

Abstract
N-myristoyltransferase (NMT) is a prospective drug target against parasitic protozoa. Herein we report the successful discovery of a series of Plasmodium vivax NMT inhibitors by high throughput screening. A high-resolution crystal structure of the hit compound in complex with NMT was obtained, allowing understanding of its novel binding mode. A set of analogues was designed and tested to define the chemical groups relevant for activity and selectivity.
INTRODUCTION
Plasmodium vivax is the most widely distributed cause of malaria in the world and is endemic in large parts of Asia and the Americas. Because P. vivax kills rarely and is not amenable to continuous in vitro culture, it has so far been relatively little studied in comparison to P. falciparum.1 Yet, the overall burden, morbidity and socioeconomic impact caused by vivax malaria make the search for efficient treatments against this infection essential.2 N-myristoyltransferase (NMT) is a ubiquitous enzyme in eukaryotes that catalyses the covalent attachment of the saturated 14-carbon fatty acid, myristate, to the N-terminal glycine of susceptible proteins by amide bond formation.3 NMT has been found to be essential in eukaryotes and the sequence variation between parasite and host NMTs should permit the development of selective inhibitors.4-6 Therefore, targeting N-myristoylation in P. vivax might constitute an attractive strategy for the treatment of vivax malaria. However, in order to validate this approach, inhibitors of P. vivax NMT (PvNMT) must first be identified. In the present paper, we report the discovery, by high throughput screening, of a new small molecule inhibitor of PvNMT: 3-butyl-4-((2-cyanoethyl)thio)-6-methoxy-2-methyl-quinoline. We describe a first study of the structure-activity relationships of this new series and establish its binding mode by resolving the high-resolution X-ray structure of this compound in complex with PvNMT.
RESULTS AND DISCUSSION
Screening
The screening assay was adapted from a recently reported 96-well plate fluorogenic assay for NMT.7 A number of potential substrates were tested, with Homo sapiens pp60src(2-16) (GSNKSKPKDASQRRR-NH2) being chosen as the most suitable substrate for the PvNMT assay, with apparent Km (Kmapp) of 5.71 ± 0.6 μM in assay conditions. The compound collection was sourced by Medical Research Council Technology, largely from commercial suppliers. In total ~59,500 compounds were screened against PvNMT. A confirmed hit rate of 0.30% was observed and, in total, 107 compounds were found to inhibit NMT activity with IC50 in the range 2–50 μM. The structure of some hit compounds and their respective activity against NMT are reported in Figure 1. At the end of the selection process, we decided to focus our efforts on compound 1 (MRT00057965), which displays micromolar IC50 against P. vivax NMT.
Figure 1. Structure and activity of some hit compounds.

Binding mode of hit compound 1
To reveal the basis of high affinity binding, we determined the first reported crystal structure of PvNMT, in a ternary complex with compound 1 and a non-hydrolysable myristoyl-coenzyme A analogue, NHM.8 X-ray diffraction data extending to a spacing of 1.55 Å were collected on synchrotron beamline ID14-4 (λ = 0.9393 Å) at the ESRF (Grenoble).
Details of structure solution and a Table of the data collection and refinement statistics can be found in Supplementary Information. The core of the structure is an 11-stranded β-sheet which is twisted so as to form an extended substrate binding groove on either side of which NHM and compound 1 are bound (Supplementary Figure 1). The mode of binding of compound 1 is well-defined by the electron density maps (Figure 2A). Compound 1 is bound in PvNMT such that ~90% of its surface area is buried, and it forms interactions with the side chains of a number of aromatic residues. Adjacent residues on one face of a β-strand, Phe103 and Phe105, pack onto opposite faces of the quinoline ring forming π-π stacking interactions. Meanwhile the phenolic ring of Tyr211 packs against the nitrile of the exocyclic 2-cyanoethylthioether group. Polar interactions are formed between the quinoline nitrogen of compound 1 and the hydroxyl of Ser319 and between the nitrile nitrogen of the ligand and the imidazole ring of His213. There are additional apolar contacts to the side chains of Leu330, Val96 and Asp98. Finally, a quintet of water molecules clusters in the neighborhood of 1, one of which forms a hydrogen bond with the sulfur of the 2-cyanoethylthioether group. The large number of interactions between the enzyme and compound 1 could certainly account for the observed inhibitory activity.
Figure 2.

A. View of 1 in chain A of PvNMT (cyan ribbon) in cylinder format and colored by atom, carbon (green) oxygen (red) nitrogen (blue) sulfur (yellow). The side chains (labeled) of surrounding protein residues are similarly colored, but with carbons in grey. Electron density (2Fo-Fc) associated with 1 contoured at 1 σ is displayed. Polar interactions with the protein and solvent are shown as dashed lines. Y211 labeled in italics is shown in two alternate conformations.
B. 1 (green) from the PvNMT ternary complex superposed with residues GLYASK of the peptide ligand from the crystal structure of the ScNMT ternary complex.9
C. 1 (green) from the PvNMT ternary complex superimposed with two inhibitors described previously for CaNMT10 (orange) and TbNMT6 (blue). The C-terminal residue of PvNMT (stick format) is labeled.
Comparison of the structure of the PvNMT complex with that of a ternary complex of Saccharomyces cerevisiae NMT (ScNMT) with myristoyl-coenzyme A and the octapeptide (GLYASKLA) 9 suggests that 1 is a competitive inhibitor that binds in the peptide binding groove of PvNMT occupying volume corresponding to that filled by Ala4 and Ser5 of the peptide in ScNMT (Figure 2B). In this superposition, the plane of the bicyclic ring in 1 is approximately perpendicular to the direction of the peptide. The binding mode of 1 (Figure 2C) differs from that described previously for potent inhibitors of Candida albicans NMT (CaNMT) and Trypanosoma brucei NMT (TbNMT).6, 10 In particular, 1 does not make any interaction with the C-terminal NMT carboxylate that is a key characteristic of those other inhibitors. Recently, Brand et al. reported cocrystal structures of hit compounds against TbNMT in complex with Leishmania major NMT (PDB accession codes 4A2Z and 4A30).11 These inhibitors present a binding mode comparable to 1 (i.e. no interaction with the NMT C-terminus and H-bonding with Ser319), but the bulkiness of the quinoline ring and the presence of the n-butyl chain in 1 appear to induce significant widening of the binding pocket.
Chemistry and Structure-Activity Relationships
A set of compounds was prepared to address the key structure-activity relationships of the quinoline scaffold. First, a series of 3-butyl-6-methoxy-2-methylquinoline derivatives was synthesized according to Scheme 1. Condensation of 4-methoxyaniline with an appropriate β-ketoester under acidic conditions led to the formation of the 4-hydroxyquinoline core 1a which could then be converted into its 4-mercaptoquinoline derivative 1c in two steps.12 1c was S-alkylated with an appropriate halogenated molecule to give the hit compound 1 and its analogs 12-16 (Table 1). Next, our efforts focused on positions 2-, 3- and 6- of the quinoline scaffold. A series of 4-hydroxyquinoline derivatives was obtained by a Conrad-Limpach or Gould-Jacobs synthesis (Scheme 2).13, 14 The 4-hydroxy position was subsequently chlorinated and substituted with 3-mercaptopropanenitrile to provide final compounds 7-11 and 20-24.
Scheme 1. Synthetic pathway to compound 1a.

aReagents and conditions: (i) PTSA·H2O, toluene, reflux, 16 h; (ii) Ph2O, reflux, 1 h; (iii) SOCl2, DMF, 80 °C, 1 h; (iv) NaSH, DMF, reflux, 4 h; (v) 3-bromopropanenitrile, K2CO3, DMF, 110 °C, 1 h; then, HCl 2 N in Et2O.
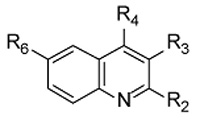
Table 1. Biochemical Activity of Tested Compounds against PvNMT and Human NMTs a.
|
Kiapp (μM) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd | R2 | R3 | R4 | R6 | PvNMT | HsNMT1 | HsNMT2 | cLog Pc | LEd | LipEe |
| 1 | Me | nBu | S-(CH2)2-CN | MeO | 1.73 ± 0.16 | 1.69 ± 0.22 | 11.39 ± 2.11 | 4.9 | 0.36 | 0.9 |
| 5b | Me | Et | S-(CH2)2-CN | MeO | 3.23 ± 0.19 | 15.63 ± 1.37 | 23.23 ± 2.53 | 3.9 | 0.37 | 1.6 |
| 6 | Me | H | S-(CH2)2-CN | MeO | > 100 | > 100 | > 100 | 2.5 | ||
| 7 | H | CO2E | S-(CH2)2-CN | MeO | 4.74 ± 0.24 | 19.43 ± 0.86 | >100 | 3.0 | 0.33 | 2.4 |
| t | ||||||||||
| 8 | H | CO2E | S-(CH2)2-CN | H | 24.42 ± 1.79 | > 100 | > 100 | 2.7 | 0.31 | 1.9 |
| t | ||||||||||
| 9 | Me | CO2E | S-(CH2)2-CN | H | 24.37 ± 0.53 | 10.80 ± 0.91 | 41.53 ± 6.23 | 3.2 | 0.30 | 1.4 |
| t | ||||||||||
| 10 | Et | CO2E | S-(CH2)2-CN | H | 60.63 ± 4.72 | 47.92 ± 7.84 | 94.17 ± 6.23 | 3.8 | 0.26 | 0.5 |
| t | ||||||||||
| 11 | nPr | CO2E | S-(CH2)2-CN | H | > 100 | > 100 | > 100 | 4.3 | ||
| t | ||||||||||
| 12 | Me | nBu | s-(ch2)2-c=ch | MeO | 11.60 ± 1.39 | > 100 | > 100 | 5.4 | 0.31 | −0.5 |
| 13 | Me | nBu | S-(CH2)3-CN | MeO | 35.23 ± 7.64 | 47.60 ± 3.51 | 46.06 ± 13.04 | 5.3 | 0.26 | −0.8 |
| 14 | Me | nBu | S-CH2-CN | MeO | > 100 | 4.6 | ||||
| 15 | Me | nBu | S-(CH2)3-OH | MeO | 17.35 ± 0.84 | > 100 | > 100 | 5.0 | 0.30 | −0.2 |
| 16 | Me | nBu | S-(CH2)2-Ph | MeO | > 100 | 6.7 | ||||
| 17 | Me | CO2E | NH-(CH2)-CN | H | >100 | 3.7 | ||||
| t | ||||||||||
| 18b | Me | nBu | S-(CH2)2-CN | H | 2.46 ± 0.16 | > 100 | > 100 | 4.6 | 0.38 | 1.0 |
| 19b | Me | nBu | S-(CH2)2-CN | Br | 21.55 ± 2.38 | > 100 | > 100 | 5.5 | 0.30 | −0.9 |
| 20 | H | CO2E | S-(CH2)2-CN | F | 7.77 ± 0.38 | 33.48 ± 9.61 | 37.52 ± 10.69 | 2.9 | 0.33 | 2.2 |
| t | ||||||||||
| 21 | H | CO2E | S-(CH2)2-CN | Me | 6.97 ± 0.54 | 27.49 ± 2.40 | > 100 | 3.2 | 0.33 | 1.9 |
| t | ||||||||||
| 22 | H | CO2E | S-(CH2)2-CN | Cl | 10.39 ± 0.80 | > 100 | > 100 | 3.5 | 0.32 | 1.5 |
| t | ||||||||||
| 23 | H | CO2E | S-(CH2)2-CN | Acetyl | > 100 | 2.0 | ||||
| t | ||||||||||
| 24 | H | CO2E | S-(CH2)2-CN | CH3-CH(OH) | > 100 | 2.8 | ||||
| t | ||||||||||
Apparent Ki values of tested compounds on the enzymatic activity of recombinant P. vivax NMT and Homo sapiens NMT isoforms 1 and 2. Each Ki is the mean ± SD from duplicates.
Purchased compound from Interbioscreen Ltd. Purity > 85% based on RP-HPLC/MS analysis.
cLog P values were calculated with ChemDraw for Excel version 12.0.2.
LE: ligand efficiency (PvNMT); LE = −RTln(Kiapp)/N where N is the number of non-hydrogen atoms of the compound.
LipE: lipophilic efficiency (PvNMT); LipE = pIC50 - cLog P.
Scheme 2. Synthetic pathways to derivatives 7-11 and 20-24a.

aReagents and conditions: (i) 140 °C, 1 h; (ii) Ph2O, reflux, 2 h; (iii) NaH, N,N-dimethylacetamide, 120 °C, 20 min; (iv) POCl3, 110 °C, 20 min; (v) 3-mercaptopropanenitrile, K2CO3, THF, reflux, 4 h.
These molecules were evaluated for their inhibitory potency against P. vivax NMT as well as for their selectivity versus human NMT isoforms 1 and 2 (HsNMT1 and HsNMT2 respectively) (Table 1).
Reducing the length of the n-alkyl chain in position 3 (compounds 1, 5, 6) resulted in a progressive loss of activity towards PvNMT as chain length was shortened. However, the replacement of this aliphatic chain by an ethyl ester (compound 7) greatly improved solubility and selectivity, particularly over HsNMT2, while retaining a good activity versus PvNMT. The n-alkyl group is enclosed in an apolar pocket though there is the potential for the ester group to form polar interactions with residue Asn365. Extension of the alkyl chain in position 2 of the quinoline scaffold (entries 9-11) led to a dramatic loss of activity against PvNMT. Consistent with these observations, the 2-methyl group in the parent compound fits snugly into a protein pocket in which the binding of bulkier substituents would lead to steric hindrance. Interestingly, compound 8 in which the 2-methyl group is deleted displayed a similar potency to 9, but with an improved selectivity over human NMTs.
Subtle modifications to the length as well as the composition of the 4-substituent chain (compounds 12-14) resulted in loss of activity. Attempts to establish π-π stacking interactions with the nearby aromatic residues Tyr211 and Tyr334, by introducing a phenyl ring in position 4 (compound 16), or to establish H-bonds with the His213 and Asn365 side chains, by introducing a hydroxyl group in place of the nitrile (compound 15), failed to improve activity. Similarly, substitution of the thioether linker with an amine (17) induced a complete loss of activity; unfortunately, the corresponding ether derivative could not be synthesized due to its chemical instability. The polarity and length of the 2-cyanoethylthioether moiety appear to be important as this group extends towards the side chains of Asn365, His213 and Tyr211. Finally a series of quinolines modified at position 6- were evaluated. While small substituents (entries 1, 18, 20-22) were tolerated by PvNMT, the presence of bigger or branched side chains (19, 23, 24) abolished inhibitory activity. In particular, the introduction of polar substituents (23, 24), with the potential to establish polar contacts with the protein carboxamide backbone, caused a dramatic loss of activity. These results suggest lack of flexibility of the protein around this position. Indeed, clashes can be predicted from the protein structure in this region, although there is potential for alterations as the methoxy group is oriented back towards the co-factor binding site and there are three water molecules within 4 Å of the methyl carbon, suggesting that certain modifications at this position might be tolerated. Interestingly, direct comparison of compounds 1 and 18 (as well as 7 and 8) shows that the removal of the 6-methoxy group induces a significant improvement of inhibition selectivity over HsNMT1 and HsNMT2. This result cannot be easily rationalized on the basis of the structure reported here, since the residues of PvNMT in contact with the inhibitor 1 are fully conserved in the human NMTs. The differences in observed activity probably arise from subtle changes in chain flexibility and residue orientation.
These optimizations led to the identification of compound 7, which exhibits micromolar activity against PvNMT, some selectivity over human NMT isoforms, and improved lead-like properties (cLog P = 3.0; LipE > 2; MW = 316) compared to the initial hit 1 (Table 1). 15
CONCLUSION
A high throughput screening campaign utilizing a fluorogenic assay has resulted in the identification of a small molecule, 1, with micromolar inhibitory activity against P. vivax NMT. Crystallization of this compound with PvNMT revealed the binding mode of this competitive inhibitor. A variety of small molecules based on the hit compound structure were synthesized and revealed some key attributes of 1. These findings constitute a starting point for the development of potent NMT inhibitors as potential therapeutics for vivax malaria, and for target validation studies.
EXPERIMENTAL SECTION
Chemistry
All chemicals were purchased from Sigma-Aldrich Ltd (UK), Acros Organics (Belgium) or VWR (UK). Reagents and solvents were used without any further purification. Purity of tested compounds was determined by RP-HPLC and was superior to 95%, unless specified. 1H and 13C NMR spectra were recorded on a Brüker AV-400 (400/100 MHz) spectrometer. Mass spectra and accurate mass data were obtained by J. Barton at the Chemistry Department Mass Spectrometry Service (Imperial College London) by electrospray ionization. The synthesis and structural characterization of all compounds is described in the Supplementary information document.
Accession code
The coordinates and structure factor files have been deposited in the Protein Data Bank under the accession code 4a95.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Prof Deborah Smith (University of York) and co-workers for helpful discussions and Marek Brzozowski for expert crystal handling. We acknowledge the European Synchrotron Radiation Facility for provision of excellent synchrotron radiation facilities.
Funding Sources
This work was supported by the Medical Research Council (MRC; grant G0900278 and U117532067), the Wellcome Trust (grant 087792) and the Biotechnology and Biological Sciences Research Council (David Phillips Research Fellowship to EWT, grant BB/D02014X/1).
Glossary
ABBREVIATIONS
- Ca
Candida albicans
- DMF
N,N-dimethylformamide
- ESRF
European Synchrotron Radiation Facility
- Hs
Homo sapiens
- NHM
S-(2-oxo)pentadecyl-coenzyme A
- NMT
N-myristoyltransferase
- PTSA
p-toluenesulfonic acid
- Pv
Plasmodium vivax
- Sc
Saccharomyces cerevisiae
- SD
standard deviation
- siRNA
small interfering RNA
- Tb
Trypanosoma brucei
REFERENCES
- 1.Mueller I, Galinski MR, Baird JK, Carlton JM, Kochar DK, Alonso PL, del Portillo HA. Key gaps in the knowledge of Plasmodium vivax, a neglected human malaria parasite. Lancet Infect. Dis. 2009;9:555–566. doi: 10.1016/S1473-3099(09)70177-X. [DOI] [PubMed] [Google Scholar]
- 2.Price RN, Tjitra E, Guerra CA, Yeung S, White NJ, Anstey NM. Vivax malaria: neglected and not benign. Am. J. Trop. Med. Hyg. 2007;77:79–87. [PMC free article] [PubMed] [Google Scholar]
- 3.Wright MH, Heal WP, Mann DJ, Tate EW. Protein myristoylation in health and disease. J. Chem. Biol. 2009;3:19–35. doi: 10.1007/s12154-009-0032-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bowyer PW, Tate EW, Leatherbarrow RJ, Holder AA, Smith DF, Brown KA. N-myristoyltransferase: a prospective drug target for protozoan parasites. ChemMedChem. 2008;3:402–408. doi: 10.1002/cmdc.200700301. [DOI] [PubMed] [Google Scholar]
- 5.Price HP, Menon MR, Panethymitaki C, Goulding D, McKean PG, Smith DF. Myristoyl-CoA:protein N-myristoyltransferase, an essential enzyme and potential drug target in kinetoplastid parasites. J. Biol. Chem. 2003;278:7206–7214. doi: 10.1074/jbc.M211391200. [DOI] [PubMed] [Google Scholar]
- 6.Frearson JA, Brand S, McElroy SP, Cleghorn LA, Smid O, Stojanovski L, Price HP, Guther ML, Torrie LS, Robinson DA, Hallyburton I, Mpamhanga CP, Brannigan JA, Wilkinson AJ, Hodgkinson M, Hui R, Qiu W, Raimi OG, van Aalten DM, Brenk R, Gilbert IH, Read KD, Fairlamb AH, Ferguson MA, Smith DF, Wyatt PG. N-myristoyltransferase inhibitors as new leads to treat sleeping sickness. Nature. 2010;464:728–732. doi: 10.1038/nature08893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goncalves V, Brannigan JA, Thinon E, Olaleye TO, Serwa R, Lanzarone S, Wilkinson AJ, Tate EW, Leatherbarrow RJ. A fluorescence-based assay for N-myristoyltransferase activity. Anal. Biochem. 2012;421:342–344. doi: 10.1016/j.ab.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brannigan JA, Smith BA, Yu Z, Brzozowski AM, Hodgkinson MR, Maroof A, Price HP, Meier F, Leatherbarrow RJ, Tate EW, Smith DF, Wilkinson AJ. N-myristoyltransferase from Leishmania donovani: structural and functional characterisation of a potential drug target for visceral leishmaniasis. J. Mol. Biol. 2010;396:985–999. doi: 10.1016/j.jmb.2009.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farazi TA, Waksman G, Gordon JI. Structures of Saccharomyces cerevisiae N-myristoyltransferase with bound myristoylCoA and peptide provide insights about substrate recognition and catalysis. Biochemistry. 2001;40:6335–6343. doi: 10.1021/bi0101401. [DOI] [PubMed] [Google Scholar]
- 10.Sogabe S, Masubuchi M, Sakata K, Fukami TA, Morikami K, Shiratori Y, Ebiike H, Kawasaki K, Aoki Y, Shimma N, D’Arcy A, Winkler FK, Banner DW, Ohtsuka T. Crystal structures of Candida albicans N-myristoyltransferase with two distinct inhibitors. Chem. Biol. 2002;9:1119–1128. doi: 10.1016/s1074-5521(02)00240-5. [DOI] [PubMed] [Google Scholar]
- 11.Brand S, Cleghorn LA, McElroy SP, Robinson DA, Smith VC, Hallyburton I, Harrison JR, Norcross NR, Spinks D, Bayliss T, Norval S, Stojanovski L, Torrie LS, Frearson JA, Brenk R, Fairlamb AH, Ferguson MA, Read KD, Wyatt PG, Gilbert IH. Discovery of a novel class of orally active trypanocidal N-myristoyltransferase inhibitors. J. Med. Chem. 2012;55:140–152. doi: 10.1021/jm201091t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoglund IPJ, Silver S, Engstrom MT, Salo H, Tauber A, Kyyronen HK, Saarenketo P, Hoffren AM, Kokko K, Pohjanoksa K, Sallinen J, Savola JM, Wurster S, Kallatsa OA. Structure-activity relationship of quinoline derivatives as potent and selective alpha(2C)-adrenoceptor antagonists. J. Med. Chem. 2006;49:6351–6363. doi: 10.1021/jm060262x. [DOI] [PubMed] [Google Scholar]
- 13.Fryer RI, Zhang P, Rios R, Gu ZQ, Basile AS, Skolnick P. Structure-Activity Relationship Studies at the Benzodiazepine Receptor (Bzr) - a Comparison of the Substituent Effects of Pyrazoloquinolinone Analogs. J. Med. Chem. 1993;36:1669–1673. doi: 10.1021/jm00063a017. [DOI] [PubMed] [Google Scholar]
- 14.Hayashi H, Miwa Y, Ichikawa S, Yoda N, Miki I, Ishii A, Kono M, Yasuzawa T, Suzuki F. 5-Ht3 Receptor Antagonists .2. 4-Hydroxy-3-Quinolinecarboxylic Acid-Derivatives. J. Med. Chem. 1993;36:617–626. doi: 10.1021/jm00057a011. [DOI] [PubMed] [Google Scholar]
- 15.Leeson PD, Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 2007;6:881–890. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.