

Abstract
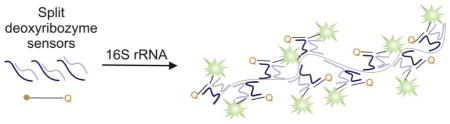
Sixty four DNA strands hybridize to 16S rRNA to form 32 deoxyribozyme catalytic cores that produce fluorescent signal. The approach allows detection of 0.6 pM 16S rRNA, or ~ 3×104 bacterial cells in a PCR-free format.
Keywords: Binary deoxyribozyme, fluorescent sensors, detection of bacteria, 16S rRNA, DNA origami
Conventional methods for the detection of pathogenic bacteria rely on either time-consuming culture growth or qPCR. These assays are sensitive to contaminations and require qualified personal or sophisticated equipment. Detection of nucleic acids directly from unamplified samples is of great importance for point-of-care (POC) diagnostic formats.[1] Such techniques would enable simple and straightforward tests that do not require expensive equipment or user expertise. Importantly, POC assays should allow fast and efficient recovery of nucleic acids from the sample and detect low concentrations of bacteria. Here, we propose a straightforward and simple assay that analyzes bacterial RNA both in samples of total RNA and in whole bacterial cells with high selectivity and low detection limit.
The idea of the assay was inspired by the DNA-origami technology.[2] In DNA origami, the phage M13mp18 DNA is folded into desired shapes with the help of hundreds of short oligonucleotide staple strands (Figure 1A). Importantly, the yield of the desired shape can reach up to 90%.[2,3] Misfolded structures are unfavourable due to the energetic sink that drives the hybridization towards the formation of only the most stable complete associate in the presence of excess amount of the staple strands.[2,3] If the produced origami shape could be easily detected by conventional methods, the approach would become adaptable for the detection of long nucleic acids, such as natural RNA molecules. In this study, we propose folding native bacterial RNA with the help of a series of functional “staple” strands into a specific nanostructure that produces fluorescent signal.
Figure 1.

The design of “deoxyribozyme-on-a-string structure” for nucleic acid analysis. A) Concept of DNA origami. B) Single binary (split) deoxyribozyme probe re-forms catalytic core in the presence of a specific analyte. C) Detection of 16S rRNA using ‘deoxyribozymes-on-a-string’ complex.
We have chosen bacterial 16S ribosomal RNA (rRNA) as a target for the proof-of-concept study. It is approximately 1500 nucleotides in length and is a component of small subunit of prokaryotic ribosome. 16S rRNA is highly conserved between different species of bacteria and archaea.[4] Along with highly conserved regions, it also contains hypervariable sequences that can provide species-specific signatures used for identification of prokaryotic organisms.[5] Moreover, multiple copies of rRNA are present in a single bacterial cell, representing an attractive target for the detection of low abundant bacteria without PCR amplification.
To develop a highly sensitive assay we took advantage of signal amplification property of deoxyribozymes, as it is shown in Figure 1B. In split or binary deoxyribozyme sensors, [6] two DNA strands containing fragments complementary to a target analyte (analyte-binding arms) and fragments complementary to a fluorophore- and quencher-labeled fluorogenic reporter substrate (F substrate) reform a catalytic core in the presence of a specific analyte. Active enzyme binds and cleaves the fluorogenic substrate, thus producing high fluorescence that can be easily detected by a conventional fluorometer.
In the present work, we combined the binary deoxyribozyme approach with the staple-assisted folding of a long nucleic acid inspired by the DNA origami to design a highly sensitive sensor for RNA detection. In the proposed assay, an RNA folded in a stable secondary structure is incubated with a multitude of functional staple strands (deoxyribozyme precursors) and a fluorogenic reporter substrate. Hybridization of the staples to the RNA unwinds its secondary structure to form “deoxyribozymes-on-a-string” complex (Figure 1C). This associate contains multiple catalytically active deoxyribozymes, which cleave the reporter substrate, thus producing fluorescent signal. The entire length of RNA molecule is used for the signal production, and each active deoxyribozyme cleaves several copies of the fluorogenic substrate due to multiple catalytic turnovers. The combination of these two factors offers high level of signal amplification.
We used binary 10–23 deoxyribozyme developed by Mokany et. al.[6b] to design 32 binary deoxyribozyme sensors that cover almost the entire sequence of E. coli 16S rRNA (Table S1). The design of the sensors was uniform; the sequences of the analyte-binding arms were the only change made for each binary 10–23 probe (Figure S1). Testing the five individual 10–23 probes with 16S rRNA transcript revealed the detection limits of 1.3-0.4 ng (16-5 pM) of 16S rRNA (Figure S2), which correlated with the limit of detection (LOD) of 5 pM for 10–23 split deoxyribozyme probe reported earlier.[6b] In order to enable the 32 deoxyribozyme probes operate together in the “deoxyribozyme-on-a-string” complex, the adjacent catalytic cores were designed to face the opposite sides of the double-stranded RNA-DNA helix by separating them by ~4.5 helical turns. The performance of the sensor was evaluated using a purified transcript of 16S rRNA. The 64 DNA staple strands formed an associate with the RNA transcript, which had an electrophoretic mobility corresponding to the predicted “deoxyribozyme-on-a-string structure” (Figure S3). The sensor was able to detect as low as ~50 pg of 16S rRNA transcript in a 150 μl-sample, or ~0.6 pM RNA analyte (Figure S4). This LOD was significantly lower than that of the individual binary 10–23 probes. This indicates that the series of binary deoxyribozyme probes bound to the RNA ‘string’ retained their catalytic functions and were able to cleave the fluorogenic substrate simultaneously according to the hypothesis shown in Figure 1C.
The sequence of 16S rRNA is highly conserved among bacteria species. For example, B. subtilis and E. coli have 79% of 16S rRNA sequence identical (Figure S5). For the reliable detection of a targeted bacterium, the sensor should be selective enough to differentiate between bacterial species. To test the selectivity of the “deoxyribozyme-on-a-string” approach, we designed the sensor targeting B. subtilis 16S rRNA (see Table S1 for sequences).
We tested the performance of both E. coli- and B. subtilis-specific sensors in the presence of total RNA from either E. coli or B. subtilis. It was found that the signal above the background can be achieved in the presence of ~0.3 ng of total bacterial RNA (Figure S6). For both sensors, an about two-fold increase in fluorescence was observed in the presence of the target RNA, while the non-target RNA triggered fluorescence almost as low as the background (Figure 2), thus confirming the high selectivity of the assay. This result is not surprising since for the catalytic core to be re-formed, a pair of deoxyribozyme strands needs to be bound to the target simultaneously. Therefore, for good selectivity it is important to avoid perfect complementarity of both strands of each non-specific sensor to the target RNA. In our design, there was no completely complementary pair of strands (Figure S5). Comparative analysis of the two bacterial 16S rRNA sequences revealed that only three deoxyribozyme strands were fully complementary to both specific and non-specific 16S rRNA molecules (see Table S1 and Figure S5). Even though some strands have analyte-binding arms perfectly complementary to both bacterial RNAs, if a considerable number of the strands are out of the complex, the equilibrium might be totally shifted towards the free folded rRNA.
Figure 2.

Selectivity of the ’deoxyribozymes-on-a-string’ sensor. E. coli- or B. subtilis-specific probes (grey or white bars, respectively) were incubated in the absence or presence of 10 ng total RNA from E. coli or from B. subtilis for 1 h. The data is an average value from two independent measurements.
RNA isolation is a simple but time-consuming procedure. Therefore, we next tested the possibility to detect 16S rRNA in a single assay starting form the whole bacterial cells. Different amounts of E. coli cells were boiled in the presence of E. coli-specific multicomponent deoxyribozyme sensor followed by incubation of the complex in the presence of the fluorogenic reporter. Fluorescence of the samples increased with increasing the cells amount (Figures 3A and S7). According to our estimation, 1×105 E. coli cells were sufficient to produce the signal above the background after 1–3 h assay. Prolonged incubation allowed reducing the detection limit to 3×104 cells, which are present in ~ 20 nL of a bacterial culture at the end of the exponential growth phase. By lowering the sample volume to 2–10 μL, which can be easily measured by the NanoDrop 3300 fluorospectrometer, for example, it is possible to decrease the limit of detection to ~4×102–2×103 cells after overnight incubation.
Figure 3.

Detection of whole bacterial cells. A) Dependence of fluorescence for E. coli-specific sensor on the amount of E. coli cells. B) Sensor selectivity. Fluorescent spectra for E. coli- and B. subtilis-specific probes in the absence or presence of E. coli cells. All spectra were recorded after 3 h incubation at 54 °C.
In conclusion, we have applied recent findings of DNA nanotechnology[2] and the advantages in the design of deoxyribozyme sensors[6] to develop a PCR-free assay that can detect specific RNA molecules in a sample of total bacterial RNA or starting from the whole cells. The limit of 16S rRNA detection was found to be 0.6 pM in case of isolated RNA transcript, or ~3×104 E. coli cells, which potentially can be further lowered to 400 cells. These limits of detection are within the range of the amount of some pathogenic bacteria in clinical samples. For example, a typical sputum sample of a Mycobacterium tuberculosis-infected individual may contain 105–106 cells/ml.[7] Using RNAse inhibitors that prevent RNA degradation in the whole-cell assay may further improve the detection limit. The assay is selective, since little or no fluorescence increase was observed in the presence of RNA from a non-targeted bacterium. The design of the sensor was straightforward and universal. Despite using multiple sensors, the reagent cost is expected to be low, since the deoxyribozyme sensors are inexpensive synthetic oligonucleotides, while the fluorogenic signal reporter is universal for all probes and, therefore, can be synthesized in bulk amount and used efficiently. The approach is potentially applicable to the detection of any folded RNA molecules including mRNA and thus can be adopted for the detection of drug-resistant bacterial strains expressing a gene responsible for the resistance. Further development of this approach may deliver a straightforward, fast and inexpensive test for POC detection of bacterial infections.
Experimental Section
For the analysis of E. coli 16S rRNA, E. coli-specific strands A1–A32 and B1–B32 (10 nM each) were annealed with isolated 16S rRNA (0.16–1.3 ng/mL) by incubating the mixtures in a buffer containing 10 mM Tris-HCl, pH 8.3, 50 mM KCl, 0.01% Triton X-100, 25 mM MgCl2 (reaction buffer) at 95°C for 5 min. The mixtures were cooled down to room temperature for 20 min. The complexes formed between 16S rRNA and the strands were isolated using centrifugal filters with MWCO 30 kDa (Millipore), washed two times with the reaction buffer and mixed with the reporter oligonucleotide (200 nM) in the reaction buffer. The final reaction mixtures were incubated at 54°C for 1–3 h or overnight. Fluorescence spectra of the samples were recorded on a Perkin-Elmer (San Jose, CA) LS-55 Luminescence Spectrometer with a Hamamatsu xenon lamp (excitation at 485 nm; emission 517 nm). More detailed experimental procedure is enclosed in Supporting Information.
Supplementary Material
Acknowledgments
The authors are grateful to Evan Cornett for 16S rRNA sequences alignment. Support from NIHGRI (R21 HG004060), NIAID R15AI10388001A1, and NSF CCF (1117205) is greatly appreciated.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the authors.
Contributor Information
Yulia V. Gerasimova, Email: yugeras@gmail.com.
Dmitry M. Kolpashchikov, Email: dmk2111@gmail.com.
References
- 1.a) Cheng MM, Cuda G, Bunimovich YL, Gaspari M, Heath JR, Hill HD, Mirkin CA, Nijdam AJ, Terracciano R, Thundat T, Ferrari M. Curr Opin Chem Biol. 2006;10:11–19. doi: 10.1016/j.cbpa.2006.01.006. [DOI] [PubMed] [Google Scholar]; b) Kim J, Yoon MY. Analyst. 2010;135:1182–1190. doi: 10.1039/c0an00030b. [DOI] [PubMed] [Google Scholar]; c) Kaittanis C, Santra S, Perez JM. Adv Drug Deliv Rev. 2010;62:408–423. doi: 10.1016/j.addr.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Kolpashchikov DM. Chem Rev. 2010;110:4709–4723. doi: 10.1021/cr900323b. [DOI] [PubMed] [Google Scholar]; e) Rosi NL, Mirkin CA. Chem Rev. 2005;105:1547–1562. doi: 10.1021/cr030067f. [DOI] [PubMed] [Google Scholar]; f) Shinde SB, Fernandes CB, Patravale VB. J Control Release. 2012;159:164–180. doi: 10.1016/j.jconrel.2011.11.033. [DOI] [PubMed] [Google Scholar]
- 2.a) Rothemund PW. Nature. 2006;440:297–302. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]; b) Sacca B, Niemeyer CM. Angew Chem. 2012;124:60–69. [Google Scholar]; Angew Chem Int Ed Engl. 2012;51:58–66. [Google Scholar]
- 3.Barish RD, Schulman R, Rothemund PW, Winfree E. Proc Natl Acad Sci USA. 2009;106:6054–6059. doi: 10.1073/pnas.0808736106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Ludwig W, Schleifer KH. FEMS Microbiol Rev. 1994;15:155–173. doi: 10.1111/j.1574-6976.1994.tb00132.x. [DOI] [PubMed] [Google Scholar]; b) Weiss LM, Zhu X, Cali A, Tanowitz HB, Wittner M. Folia Parasitol. 1994;41:81–90. [PubMed] [Google Scholar]; c) Staley JT. J Ind Microbiol Biotechnol. 2009;36:1331–1336. doi: 10.1007/s10295-009-0642-8. [DOI] [PubMed] [Google Scholar]
- 5.a) Watanabe Y, Fujihara M, Obara H, Nagai K, Harasawa R. J Vet Med Sci. 2011;73:1657–1661. doi: 10.1292/jvms.11-0293. [DOI] [PubMed] [Google Scholar]; b) Indra A, Blaschitz M, Kernbichler S, Reischl U, Wewalka G, Allerberger F. J Med Microbiol. 2010;59:1317–1323. doi: 10.1099/jmm.0.020792-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Haugland RA, Varma M, Sivaganesan M, Kelty C, Peed L, Shanks OC. Syst Appl Microbiol. 2010;33:348–357. doi: 10.1016/j.syapm.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 6.a) Kolpashchikov DM. Chembiochem. 2007;8:2039–2042. doi: 10.1002/cbic.200700384. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mokany E, Bone SM, Young PE, Doan TB, Todd AV. J Am Chem Soc. 2010;132:1051–1059. doi: 10.1021/ja9076777. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gerasimova YV, Cornett E, Kolpashchikov DM. Chembiochem. 2010;11:811–817. doi: 10.1002/cbic.201000006. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Mokany E, Tan YL, Bone SM, Fuery CJ, Todd AV. Clin Chem. 2013;59:419–426. doi: 10.1373/clinchem.2012.192930. [DOI] [PubMed] [Google Scholar]
- 7.Kolwijck E, Mitchell M, Venter A, Friedrich SO, Dawson R, Diacon AH. J Clin Microbiol. 2013;51:1094–1098. doi: 10.1128/JCM.02751-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.