

Highlights
-
•
Histidine-tagged bovine enterokinase was refolded from bacterial inclusion bodies.
-
•
Refolding yields satisfy high demands of protein crystallography projects.
-
•
Enterokinase specifically cleaved artificial propeptides from target proteins.
Abbreviations: BENAC, benzamidine affinity chromatography; EK, enterokinase; GST, glutathione-S-transferase; IB, inclusion body; IEC, ion exchange chromatography; IMAC, mmobilized metal ion affinity chromatography; KLK, kallikrein-related peptidase; SUMO, small ubiquitin-like modifier; TEV, tobacco etch virus; uPA, urokinase-type plasminogen activator
Keywords: Biotechnology, Enteropeptidase, Inclusion bodies, In vitro folding, Serine proteases
Abstract
Enterokinase, a two-chain duodenal serine protease, activates trypsinogen by removing its N-terminal propeptide. Due to a clean cut after the non-primed site recognition sequence, the enterokinase light chain is frequently employed in biotechnology to separate N-terminal affinity tags from target proteins with authentic N-termini. In order to obtain large quantities of this protease, we adapted an in vitro folding protocol for a pentahistidine-tagged triple mutant of the bovine enterokinase light chain. The purified, highly active enzyme successfully processed recombinant target proteins, while the pentahistidine-tag facilitated post-cleavage removal. Hence, we conclude that producing enterokinase in one's own laboratory is an efficient alternative to the commercial enzyme.
1. Introduction
Mammalian enterokinase (EK) (EC 3.4.21.9, MEROPS identifier S01.156), also known as enteropeptidase, is a serine protease found in the brush border membrane of duodenal enterocytes (Imamura and Kitamoto, 2003). It removes the N-terminal propeptide from trypsinogen, thus releasing trypsin (Yamashina, 1956). Since trypsin in turn activates a range of digestive enzymes, congenital EK deficiency leads to severe intestinal malabsorption (Holzinger et al., 2002). The EK zymogen from Bos taurus (UniProt accession P98072) comprises a 115 kDa single chain. Duodenase cleaves this proform between Lys800 and Ile801, which yields an N-terminal heavy chain and a C-terminal light chain (Zamolodchikova et al., 2000). These chains remain covalently linked by a disulfide bond. The multidomain heavy chain anchors EK in the cell membrane and thereby increases its specificity for trypsinogen (Lu et al., 1997). The light chain contains a single protease domain belonging to the chymotrypsin fold (Lu et al., 1999). It retains its activity and in vitro substrate specificity after separation from the heavy chain (Light and Fonseca, 1984) or heterologous expression (Collins-Racie et al., 1995; Vozza et al., 1996).
The trypsinogen propeptide harbors a sequence of four aspartates followed by a single lysine (P5-DDDDK-P1, applying the Schechter and Berger, 1967 nomenclature), which form the canonical EK recognition sequence. At least two structural determinants of the EK substrate binding site account for this unique specificity: On the one hand, Asp189 and Ser190 (chymotrypsinogen numbering scheme is used throughout the text for residues of the EK light chain) optimize the S1 pocket for recognition of lysine side chains (Lu et al., 1999; Sichler et al., 2002). On the other hand, the KRRK motif in the 99-loop confers a positive charge to the S2 and S4 pocket, which formally complements the charge of the carboxylate groups of the tetra-aspartate sequence (Lu et al., 1999; Schöpfel et al., 2011). Unlike the narrow nonprimed-site specificity, EK is relatively promiscuous on the primed sites, accepting a wide range of residues in the P1′ and P2′ position (Hosfield and Lu, 1999).
Its ability to cut cleanly after the P1 residue and commercial availability have rendered the EK light chain a hardy perennial in preparative protein biochemistry, where it is frequently used to separate an affinity tag from a target protein (Young et al., 2012). Microgram quantities of EK may enable the purification of sufficient amounts of target protein for functional studies (Debela et al., 2006). However, the purification of target proteins on a scale suitable for X-ray crystallography often consumes correspondingly large amounts of EK, sometimes up to the milligram range. Consequently, structural biology laboratories may face significant costs when buying EK from commercial suppliers (Fong et al., 2010). Furthermore, downstream applications sensitive to contaminating proteins demand that even traces of EK are removed following the cleavage reaction. Removal commonly proceeds via resin-coupled antibodies and precludes reuse of either EK or resin. In order to overcome these limitations, we set out to establish an EK expression and purification protocol that should (1) yield amounts sufficient for routine structural biology work, (2) facilitate post-digestion removal of the protease and (3) require a reasonable number of working hours.
2. Materials and methods
2.1. Materials
Enzymes and gel markers were purchased from Thermo Scientific (Waltham, MA, USA). The pET-41a-GST_EK vector, which encodes Chinese Northern Yellow Bovine EK with an N-terminal GST-tag (Tan et al., 2007), was kindly provided by Haidong Tan. Bz-PFR-pNA, Suc-AAPR-pNA and Tos-GPR-pNA were obtained from Bachem (Weil am Rhein, Germany), Chromozym X (Mc-D-Nle-GR-pNA) was bought from Roche (Grenzach-Wyhlen, Germany). Recombinant porcine EK was provided by GenScript (Piscataway, NJ, USA). Chromatography resins were obtained from GE Healthcare (Uppsala, Sweden). Chemicals, of the highest purity available, were either from Merck (Darmstadt, Germany), AppliChem (Darmstadt, Germany) or Sigma–Aldrich (St. Louis, MO, USA).
2.2. Cloning
Plasmid pET-15b-EK_C122S_His5 was prepared from pET-41a-GST_EK by three steps of round-the-horn site-directed mutagenesis (Moore, 2012). First, primers 1F and 1R (Table 1) removed the sequence encoding the GST-tag. Second, primers 2F and 2R introduced a C122S mutation. Finally, primers 3F and 3R generated an insert that was cloned between the Nco I and Xho I sites of pET-15b. PCR was conducted on a Mastercycler ep gradient (Eppendorf; Hamburg, Germany). Sequencing at Eurofins MWG (Ebersberg, Germany) confirmed that each clone was correct. pET-15b-EK_C122S_His5 is available from Addgene (ID 49048).
Table 1.
PCR primers used for cloning pET-15b-EK_C122S_His5. Restriction sites are underlined.
| Primer | Sequence (5′ to 3′) | Restriction site |
|---|---|---|
| 1F | TATAGGGGACATATGTATATCTCCTTC | n/a |
| 1R | GATGACGACGACAAGATTGTCGG | n/a |
| 2F | GCTAATAGGCTGTATATAATCTGTGTAG | n/a |
| 2R | TTACCAGAAGAAAATCAAGTTTTTC | n/a |
| 3F | ATGCCTCGAGCTAATGATGATGATGATGTAGAAAACTTTGTATCCACTCT | Xho I |
| 3R | ATGCCCATGGGCCCTATAGATGACGACGACAAGATTG | Nco I |
2.3. Protein expression
E. coli BL21(DE3) cells transformed with the pET-15b-EK_C122S_His5 plasmid were grown over-night in 200 mL LB medium supplemented with 100 μg/mL ampicillin. Twelve shakers with 600 mL culture were inoculated 1:40 (v/v) with the preculture and grown (37 °C, 230 rpm) until an OD600 of 1.2 was reached. Protein expression was induced with 0.5 mM IPTG for 4 h at 37 °C. Cells were harvested by centrifugation (15 min, 4000 × g, 4 °C).
2.4. Inclusion body isolation
Harvested cells were resuspended in 50 mM Tris pH 7.0 (30 mL buffer per 10 g cell paste) and lyzed by sonication (3 min × 5 min with 50% duty time and 95% power, interrupted by chilling on ice for at least 5 min). Insoluble matter was washed twice with 50 mM Tris pH 7.0, 500 mM NaCl, 20 mM EDTA, 2% (v/v) Triton X-100, and once with 50 mM Tris pH 7.0, 20 mM EDTA. The washed IBs were solubilized 1:20 (w/v) in 7.5 M guanidine-HCl pH 9, 50 mM Tris, 100 mM β-mercaptoethanol for 24 h. After adjusting the pH to 3.5–4, the solution was dialyzed against 5 mM citrate pH 3.5–4.0. Precipitate was collected by centrifugation (20 min, 18 000 × g, room temperature) and solubilized 1:10 (w/v) in 7.5 M guanidine-HCl, 50 mM Tris pH 4.0–4.5 for several hours.
2.5. In vitro folding
In vitro folding was initiated by dropwise dilution of the IB solution into the 100-fold volume of 500 mM arginine, 50 mM Tris, 20 mM CaCl2, 1 mM EDTA, 5 mM cysteine-HCl, 0.5 mM cystine (from a 400 mM cystine stock dissolved in 2 M HCl), pH 8.3 at room temperature (Rudolph and Lilie, 1996; Zögg and Brandstetter, 2009). After 72 h at 16 °C, the solution was concentrated 20-fold either by tangential flow filtration (Centramate; Pall; Port Washington, NY, USA) or by dialysis against PEG 35000. For the latter protocol, several dialysis tubes (35 mm diameter, molecular weight cutoff 10 kDa) containing the in vitro folding solution were incubated in PEG 35000 flakes for at most 16 h at 4 °C. The concentrate was dialyzed against 50 mM Tris pH 7.5, 50 mM NaCl. Precipitate was removed by centrifugation (43 000 × g, 20 min, room temperature) and filtration (membrane pore size: 0.2 μm).
2.6. Protein purification
The protein solution was loaded onto a Q sepharose column equilibrated in IEC buffer (50 mM Tris–HCl, pH 7.5) for negative purification. The ratio of load to resin was about 50:1 (v/v) in this and all following chromatography steps. The flow-through contained about 75% of correctly folded EK as shown by SDS-PAGE. The remainder was eluted from the column with 3 resin volumes of IEC buffer supplemented with 150 mM NaCl.
IEC was followed by either BENAC or IMAC. In the first case, flow-though and elution from IEC were combined and brought to 500 mM NaCl. The solution was loaded onto a benzamidine sepharose column equilibrated in BENAC buffer (50 mM Tris–HCl, 500 mM NaCl, pH 7.5). After washing with 8 resin volumes of BENAC buffer, bound EK was eluted with 3 × 2.5 resin volumes of BENAC buffer supplemented with 25, 50 and 100 mM benzamidine, respectively.
In the second case, flow-though and elution from IEC were combined and brought to 500 mM NaCl, 10 mM imidazole. The solution was loaded onto a Ni2+ sepharose column equilibrated in IMAC buffer (50 mM Tris–HCl, 500 mM NaCl, 10 mM imidazole, pH 7.5). After washing with 8 resin volumes of IMAC buffer, bound EK was eluted with 3 × 2.5 resin volumes of IMAC buffer supplemented with 250 mM imidazole.
The BENAC or IMAC eluates were concentrated to 80–100 μg/mL in Amicon Ultra-15 Centrifugal Filter Units, molecular weight cutoff 10 kDa (Millipore; Billerica, MA, USA), while the buffer was exchanged to 50 mM Tris pH 7.5, 50 mM NaCl, 10% (v/v) glycerol. EK aliquots were stored at −80 °C.
An overview of the EK production workflow is given in Fig. 1.
Fig. 1.
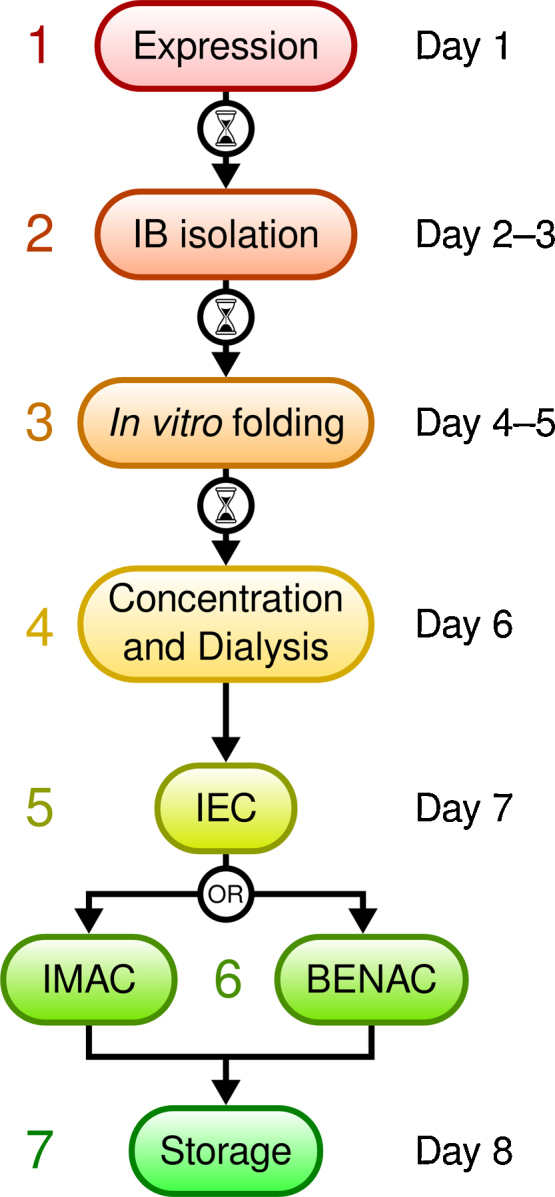
Scheme of the EK production workflow. Protein production can be modularly organized with possible break points indicated by an hourglass symbol.
2.7. Kinetic measurements
EK activity was measured in 100 μL buffer (50 mM Tris–HCl pH 7.5, 50 mM NaCl, 10% (v/v) DMSO) containing 800 ng (300 nM) enzyme and 500 μM chromogenic substrate. Time-dependent substrate cleavage corresponded to changes in absorbance at 405 nm. Protein concentrations were determined by absorbance at 280 nm, using computed extinction coefficients and molecular weights. Protein substrates were digested by EK in 50 mM Tris pH 8.0, 250 mM NaCl. Molar ratios of EK to target protein were varied. Digestion reactions proceeded for 15 h at room temperature and were monitored by SDS-PAGE.
3. Results and discussion
3.1. Protein production
In general, prokaryotes fail to express serine proteases belonging to the chymotrypsin fold in a soluble form. Such proteins contain several cysteines, which refuse to form disulfide bonds in the reducing environment of the prokaryotic cytosol (Rudolph and Lilie, 1996). Still, several groups have reported that E. coli cells express the EK light chain in a soluble form if it is fused to an N-terminal partner such as thioredoxin (Yuan and Hua, 2002), GST (Tan et al., 2007) or protein disulfide isomerase (Chun et al., 2011). However, in our hands, expression of GST-tagged EK yielded mostly IBs. Negligible amounts of the protein were soluble, yet inactive. Furthermore, these IBs resisted refolding, most likely due to the large N-terminal fusion partner that was unable to fold spontaneously. All in all, we consider prokaryotic expression of soluble EK an inherently difficult approach. This approach is presumably highly sensitive to external variables such as the choice of expression medium, expression levels, or the overall oxidation state. By contrast, chymotrypsin-related proteases are promising candidates for in vitro folding by dialysis or dilution. Therefore, we decided to apply an in vitro folding protocol well-established for coagulation factors (Zögg and Brandstetter, 2009) to EK IBs after overexpression.
Our newly designed construct EK_C122S_His5 comprised the following elements: (1) an N-terminal artificial propeptide (MGPIDDDDK), which contained an EK recognition sequence that facilitated autoactivation; (2) the light chain of bovine EK from Ile16 to His243 containing the mutations P92R, E185D and C122S; the latter mutation eliminated a surface cysteine, which in full-length EK forms a disulfide bond with the heavy chain; the mutations P92R and E185D presumably represent the EK variant that naturally occurs in the Chinese Northern Yellow breed, with no reported effect on enzymatic activity, consistent with their solvent-exposed position (Tan et al., 2007); and (3) four additional histidine residues at the C-terminus, which constituted a pentahistidine tag together with His243. This tag facilitated post-cleavage removal via IMAC. Since its affinity to a metal ion resin was sufficient, we did not add a sixth histidine to the tag in order to further enhance its affinity (Knecht et al., 2009). Following in vitro folding and autocatalyzed activation, monomeric EK_C122S_His5 was separated from incorrectly folded and aggregated species via negative IEC followed by either BENAC or IMAC (Fig. 2A and B). We considered the latter two purification techniques equivalent, since either technique resulted in protein that was more than 95% pure. We also successfully tested a BENAC-only purification protocol (cf. Fig. 2A). However, removal of soluble aggregates with IEC increased chromatography flow rates and the lifespan of the benzamidine-coupled resin. The final yield of properly folded, monomeric and active EK was several milligrams per liter of bacterial culture, corresponding to approximately 0.1% (w/w) of solubilized IBs (Table 2). EK was routinely concentrated to 80–100 μg/mL and stored at temperatures of −20 °C (short term) or −80 °C (long term). Nevertheless, the peptidase resisted autodegradation for several weeks even when stored at room temperature (Fig. 2C) and was stable at up to 5 mg/mL. In total, EK production consumed 15–20 working hours over a period of about eight days. The preparation procedure can be interrupted after steps 1, 2 and 3 without compromising the preparation quality or yield (Fig. 1).
Fig. 2.

Purification of EK and stability of the enzyme. (A) BENAC and (B) IMAC of EK_C122S_His5 after in vitro folding and autoactivation. L, load; F, flow-through; W1 and W2, washes; E1–3, elutions; M, molecular mass marker. All SDS-PAGE samples were prepared and run under nonreducing conditions. (C) EK stability during storage for 32 days at room temperature as judged from its activity against Bz-PFR-pNA.
Table 2.
Yields during purification of EK_C122S_His5. Step numbers refer to Fig. 1.
| Step | Amounta | Yield (per L culture)a | Yield (%)b |
|---|---|---|---|
| 1 Cell pellet wet weight | 35 ± 4 g | 4.9 ± 0.5 g | – |
| 2 IBs (after third wash) | 18 ± 2 g | 2.6 ± 0.2 g | – |
| 2 IBs (after dialysis against citrate buffer) | 16 ± 1 g | 2.2 ± 0.1 g | 100 |
| 6 BENAC eluate | 28 ± 8 mg | 3.9 ± 1.1 mg | 0.2 |
| 7 Final product | 12 ± 3 mg | 1.7 ± 0.4 mg | 0.1 |
Mean values and standard deviations from three folding experiments.
Relative to weight of solubilized IBs (after dialysis against citrate buffer).
3.2. Enzymatic activity against small-molecule substrates and proteinaceous targets
Although EK has been reported to be relatively specific for the recognition sequence DDDDK↓ (Kim et al., 2008; Maroux et al., 1971), EK_C122S_His5 cleaved several chromogenic substrates with arginine instead of lysine in P1 and lacking any acidic non-primed residues. Its specific activities against these substrates were similar to the values measured with commercially available porcine EK (Fig. 3A). Differences, as observed for Chromozym X, may result from N-glycosylation of the yeast-expressed porcine enzyme at Asn75, Asn113 and Asn175 (Vozza et al., 1996), which was absent in refolded EK. As anticipated, the protease selectively cleaved artificial propeptides containing an EK recognition sequence from human KLK4 and from the β-chain of uPA (Fig. 3B and C). In the case of KLK4, a molar ratio of 1:1000 (EK to KLK4) ensured complete cleavage of the target protein within 15 h. This observation demonstrates the high proteolytic efficiency of EK, since milligram amounts may cleave grams of recombinant protein.
Fig. 3.

Enzymatic activity of refolded EK and specific cleavage of EK targets. (A) Specific activities of EK_C122S_His5 and porcine EK (GenScript, Piscataway, NJ). Each value represents the mean of three measurements. (B) KLK4 was incubated without EK (∅) or with the protease at molar ratios ranging from 1:250 to 1:4000. (C) pro-uPA was incubated with EK at a molar ratio of 1:500. All SDS-PAGE samples were prepared under nonreducing conditions. M, molecular mass marker.
Important alternatives to EK include TEV and SUMO protease. At first sight, these enzymes overcome the potential drawbacks of EK, namely its intricate preparation and occasional unspecific cleavages. Soluble, high-yield expression in E. coli followed by simple purification has been reported for both TEV (Berg et al., 2006) and SUMO protease (Reverter and Lima, 2009). TEV protease exclusively cuts at the sequence P6-ExxYxQ↓G/S (Tözsér et al., 2005). SUMO protease recognizes the tertiary structure of the SUMO tag instead of a linear epitope and cleaves C-terminal of a conserved diglycine motif (Malakhov et al., 2004). Nonetheless, one should stay aware of putative pitfalls associated with these proteases: Efficient processing by TEV protease requires reducing buffer conditions and sometimes a molar ratio of 1:10 or higher. In addition, β-branched side chains in P1′ (especially isoleucine and valine) strongly interfere with digestion (Kapust et al., 2002). This limitation renders TEV protease impractical for the activation of chymotrypsin-like serine proteases, which generally require an isoleucine, leucine or valine at their mature N-terminus (Song et al., 2002). The large SUMO tag (about 100 residues) might decrease in vitro folding yields if its ability to fold spontaneously is restricted in the optimal refolding buffer for the target protein. By contrast, the shortest EK-removable tag is the eight-residue FLAG tag with its internal EK cleavage site, which lacks secondary structure (Hopp et al., 1988). Moreover, the active site cysteine of SUMO protease as well as TEV (or prescission) protease typically requires 1–2 mM dithiothreitol in the cleavage buffer (Peroutka et al., 2011), complicating the tag-removal from cysteine-containing target proteins, e.g., chymotrypsin-like proteases. Intriguingly, relying on the structure of the SUMO tag constitutes the main advantage of SUMO protease as well as its major drawback with respect to EK. On the one hand, exclusive recognition of an intact tertiary structure confers absolute substrate specificity to SUMO protease. On the other hand, this peculiarity renders SUMO protease markedly susceptible to a range of detergents (Vergis and Wiener, 2011). Hence, it will refuse to process most SUMO-tagged recombinant membrane proteins, which commonly require detergents to remain soluble during purification. In contrast, chymotrypsin-related serine proteases like EK, thrombin and factor Xa retain their activity even in detergent-containing buffers. Taken together, commonly used proteases like SUMO, TEV, or prescission are complementary rather than alternative tools to EK in the protein engineering box.
4. Conclusion
In summary, we expressed pentahistidine-tagged EK in E. coli and refolded the protein from bacterial IBs. Purified EK specifically cleaved N-terminal tags from recombinant target proteins. Thus, we conclude that despite the emergence of other highly specific proteases, do-it-yourself EK will remain an essential tool for protein biochemists.
Acknowledgments
We are grateful to Haidong Tan and Zongbao Zhao for providing the enterokinase plasmid. Also, we appreciate the help of Nina Dathan, who optimized the PEG dialysis protocol. Finally, we would like to thank Martina Wiesbauer for providing protein samples, and the Austrian Science Fund (FWF) for funding (project I631-B11).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.jbiotec.2013.10.022.
Appendix A. Supplementary data
The following are the supplementary data to this article:
References
- Berg S., van den, Löfdahl P.-A., Härd T., Berglund H. Improved solubility of TEV protease by directed evolution. J. Biotechnol. 2006;121:291–298. doi: 10.1016/j.jbiotec.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Chun H., Joo K., Lee J., Shin H.-C. Design and efficient production of bovine enterokinase light chain with higher specificity in E. coli. Biotechnol. Lett. 2011;33:1227–1232. doi: 10.1007/s10529-011-0562-3. [DOI] [PubMed] [Google Scholar]
- Collins-Racie L.A., McColgan J.M., Grant K.L., DiBlasio-Smith E.A., McCoy J.M., LaVallie E.R. Production of recombinant bovine enterokinase catalytic subunit in Escherichia coli using the novel secretory fusion partner DsbA. Biotechnology (N. Y.) 1995;13:982–987. doi: 10.1038/nbt0995-982. [DOI] [PubMed] [Google Scholar]
- Debela M., Magdolen V., Schechter N., Valachova M., Lottspeich F., Craik C.S., Choe Y., Bode W., Goettig P. Specificity profiling of seven human tissue kallikreins reveals individual subsite preferences. J. Biol. Chem. 2006;281:25678–25688. doi: 10.1074/jbc.M602372200. [DOI] [PubMed] [Google Scholar]
- Fong B.A., Wu W.-Y., Wood D.W. The potential role of self-cleaving purification tags in commercial-scale processes. Trends Biotechnol. 2010;28:272–279. doi: 10.1016/j.tibtech.2010.02.003. [DOI] [PubMed] [Google Scholar]
- Holzinger A., Maier E.M., Bück C., Mayerhofer P.U., Kappler M., Haworth J.C., Moroz S.P., Hadorn H.-B., Sadler J.E., Roscher A.A. Mutations in the proenteropeptidase gene are the molecular cause of congenital enteropeptidase deficiency. Am. J. Hum. Genet. 2002;70:20–25. doi: 10.1086/338456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopp T.P., Prickett K.S., Price V.L., Libby R.T., March C.J., Pat Cerretti D., Urdal D.L., Conlon P.J. A short polypeptide marker sequence useful for recombinant protein identification and purification. Bio/Technology. 1988;6:1204–1210. [Google Scholar]
- Hosfield T., Lu Q. Influence of the amino acid residue downstream of (Asp)4Lys on enterokinase cleavage of a fusion protein. Anal. Biochem. 1999;269:10–16. doi: 10.1006/abio.1998.3084. [DOI] [PubMed] [Google Scholar]
- Imamura T., Kitamoto Y. Expression of enteropeptidase in differentiated enterocytes, goblet cells, and the tumor cells in human duodenum. Am. J. Physiol. Gastrointest. Liver Physiol. 2003;285:G1235–G1241. doi: 10.1152/ajpgi.00198.2003. [DOI] [PubMed] [Google Scholar]
- Kapust R.B., Tözsér J., Copeland T.D., Waugh D.S. The P1′ specificity of tobacco etch virus protease. Biochem. Biophys. Res. Commun. 2002;294:949–955. doi: 10.1016/S0006-291X(02)00574-0. [DOI] [PubMed] [Google Scholar]
- Kim Y.-T., Nishii W., Matsushima M., Inoue H., Ito H., Park S.J., Takahashi K. Substrate specificities of porcine and bovine enteropeptidases toward the peptide Val-(Asp)4-Lys-Ile-Val-Gly and its analogs. Biosci. Biotechnol. Biochem. 2008;72:905–908. doi: 10.1271/bbb.70732. [DOI] [PubMed] [Google Scholar]
- Knecht S., Ricklin D., Eberle A.N., Ernst B. Oligohis-tags: mechanisms of binding to Ni2+-NTA surfaces. J. Mol. Recognit. 2009;22:270–279. doi: 10.1002/jmr.941. [DOI] [PubMed] [Google Scholar]
- Light A., Fonseca P. The preparation and properties of the catalytic subunit of bovine enterokinase. J. Biol. Chem. 1984;259:13195–13198. [PubMed] [Google Scholar]
- Lu D., Fütterer K., Korolev S., Zheng X., Tan K., Waksman G., Sadler J.E. Crystal structure of enteropeptidase light chain complexed with an analog of the trypsinogen activation peptide. J. Mol. Biol. 1999;292:361–373. doi: 10.1006/jmbi.1999.3089. [DOI] [PubMed] [Google Scholar]
- Lu D., Yuan X., Zheng X., Sadler J.E. Bovine proenteropeptidase is activated by trypsin, and the specificity of enteropeptidase depends on the heavy chain. J. Biol. Chem. 1997;272:31293–31300. doi: 10.1074/jbc.272.50.31293. [DOI] [PubMed] [Google Scholar]
- Malakhov M.P., Mattern M.R., Malakhova O.A., Drinker M., Weeks S.D., Butt T.R. SUMO fusions and SUMO-specific protease for efficient expression and purification of proteins. J. Struct. Funct. Genom. 2004;5:75–86. doi: 10.1023/B:JSFG.0000029237.70316.52. [DOI] [PubMed] [Google Scholar]
- Maroux S., Baratti J., Desnuelle P. Purification and specificity of porcine enterokinase. J. Biol. Chem. 1971;246:5031–5039. [PubMed] [Google Scholar]
- Moore S. 2012. Round-the-horn site-directed mutagenesis [WWW Document] http://openwetware.org/wiki/%27Round-the-horn_site-directed_mutagenesis (accessed 02.07.13) [Google Scholar]
- Peroutka R.J., III, Orcutt S.J., Strickler J.E., Butt T.R. SUMO fusion technology for enhanced protein expression and purification in prokaryotes and eukaryotes. Methods Mol. Biol. 2011;705:15–30. doi: 10.1007/978-1-61737-967-3_2. [DOI] [PubMed] [Google Scholar]
- Reverter D., Lima C.D. Preparation of SUMO proteases and kinetic analysis using endogenous substrates. Methods Mol. Biol. 2009;497:225–239. doi: 10.1007/978-1-59745-566-4_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph R., Lilie H. In vitro folding of inclusion body proteins. FASEB J. 1996;10:49–56. [PubMed] [Google Scholar]
- Schechter I., Berger A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- Schöpfel M., Tziridis A., Arnold U., Stubbs M.T. Towards a restriction proteinase: construction of a self-activating enzyme. ChemBioChem. 2011;12:1523–1527. doi: 10.1002/cbic.201000787. [DOI] [PubMed] [Google Scholar]
- Sichler K., Hopfner K.P., Kopetzki E., Huber R., Bode W., Brandstetter H. The influence of residue 190 in the S1 site of trypsin-like serine proteases on substrate selectivity is universally conserved. FEBS Lett. 2002;530:220–224. doi: 10.1016/s0014-5793(02)03495-6. [DOI] [PubMed] [Google Scholar]
- Song H.-W., Choi S.-I., Seong B.L. Engineered recombinant enteropeptidase catalytic subunit: effect of N-terminal modification. Arch. Biochem. Biophys. 2002;400:1–6. doi: 10.1006/abbi.2001.2737. [DOI] [PubMed] [Google Scholar]
- Tan H., Wang J., Zhao Z.K. Purification and refolding optimization of recombinant bovine enterokinase light chain overexpressed in Escherichia coli. Protein Expr. Purif. 2007;56:40–47. doi: 10.1016/j.pep.2007.07.006. [DOI] [PubMed] [Google Scholar]
- Tözsér J., Tropea J.E., Cherry S., Bagossi P., Copeland T.D., Wlodawer A., Waugh D.S. Comparison of the substrate specificity of two potyvirus proteases. FEBS J. 2005;272:514–523. doi: 10.1111/j.1742-4658.2004.04493.x. [DOI] [PubMed] [Google Scholar]
- Vergis J.M., Wiener M.C. The variable detergent sensitivity of proteases that are utilized for recombinant protein affinity tag removal. Protein Expr. Purif. 2011;78:139–142. doi: 10.1016/j.pep.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vozza L.A., Wittwer L., Higgins D.R., Purcell T.J., Bergseid M., Collins-Racie L.A., LaVallie E.R., Hoeffler J.P. Production of a recombinant bovine enterokinase catalytic subunit in the methylotrophic yeast Pichia pastoris. Biotechnology (N. Y.) 1996;14:77–81. doi: 10.1038/nbt0196-77. [DOI] [PubMed] [Google Scholar]
- Yamashina I. The action of enterokinase on trypsinogen. Acta Chem. Scand. 1956;10:739–743. doi: 10.1016/0006-3002(56)90329-8. [DOI] [PubMed] [Google Scholar]
- Young C.L., Britton Z.T., Robinson A.S. Recombinant protein expression and purification: a comprehensive review of affinity tags and microbial applications. Biotechnol. J. 2012;7:620–634. doi: 10.1002/biot.201100155. [DOI] [PubMed] [Google Scholar]
- Yuan L.-D., Hua Z.-C. Expression, purification, and characterization of a biologically active bovine enterokinase catalytic subunit in Escherichia coli. Protein Expr. Purif. 2002;25:300–304. doi: 10.1016/s1046-5928(02)00012-8. [DOI] [PubMed] [Google Scholar]
- Zamolodchikova T.S., Sokolova E.A., Lu D., Sadler J.E. Activation of recombinant proenteropeptidase by duodenase. FEBS Lett. 2000;466:295–299. doi: 10.1016/s0014-5793(00)01092-9. [DOI] [PubMed] [Google Scholar]
- Zögg T., Brandstetter H. Structural basis of the cofactor- and substrate-assisted activation of human coagulation factor IXa. Structure. 2009;17:1669–1678. doi: 10.1016/j.str.2009.10.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.