under high-EPS conditions may perturb the function and survival of cells.
Drug re-profiling has emerged as a new strategy for drug discovery and development and a way to identify new treatments for diseases [35]. In this strategy, the pharmacological action of existing medicines whose safety and pharmacokinetics have already been confirmed clinically and whose use has been approved is examined comprehensively at the molecular level and the results are adopted for the development of new medicines. The results can also be applied to the development of existing drugs for use as medicines for the treatment of other diseases. The present study may lead to breakthroughs in drug discovery and development. We showed here that EPS induces GSH synthesis and apoptosis, suggesting both beneficial and damaging effects of EPS. The therapeutic EPS dose might be a new strategy useful for improving oxidative stress-related diverse diseases including neurodegenerative diseases and atherosclerosis as well as diabetes.
In summary, we demonstrated for the first time that EPS at near-plasma concentration increases intracellular GSH levels through transcription regulation. EPS can activate Nrf2, which induces GSH biosynthesis, resulting in enhanced resistance to oxidative stress. As oxidative stress and low GSH levels are the key contributors to aging and chronic diseases, EPS may be effective in preventing and/or attenuating the progress of those processes.
Acknowledgments
This study was supported in part by the Education and Research Grant from Hokkaido Pharmaceutical University and the Akiyama Life Science Foundation.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Steele J.W., Faulds D., Goa K.L. Epalrestat: a review of its pharmacology, and therapeutic potential in late-onset complications of diabetes mellitus. Drugs Aging. 1993;3:532–555. doi: 10.2165/00002512-199303060-00007. [DOI] [PubMed] [Google Scholar]
- 2.Ramirez M.A., Borja N.L. Epalrestat: an aldose reductase inhibitor for the treatment of diabetic neuropathy. Pharmacotherapy. 2008;28:646–655. doi: 10.1592/phco.28.5.646. [DOI] [PubMed] [Google Scholar]
- 3.Morrell C.N. Reactive oxygen species: finding the right balance. Circ. Res. 2008;103:571–572. doi: 10.1161/CIRCRESAHA.108.184325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sian J., Dexter D.T., Lees A.J., Daniel S., Jenner P., Marsden C.D. Glutathione-related enzymes in brain in Parkinson's disease. Ann. Neurol. 1994;36:356–361. doi: 10.1002/ana.410360306. [DOI] [PubMed] [Google Scholar]
- 5.Schulz J.B., Lindenau J., Seyfried J., Dichgans J. Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem. 2000;267:4904–4911. doi: 10.1046/j.1432-1327.2000.01595.x. [DOI] [PubMed] [Google Scholar]
- 6.Shurtz-Swirski R., Sela S., Herskovits A.T., Shasha S.M., Shapiro G., Nasser L., Kristal B. Involvement of peripheral polymorphonuclear leukocytes in oxidative stress and inflammation in type 2 diabetic patients. Diabetes Care. 2001;24:104–110. doi: 10.2337/diacare.24.1.104. [DOI] [PubMed] [Google Scholar]
- 7.Rosenblat M., Coleman R., Aviram M. Increased macrophage glutathione content reduces cell-mediated oxidation of LDL and atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis. 2002;163:17–28. doi: 10.1016/s0021-9150(01)00744-4. [DOI] [PubMed] [Google Scholar]
- 8.Kevil C.G., Pruitt H., Kavanagh T.J., Wilkerson J., Farin F., Moellering D., Darley-Usmar V.M., Bullard D.C., Patel R.P. Regulation of endothelial glutathione by ICAM-1: implications for inflammation. FASEB J. 2004;18:1321–1323. doi: 10.1096/fj.03-1401fje. [DOI] [PubMed] [Google Scholar]
- 9.Valko M., Leibfritz D., Moncol J., Cronin M.T., Mazur M., Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 10.Meister A. Selective modification of glutathione metabolism. Science. 1983;220:472–477. doi: 10.1126/science.6836290. [DOI] [PubMed] [Google Scholar]
- 11.Kensler T.W., Wakabayashi N., Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 12.Kalyanaraman B. Teaching the basics of redox biology to medical and graduate students: oxidants, antioxidants and disease mechanisms. Redox Biol. 2013;1:244–257. doi: 10.1016/j.redox.2013.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kansanen E., Kuosmanen S.M., Leinonen H., Levonen A.L. The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1:45–49. doi: 10.1016/j.redox.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parthasarathy S., Barnett J., Fong L.G. High-density lipoprotein inhibits the oxidative modification of low-density lipoprotein. Biochim. Biophys. Acta. 1990;1044:275–283. doi: 10.1016/0005-2760(90)90314-n. [DOI] [PubMed] [Google Scholar]
- 15.Tsukamoto M., Tampo Y., Sawada M., Yonaha M. Paraquat-induced oxidative stress and dysfunction of the glutathione redox cycle in pulmonary microvascular endothelial cells. Toxicol. Appl. Pharmacol. 2002;178:82–92. doi: 10.1006/taap.2001.9325. [DOI] [PubMed] [Google Scholar]
- 16.Kawasaki N., Tanimoto T., Tanaka A. Characterization of aldose reductase and aldehyde reductase from rat testis. Biochim. Biophys. Acta. 1989;996:30–36. doi: 10.1016/0167-4838(89)90090-3. [DOI] [PubMed] [Google Scholar]
- 17.Beyer-Mears A., Cruz E. Reversal of diabetic cataract by sorbinil, an aldose reductase inhibitor. Diabetes. 1985;34:15–21. doi: 10.2337/diab.34.1.15. [DOI] [PubMed] [Google Scholar]
- 18.Terao K., Niki E. Damage to biological tissues induced by radical initiator 2,2'-azobis(2-amidinopropane) dihydrochloride and its inhibition by chain-breaking antioxidants. Free Radic. Biol. Med. 1986;2:193–201. doi: 10.1016/s0748-5514(86)80070-8. [DOI] [PubMed] [Google Scholar]
- 19.Kappus H., Sies H. Toxic drug effects associated with oxygen metabolism: redox cycling and lipid peroxidation. Experientia. 1981;37:1233–1241. doi: 10.1007/BF01948335. [DOI] [PubMed] [Google Scholar]
- 20.Hotta N., Akanuma Y., Kawamori R., Matsuoka K., Oka Y., Shichiri M., Toyota T., Nakashima M., Yoshimura I., Sakamoto N., Shigeta Y. Long-term clinical effects of epalrestat, an aldose reductase inhibitor, on diabetic peripheral neuropathy: the 3-year, multicenter, comparative aldose reductase inhibitor-diabetes complications trial. Diabetes Care. 2006;29:1538–1544. doi: 10.2337/dc05-2370. [DOI] [PubMed] [Google Scholar]
- 21.Hotta N., Kawamori R., Fukuda M., Shigeta Y. Aldose reductase inhibitor-diabetes complications trial study group, long-term clinical effects of epalrestat, an aldose reductase inhibitor, on progression of diabetic neuropathy and other microvascular complications: multivariate epidemiological analysis based on patient background factors and severity of diabetic neuropathy. Diabet. Med. 2012;29:1529–1533. doi: 10.1111/j.1464-5491.2012.03684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ono Pharmaceutical Co., Ltd., Kinedak (epalrestat) Package Insert, Osaka, Japan (2009).
- 23.Steele M.L., Fuller S., Patel M., Kersaitis C., Ooi L., Münch G. Effect of Nrf2 activators on release of glutathione, cysteinylglycine and homocysteine by human U373 astroglial cells. Redox Biol. 2013;1:441–445. doi: 10.1016/j.redox.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kashima K., Sato N., Sato K., Shimizu H., Mori M. Effect of epalrestat, an aldose reductase inhibitor, on the generation of oxygen-derived free radicals in neutrophils from streptozotocin-induced diabetic rats. Endocrinology. 1998;139:3404–3408. doi: 10.1210/endo.139.8.6152. [DOI] [PubMed] [Google Scholar]
- 25.Hamada Y., Nakamura J., Naruse K., Komori T., Kato K., Kasuya Y., Nagai R., Horiuchi S., Hotta N. Epalrestat, an aldose reductase inhibitor, reduces the levels of Nepsilon-(carboxymethyl)lysine protein adducts and their precursors in erythrocytes from diabetic patients. Diabetes Care. 2000;23:1539–1544. doi: 10.2337/diacare.23.10.1539. [DOI] [PubMed] [Google Scholar]
- 26.Ohmura C., Watada H., Azuma K., Shimizu T., Kanazawa A., Ikeda F., Yoshihara T., Fujitani Y., Hirose T., Tanaka Y., Kawamori R. Aldose reductase inhibitor, epalrestat, reduces lipid hydroperoxides in type 2 diabetes. Endocr. J. 2009;56:149–156. doi: 10.1507/endocrj.k08e-237. [DOI] [PubMed] [Google Scholar]
- 27.Eckersley L. Role of the Schwann cell in diabetic neuropathy. Int. Rev. Neurobiol. 2002;50:293–321. doi: 10.1016/s0074-7742(02)50081-7. [DOI] [PubMed] [Google Scholar]
- 28.Vincent A.M., Kato K., McLean L.L., Soules M.E., Feldman E.L. Sensory neurons and Schwann cells respond to oxidative stress by increasing antioxidant defense mechanisms. Antioxid. Redox Signal. 2009;11:425–438. doi: 10.1089/ars.2008.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim H.J., Vaziri N.D. Contribution of impaired Nrf2–Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am. J. Physiol. – Renal Physiol. 2010;298:F662–671. doi: 10.1152/ajprenal.00421.2009. [DOI] [PubMed] [Google Scholar]
- 30.Okita Y., Kamoshida A., Suzuki H., Itoh K., Motohashi H., Igarashi K., Yamamoto M., Ogami T., Koinuma D., Kato M. Transforming growth factor-β induces transcription factors MafK and Bach1 to suppress expression of the heme oxygenase-1 gene. J. Biol. Chem. 2013;288:20658–20667. doi: 10.1074/jbc.M113.450478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Magesh S., Chen Y., Hu L. Small molecule modulators of Keap1–Nrf2–ARE pathway as potential preventive and therapeutic agents. Med. Res. Rev. 2012;32:687–726. doi: 10.1002/med.21257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang C., Yan R., Luo D., Watabe K., Liao D.F., Cao D. Aldo-keto reductase family 1 member B10 promotes cell survival by regulating lipid synthesis and eliminating carbonyls. J. Biol. Chem. 2009;284:26742–26748. doi: 10.1074/jbc.M109.022897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Asano T., Saito Y., Kawakami M., Yamada N. Fidarestat (SNK-860), a potent aldose reductase inhibitor, normalizes the elevated sorbitol accumulation in erythrocytes of diabetic patients. J. Diabetes Complications. 2002;16:133–138. doi: 10.1016/s1056-8727(01)00175-1. [DOI] [PubMed] [Google Scholar]
- 34.Shoeb M., Ramana K.V., Srivastava S.K. Aldose reductase inhibition enhances TRAIL-induced human colon cancer cell apoptosis through AKT/FOXO3a-dependent upregulation of death receptors. Free Radic. Biol. Med. 2013;63:280–290. doi: 10.1016/j.freeradbiomed.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mizushima T. Drug discovery and development focusing on existing medicines: drug re-profiling strategy. J. Biochem. 2011;149:499–505. doi: 10.1093/jb/mvr032. [DOI] [PubMed] [Google Scholar]