

Abstract
A new series of 4-aminopyridyl-based lead inhibitors targeting Trypanosoma cruzi CYP51 (TcCYP51) has been developed using structure-based drug design as well as structure-property relationship (SPR) analyses. The screening hit starting point, LP10 (KD ≤ 42 nM; EC50 of 0.65 µM), has been optimized to give the potential leads 14t, 27i, 27q, 27r, and 27t, that have low nanomolar binding affinity to TcCYP51 and significant activity against T. cruzi amastigotes cultured in human myoblasts (EC50 = 14–18 nM for 27i and 27r). Many of the optimized compounds have improved microsome stability, and most are selective against human CYPs 1A2, 2D6 and 3A4 (<50% inhibition at 1 µM). A rationale for the improvement of microsome stability and selectivity of inhibitors against human metabolic CYP enzymes is presented. In addition, the binding mode of 14t with the T. brucei CYP51 (TbCYP51) ortholog has been characterized by x-ray structure analysis.
Keywords: Chagas disease, non-azole CYP51 inhibitors, structure guided drug design, structure activity and property relationships, x-ray structure
INTRODUCTION
Chagas disease, or American trypanosomiasis is a chronic tropical infection caused by the protozoan parasite Trypanosoma cruzi. The infection can be lethal if untreated. Chagas disease is the leading cause of heart failure in Latin America.1 Although first described a century ago,2 it is still a major public health challenge in South America. Furthermore, many cases have been reported in North America, Europe, and Asia due to human population movements, migration of the triatomine insect vectors, HIV-coinfections, and contaminated blood transfusion.1b
Currently nifurtimox and benznidazole are the only drugs approved for treatment of Chagas disease.3 Although these drugs, which date from the late 1960s, show considerable efficacy in the acute stage of Chagas disease, their efficacy is debated in the chronic stage, which involves chronic Chagas cardiomyopathy, leading to congestive heart failure, thromboembolic phenomena, severe arrhythmias, and sudden unexpected death.3b, 4 Moreover, these old drugs are associated with frequent side effects such as dermatitis, gastrointestinal, and neurologic toxicities, and even a rare case of bone marrow suppression.1a Therefore, the need exists to develop new therapeutics bearing better safety profiles and improved efficacy to treat T. cruzi infections and prevent cardiovascular Chagas disease.
Sterol biosynthesis is a recognized target for the development of new therapeutic agents to treat T. cruzi infections.5 Sterol 14-demethylase (CYP51) has been successfully targeted for combating pathogenic fungal infections with azole drugs such as fluconazole, ketoconazole, and posaconazole, among others.6 CYP51 catalyzes the oxidative removal of the 14-methyl group of lanosterol and produces Δ14,15-unsaturated intermediates in ergosterol biosynthesis.5 Due to the similarity of sterols and their biosynthesis pathways in fungi and T. cruzi, the anti-parasitic effects of these azole drugs against T. cruzi in infected mammalian cells have been observed.7 Therefore, clinical trials of posaconazole and other antifungal agents in combination with benznidazole are underway for treatment of chronic Chagas disease.8 Recently, tipifarnib, a class of farnesyl transferase inhibitors, has been repurposed as an anti-parasitic agent in the laboratory setting.9 Hit-to-lead optimization of a new “NEU” series, identified via a HTS campaign at Northeastern University, has been achieved (Fig 1).10 In addition, x-ray co-crystal structures of T. cruzi CYP51 (TcCYP51) with bound posaconazole, fluconazole, VNF, and NEU321 have been determined for use in structure-based design approaches to the development of anti-Chagas agents.10–11 However, all of these lead compounds are azoles, and there is an emerging issue of the rapid appearance of laboratory-induced resistance to azoles in T. cruzi cell culture.12 Thus, the development of therapeutics with different scaffolds targeting TcCYP51 is an important undertaking.
Figure 1. Inhibitors of TcCYP51.

(A) Azole type CYP51 inhibitors. (B) Pyridinyl type CYP51 inhibitors.
Recently, non-azole hits of the LP10 series were identified from a HTS campaign at UC San Francisco.13 The binding modes of these non-azole hits were characterized, and their co-crystal structures with M. tuberculosis CYP51 (MtCYP51) were determined.13a A 60% cure rate was attained in a mouse model of T. cruzi infection using the non-azole CYP51 inhibitor LP10.14 Accordingly, in an effort to develop more potent non-azole CPY51 inhibitor leads as anti-Chagas agents, we embarked on the optimization of LP10 by using structure-based drug design considerations in conjunction with in vitro DMPK analysis (microsome stability and CYP inhibition) to drive rounds of inhibitor optimization. In particular, we strived to increase compound stability in human, rat, and mouse liver microsome preparations, while retaining or increasing inhibition potency toward T. cruzi in infected mammalian cells, in order to identify candidates with properties appropriate to advance into animal models of T. cruzi infection. In addition, selectivity against human cytochrome P450 enzymes CYP1A2, CYP2C9, CYP2D6, and CYP3A4, the most important CYPs involved in drug metabolism and drug-drug interactions15, has also been monitored and used for compound prioritization in our structure optimization efforts.
Results and Discussion
Identification of the most active enantiomer of LP10
The original hit (LP10) is a mixture of two racemic diastereomers: S- and R-isomers at the tryptophan unit and cis/trans-isomers within the methylcyclohexane ring. We began by identifying the most active enantiomer of the LP10 scaffold as a starting point for structure optimization. To eliminate the impact of cis/trans isomerization in the methylcyclohexane ring, we evaluated two enantiomerically pure LP10 analogs with a simple cyclohexyl unit (Table 1). The S-isomer 1 has ~30-fold better binding affinity (KD) toward T. cruzi CYP51 than the R-isomer 2. This result is consistent with the co-crystal structures of LP9 (the methionine analog of LP10) and of LP11 (the valine analog of LP10) with MtCYP51, which have S-enantiomers in the bound structures.13a In addition, the S-isomer 1 had two-fold higher potency (EC50) against T. cruzi in infected cells than the R-isomer. Both isomers had similar microsome stability and both were potent inhibitors of human CYP enzymes 2C9, 2D6 and 3A4 (≥90% inhibition at 10 µM). Accordingly, we pursued S-enantiomers of LP10 analogs in the development of non-azole CYP51 leads.
Table 1.
Biochemical and cell-based activities, microsome stability and CYP inhibition properties of LP10 and analogs 1 and 2
| compound | KD (nM) |
EC50 (µM) |
Microsome stability t1/2 (min)a |
% inhibition of human CYPs at 10 µM (unless indicated otherwise) |
||||
|---|---|---|---|---|---|---|---|---|
| h | m | 1A2 | 2C9 | 2D6 | 3A4 | |||
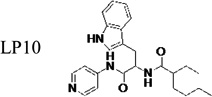 |
≤42 | 0.65 | - | - | - | - | - | - |
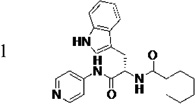 |
≤5 c | 0.68 | 3.8 | 4.2 | 41 (23)d | 99 (92)d | 93 (77)d | 96 (75)d |
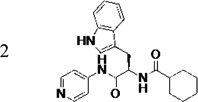 |
140 | 1.5 | 6.5 | 6.9 | 55 (21)d | 99 (92)d | 96 (62)d | 99 (77)d |
| Sutentb | - | - | 30 | 11 | - | - | - | - |
| Furafylline (40 µM)b | - | - | - | - | 86 | 5 | 4 | 8 |
| Sulfaphenazoleb | - | - | - | - | 20 | 92 | 7 | 21 |
| Quinidineb | - | - | - | - | 23 | 9 | 90 | 36 |
| Ketoconazole (1 µM)b | - | - | - | - | 22 | 22 | 4 | 95 |
Stability of compounds in human (h) and mouse (m) liver microsomes, using sunitinib as a reference control.
Reference compounds for microsome stability and human CYP inhibition
KD of ≤5 nM (a hundredth of the target concentration) was estimated from the titration curves at 0.5 µM TcCYP51 for the tightest binding inhibitors, if a plateau was reached at the stoichiometric enzyme-inhibitor ratio.
Values in parentheses are % inhibition of the indicated human CYPs at 1 µM.
Structure activity relationship of initial LP10 analogs
At the outset of these studies, the x-ray structure of TcCYP51 with bound LP10 was not available. Thus, design of initial analog sets was guided by our previous studies on MtCYP51 demonstrating that the hydrophobic contacts experienced by the indole ring accounts for ca. 100-fold increased binding affinity of LP10 toward TcCYP51 (KD ≤ 42 nM), compared to LP9 and LP11 (KD = 6,900 and 9,200 nM, respectively) which have smaller (methylthio)ethyl and isopropyl groups (methionine and valine side chains), respectively.13a
First, we explored the role of the aromatic nitrogen atom in drug-target interactions. Introduction of a methoxy substituent at the 2-position of the 4-acylaminopyridine unit (3a in Table 2) resulted in loss of binding affinity toward TcCYP51; 3a was also inactive against T. cruzi in infected cells even at 10 µM, the highest concentration tested. Similarly, 3b with a 3,5-dimethylisoxazole unit as a pyridine replacement did not bind to CYP51 and was not active against the parasite in cell-based assays. These results indirectly confirm the binding mode of LP10 that requires coordination of the pyridine nitrogen to the heme iron. Thus, a functional group that hinders the pyridine nitrogen will destabilize the interaction with the heme iron, resulting in a loss of biochemical and cell-based activity. Consistent with this analysis, the ability of 3a and 3b to inhibit human CYP enzymes was also significantly decreased compared to LP10.
Table 2.
Biochemical and cell-based activities, microsome stability and CYP inhibition properties of inhibitors 3.a
 |
KD (nM) |
EC50 (µM) |
Microsome stability t1/2 (min) |
%inhibition of human CYPs (at 10 µM) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | h | rat | m | 1A2 | 2C9 | 2D6 | 3A4 | |||
| 3a | n/bb | n/ec | 5 | 2 | 2 | 25 | 16 | 30 | 50 | |||
| 3b | n/b | n/e | 4 | 4 | 2 | 24 | 39 | 5 | 22 | |||
| 3c | n/b | n/e | >120 | 59 | 47 | 0 | 25 | 37 | 25 | |||
| 3d | n/b | n/e | 97 | 53 | 51 | 2 | 25 | 63 | 19 | |||
| 3e | n/b | n/e | 45 | 32 | 11 | 6 | 61 | 65 | 54 | |||
| 3f | n/b | n/e | 16 | 39 | 7 | −26 | 13 | 69 | 23 | |||
| 3g | <5 | - | - | - | - | - | - | - | - | |||
| 3h | 91 | - | - | - | - | - | - | - | - | |||
| 3i | <5 | 3.0 | 3 | 3 | 2 | 22 | 96 | 91 | 96 | |||
| 3j | 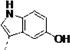 |
220 | n/e | 86 | 26 | 21 | 12 | 98 | 89 | 94 | ||
| 3k | 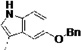 |
<5 | 0.58 | 4 | 3 | 2 | 76 | 99 | 98 | 99 | ||
| 3l | 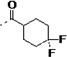 |
792 | 3.2 | 6 | 7 | 5 | 14 | 98 | 86 | 94 | ||
| 3m | <5 | 0.90 | 5 | 3 | 2 | 26 | 99 | 93 | 98 | |||
| 3n | <5 | 0.61 | 6 | 3 | 2 | 44 | 98 | 91 | 99 | |||
| 3o | <5 | 0.30 | 4 | 3 | 3 | 64 | 99 | 96 | 97 | |||
| 3p | 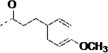 |
12 | 1.6 | 4 | 4 | 2 | 33 | 99 | 91 | 99 | ||
| 3q | 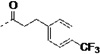 |
14 | 0.85 | 3 | 3 | 3 | 54 | 99 | 96 | 99 | ||
| 3r | <5 | 0.16 | 6 | 3 | 3 | 51 | 96 | 84 | 86 | |||
Second, analogs with secondary or tertiary amine units, such as 3c, 3d, 3e, and 3f, were also found to have substantially reduced biochemical and cell-based potency, presumably because they are unable to bind in the TcCYP51 active site which accommodates the highly lipophilic natural substrate, lanosterol. However, the microsome stability of 3c, 3d, 3e, and 3f was significantly increased (30–120 min half-life), and inhibition of human CYP enzymes by these compounds was greatly decreased relative to LP10. Therefore, balancing the charge or polarity distribution of inhibitor analogs was viewed as an important factor to address in the development of more active analogs.
Third, the indole ring of LP10 was replaced with several isosteres. While 3g and 3i possessing 3-benzothiophene and N-methyl indole have similar binding affinity to CYP51 as compared to 1 (KD ≤5 nM), analogs 3h and 3j possessing 1-naphthyl and 5-hydroxy indole units have substantially decreased binding affinity (KD = 91 and 220 nM, respectively). In contrast, introduction of larger hydrophobic substituents at the 5-position of the indole (as in analog 3k) did not reduce binding affinity to TcCYP51, suggesting that room in the binding site is available to accommodate the bulky substituents at the 5-position of the indole ring (discussed subsequently in a context of the x-ray structure) without the loss of binding affinity and inhibition potency.
Finally, in order to explore the site of cyclohexane ring binding in TcCYP51, the cyclohexanecarboxamide unit was replaced with other aliphatic carboxylamides containing a terminal phenyl ring. We found that analogs 3m – 3r have similar binding affinity to TcCYP51 and comparable inhibition potency against T. cruzi in infected cells, compared to those of 1. However, all of these non-azole inhibitors were rapidly degraded by liver microsome preparations in our standard stability assay using 1 mg/mL hepatic microsomal protein (t1/2 = < 6 min).
Based on the results of this early stage SAR effort (Table 2), plans to increase inhibitor potency of analogs of 1 were formulated and included strategic decisions to: (1) retain the 4-acylamimopyridine moiety as a heme binding unit; (2) retain the indole ring or extend the 5-position of the indole; and, most importantly, (3) to identify replacements for the methylcyclohexane ring to increase microsome stability and decrease inhibition of drug-metabolizing CYP enzymes.
Synthesis of LP10 analogs (1–3r)
Enantiomeric pure 1 was synthesized by the sequence summarized in Scheme 1. Briefly, the S-enantiomer of N-Boc tryptophan (L-tryptophan) was coupled with 4-aminopyridine to produce 5. Treatment of 5 with 4N HCl in dioxane followed by treatment of the deprotected amine with cyclohexanecarbonyl chloride produced enantiomerically pure 1. A similar sequence, starting from R-N-Boc tryptophan, was used to synthesize 2 (not shown). Analogs 3a and 3b were synthesized by replacing 4-aminopyridine with 4-amino-2-methoxypyridine and 4-amino-3,5-dimethylisoxazole, respectively. Acylation of 6 with N-Boc-isonipecotic acid followed by treatment with TFA to effect deprotection of the Boc unit provided 3c, isolated as the HCl salt. Amine salt 3f was obtained by the alkylation of 6 and benzyl bromide. Various carboxylic acids were coupled with 6 using pentafluorophenyl trifluoroacetate as the dehyrdrating agent16 to generate 3d, 3e, and 3m – 3r. Replacements for the indole moiety of 1 were explored by using commercially available 5-hydroxytryptophan 10 as a starting material. The carboxyl and amine groups were blocked as the methyl ester and t-butyl carbamate (Boc), respectively, and 11 was treated with benzyl bromide and Cs2CO3 in acetone to produce 12. Hydrolysis of the methyl ester unit of 12 produced 13, from which 3k was obtained by following the procedure for the synthesis of 1.
Scheme 1.

Reagents and conditions: (a) PyBOP, HOBt, NEt3, CH2Cl2, 4-aminopyridine, 0 °C to room temp., 1h, 94%. (b) 4N HCl in dioxane, dioxane, room temp., 12h, >90% (crude) (c) cyclohexancarbonyl chloride, NEt3, CH2Cl2, 0 °C to room temp., 1h, 94%. (d) PyBOP, HOBt, NEt3, CH2Cl2, 4-amino-2-methoxypyridine or 4-amino-3,5-dimethylisoxazole, 0 °C to room temp., 1h, 84%. (e) pentafluorophenyl trifluoroacetate, 1-Boc-isonipecotic acid, NEt3, CH2Cl2, 0 °C to room temp., 1h, 53%. (f) trifluoroacetic acid, CH2Cl2, room temp., 1h, 47%. (g) benzylbromide, NEt3, CH2Cl2, room temp., 12h, 30%. (h) pentafluorophenyl trifluoroacetate, NEt3, CH2Cl2, alkyl carboxylic acids, 0 °C to room temperature, 1h, ~80%. (i) SOCl2, CH3OH, 0 °C to room temp., 12h, then (Boc)2O, NEt3, CH2Cl2, 0 °C to room temp., 6h, 82%. (j) benzylbromide, Cs2CO3, acetone, room temp., 12h, 77%. (k) 10% NaOH, CH3OH, 0 °C, 2h, 97%. (l) H2, Pd/C, CH3OH/THF, room temp., 24h, 29%.
Cyclohexyl ring replacement
Based on the initial SAR/SPR analysis, a series of inhibitors were synthesized by using various carboxylic acids to replace the cyclohexylcarboxamide unit of LP10 (Scheme 2 and Table 3). The objective was to increase microsome stability and selectivity against human CYP enzymes, while retaining or increasing inhibition potency against T. cruzi in infected cells. First, a benzamide was used to replace the cyclohexylcarboxamide moiety of 1. The binding affinity (KD = ≤5 nM), T. cruzi inhibition potency (EC50 = 0.39 nM), microsome stability and inhibition of human CYPs of 14a were similar to those of 1 (Table 3). The successful replacement of the cyclohexylcarboxamide encouraged us to explore additional substituted benzamide derivatives. Because the phenyl ring of the benzamide is a potential site of metabolism by human CYP enzymes,17 we anticipated that substitution at the appropriate “soft sites” of the aryl ring could block metabolic oxidation reactions and lead to inhibitors with enhanced microsome stability, while at the same time exploring available space in the active site and hopefully enhancing inhibitor potency against CYP51.
Scheme 2.

(a) PyBOP, HOBt, NEt3, CH2Cl2, benzoic acids or naphthoic acids, 0 °C to room temp., 1h, typically 80%.
Table 3.
Biochemical and cell-based activities, microsome stability and CYP inhibition properties of inhibitors 14.a
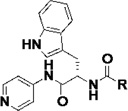 |
KD (nM) |
EC50 (µM) |
Microsome stability t1/2 (min) |
%inhibition of human CYPs (at 1 µM, unless indicated otherwise) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| R | h | Rat | m | 1A2 | 2C9 | 2D6 | 3A4 | |||
| 3r | <5 | 0.16 | 6 | 3 | 3 | 51b | 96b | 84b | 86b | |
| 14a | <5 | 0.39 | 4 | 9 | 4 | 18b | 95b | 88b | 90b | |
| 14b | <5 | 0.11 | 12 | 6 | 3 | 19b | 98b | 90b | 98b | |
| 14c | <5 | 0.25 | 8 | 9 | 5 | 48b | 99b | 97b | 98b | |
| 14d | <5 | 0.88 | 5 | 7 | 5 | 78b | 99b | 98b | 91b | |
| 14e | <5 | 0.66 | 7 | 4 | 4 | 58b | 99b | 95b | 91b | |
| 14f | 38 | 0.35 | 8 | 8 | 6 | 46b | 99b | 92b | 92b | |
| 14g | 80 | 0.59 | 6 | 11 | 4 | 28b | 96b | 95b | 97b | |
| 14h | 95 | 0.21 | 6 | 9 | 3 | 29 | 86 | 64 | 80 | |
| 14i | 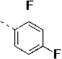 |
<5 | 0.33 | 25 | 6 | 4 | 32 | 89 | 59 | 94 |
| 14j | 60 | 0.036 | 9 | 8 | 4 | 42 | 94 | 86 | 90 | |
| 14k | 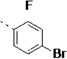 |
30 | 0.13 | 12 | 4 | 4 | 34 | 93 | 76 | 94 |
| 14l | <5 | 0.26 | 11 | 9 | 8 | 76b | 99b | 96b | 95b | |
| 14m | 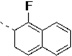 |
<5 | 0.54 | 18 | 7 | 6 | 47 | 97 | 88 | 91 |
| 14n | <5 | 0.33 | 14 | 9 | 8 | 18 | 94 | 86 | 65 | |
| 14o | 50 | 0.46 | 29 | 23 | 18 | 41 | 95 | 73 | 77 | |
| 14p | 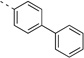 |
<5 | 0.55 | 19 | 19 | 23 | 67b | 99b | 95b | 92b |
| 14q | 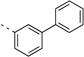 |
<5 | 0.89 | 10 | 6 | 5 | 80b | 99b | 98b | 92b |
| 14r | 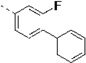 |
50 | 0.37 | 16 | 15 | 25 | 36 | 87 | 61 | 56 |
| 14s |  |
<5 | 0.30 | 16 | 20 | 24 | 32 | 82 | 58 | 68 |
| 14t |  |
<5 | 0.19 | 17 | 25 | 36 | 31 | 85 | 54 | 73 |
See notes to Table 1
CYP inhibition for this compound was performed at 10 µM
Fluoro, chloro, bromo and other substituents were added to the benzamide unit in attempts to block the potential soft site(s) on the phenyl ring of 14a. Of the set of inhibitors 14b – 14k that were synthesized in this effort, several had increased activity against T. cruzi in cell culture (14b, 14h, 14j, 14k) and while retaining microsome stability. Acyl groups with larger naphthyl and biaryl units were used in attempts to further improve the microsome stability (14l – 14t). Gratifyingly, many of these inhibitors had increased microsome stability, while retaining inhibitor potency. For instance, substitution of a fluorine or a bromine at the 6-position of the naphthyl ring led to 14n and 14o with enhanced microsome stability compared to 1. Analogs with biaryl units also had enhanced microsome stability (14p, 14r, 14s, and 14t) compared to 1 and 14a. Of these, analog 14t was of particular interest as it displayed good inhibition potency (EC50 = 0.19 µM) against T. cruzi in infected cells and moderate (but improved) microsome stability (t1/2 = 17/25/36 min) against human/rat/mouse liver microsomes.
X-ray structure of 14t complexed with TbCYP51
Although inhibitor design ultimately targeted TcCYP51, the best co-crystals with 14t that diffracted to 2.67 Å were obtained for the TbCYP51 ortholog (85% sequence identity). The majority of the first tier active site residues are identical between these two parasite CYP51 enzymes, with the exception of substrate-specific F105, which is isoleucine in the T. cruzi counterpart. With this difference, the structural information obtained for the TbCYP51-14t complex facilitated further rounds of structure-based design of TcCYP51 inhibitors.
Inhibitor 14t bound in the active site of CYP51 has several interesting features. First, the pyridine nitrogen of the 4-acylaminopyridine unit is coordinated to the heme iron (Fig 2), as expected from the series of the co-structures for the LP10 analogs bound to MtCYP5118. Second, the indole ring of 14t (PDB small molecule code 18I) occupies the hydrophobic area enclosed largely by the heme macrocycle and the π-electron rich residues Y103, M106, F110, Y116, F290 plus A287 (Fig. 2A); this is the same area where the 2,4-difluorophenyl unit of posaconazole binds (Fig. 2B). Variable F105 is >5 Å away from the indole ring and within 4 Å of the carbonyl group adjacent to the biaryl moiety of 14t, suggesting additional hydrophobic contacts with the inhibitor in TbCYP51 which are missing from T. cruzi ortholog. Last, the biaryl ring of 14t projects towards the solvent exposed area, as does the tail part of the posaconazole, 19 but via a different hydrophobic tunnel between the FG-loop (residues 205–210) and the hairpin of the two-stranded β-sheet at the protein C-terminus (residues 459–461) (Fig. 2C).
Figure 2. X-ray co-crystal structure of the Tb CYP51-14t complex.

(A) Electron density map (blue mesh) contoured at 1.0 σ delineates the positions of 14t (yellow sticks) in the active site. In purple are amino acid residues providing hydrophobic contacts within 5 Å to the indol moiety of 14t plus F105. Heme is displayed as grey sticks. (B) View of the 14t-bound CYP51 clipped by a plane through the binding site compares the binding modes of 14t (yellow) and posaconazole (cyan). The structure of TbCYP51 complexed with posaconazole (PDB code: 2×2N) is superimposed on that of with 14t. The active site surface is colored by hydrophobicity from orange (lipophilic) to blue (hydrophylic). (C) View of bound inhibitors from the entrance to the active site. The enzyme is represented by a gray surface. The hydrophobic units of posaconazole and 14t occupy different hydrophobic tunnels in corresponding co-crystal structures. The images here and otherwise were generated using PYMOL29 or CHIMERA30.
A hydrophobic cavity accommodating the indole ring of 14t extends further toward F110 (Fig. 3A) to provide space sufficient to bind a substituted benzyl ring of NEE (PDB ID 4H6O) (Fig. 3B ) or a rigid biaryl moiety of VNF (PDB ID 3KSW) (Fig. 3C). In addition to F110, the cavity is enclosed by Y116, providing stacking interactions with the 4-chloro-3,5-dimethylbenzyl unit of NEE, as well as by the aliphatic hydrophobic residues A115, M123, L127, L130, A287, and hydrophilic neutral Q126. Presumably, the bulky substituents at the 5-position of the indole ring in analog 3k bind in this cavity.
Figure 3. Comparison of the 14t binding mode with NEE and VNF.

View of the 14t-bound TbCYP51 clipped by a plane through the binding site. A hydrophobic cavity accommodating the indole ring of 14t (yellow) extends toward F110 (A). This extension accommodates a substituted benzyl ring in NEE (purple) (PDB ID 4H6O) (B) or biaryl moiety of VNF (pink) (PDB ID 3KSW) in the corresponding TcCYP51 co-crystal structures. (C). Both structures were superimposed on the 14t-bound TbCYP51.
Synthesis of aryl carboxylic acids
The biaryl carboxylic acid intermediates were prepared by palladium-mediated coupling reactions of commercially available 4-bromo-2-fluorobenzoic acid 15 with various aryl boronic acids (Scheme 3). This reaction was performed under microwave irradiation (100 °C for 1 h) and provided products 16 in >90% yields. Intermediate 17 was obtained by the Heck reaction of 15 and 1-chloro-4-vinylbenzene in the presence of Pd(OAc)2. Nucleophilic aromatic substitution of 15 with aniline yielded biarylamine 19. Phenylacetylene was coupled with ester 18 to provide the biaryl acetylene 21a, which was hydrolyzed under basic conditions to afford 21b. Treatment of methyl 4-bromo-2-fluorobenzoate (18) with morpholine or N-Boc-piperazine at 50° C in toluene gave 22 and 23; by-products were obtained when this reaction was performed at higher temperature (ca. 100 °C). The N-Boc protecting group of 23 was removed by treatment with TFA. Aryl sulfonylamide 25b and N-benzylpiperazine 26b were obtained by the reactions of amine 24 with benzenesulfonyl chloride and benzyl bromide followed by ester hydrolysis under basic conditions, respectively. All benzoic acid derivatives were coupled with indole derivative 6 to generate the inhibitors 27 (Table 4) by using the reaction conditions in Scheme 2.
Scheme 3.

(a) Aryl boronic acid, 5 mol% Pd2(dba)3, 10 mol% PCy3, 2M K3PO4, dioxane, 100 °C (microwave), 1h, ~90%. (b) 1-chloro-4-vinylbenzene, 5 mol% Pd(OAc)2, 10 mol% P(o-tolyl)3, Et3N, DMF, 100 °C (microwave), 2h, 73%. (c) SOCl2, CH3OH, room temp. 12 h, 96%. (d) aniline, 5 mol% Pd(OAc)2, 10 mol% BINAP, Cs2CO3, toluene, 100 °C (microwave), 2h, 73%. (e) 10% NaOH (aq), CH3OH, 50 °C, 1h, 91%. (f) ethynylbenzene, 5 mol% Pd(OAc)2, 10 mol% BINAP, Et3N, CuI, toluene, 110 °C (microwave), 2h, 77%. (g) N-boc-piperazine or morpholine, Pd(OAc)2, P(o-tolyl)3, Cs2CO3, toluene, 50 °C, 48h, 91%. (h) trifluoroacetic acid, CH2Cl2, room temp. 1 h, >90% (crude). (i) phenylsulfonyl chloride, Et3N, CH2Cl2, 0 °C to room temp. 1h, 81%. (j) 4-fluoro-benzyl bromide, Et3N, CH2Cl2, room temp. 1h, 88%.
Table 4.
Biochemical and cell-based activities, microsome stability and CYP inhibition properties of inhibitors 27.a
 |
KD nM |
EC50 µM |
Microsome stability t1/2 (min) |
%inhibition of human CYPs (at 1 µM, unless indicated otherwise) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| R | h | Rat | m | 1A2 | 2C9 | 2D6 | 3A4 | |||
| 14t | <5 | 0.19 | 17 | 25 | 36 | 31 | 85 | 54 | 73 | |
| 27a | <5 | 0.57 | 19 | 12 | 20 | 14 | 87 | 68 | 41 | |
| 27b | <5 | 0.28 | 18 | 13 | 23 | 14 | 85 | 58 | 63 | |
| 27c |  |
<5 | 1.0 | 18 | 41 | 50 | 0 | 71 | 20 | 43 |
| 27d | <5 | 0.28 | 12 | 15 | 22 | 3 | 77 | 34 | 58 | |
| 27e | <5 | 0.97 | 35 | 22 | 27 | 0 | 75 | 37 | 14 | |
| 27f | 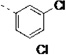 |
<5 | 0.98 | 21 | 36 | 53 | 10 | 80 | 34 | 41 |
| 27g | <5 | 0.28 | 16 | 10 | 19 | 33 | 92 | 70 | 74 | |
| 27h | <5 | 0.97 | 22 | 20 | 32 | 48 | 93 | 75 | 54 | |
| 27i | <5 | 0.014 | 23 | 32 | 14 | 10 | 91 | 38 | 69 | |
| 27j | <5 | 0.22 | 6 | 14 | 22 | 15 | 87 | 32 | 57 | |
| 27k | <5 | 0.23 | 34 | 125 | 83 | 0 | 85 | 16 | 21 | |
| 27l | <5 | 0.25 | 31 | 32 | 29 | 11 | 94 | 68 | 92 | |
| 27m | <5 | 0.20 | 20 | 19 | 17 | 29 | 91 | 57 | 90 | |
| 27n | 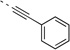 |
<5 | 0.98 | >120 | 67 | 104 | 23 (26)b | 88 (96)b | 28 (68)b | 28 (46)b |
| 27o | <5 | 0.98 | 70 | 67 | 53 | 21 (25)b | 90 (94)b | 63 (85)b | 25 (33)b | |
| 27p | 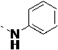 |
<5 | 0.47 | 28 | 31 | 25 | 12 | 94 | 83 | 79 |
| 27q | <5 | 0.057 | 13 | 19 | 7 | 10 (44)b | 74 (98)b | 66 (89)b | 54 (94)b | |
| 27r | 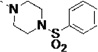 |
<5 | 0.018 | 7 | 19 | 3 | 37 | 97 | 85 | 61 |
| 27s | 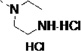 |
<5 | 0.46 | 15 | 36 | 41 | 5 (21)b | 16 (71)b | 22 (81)b | 39 (82)b |
| 27t | - | 0.039 | 4 | 26 | 20 | 0 | 87 | 51 | 61 | |
See notes to Table 1
Values in parentheses are % inhibition of the indicated human CYPs at 10 µM
Structure and property guided optimization of 14t
The terminal phenyl ring of 14t was extensively modified since it is oriented toward the solvent accessible area and opportunities existed to enhance microsome stability and minimize inhibition of human CYP enzymes through such modifications. Indeed, the substituted biaryl derivatives 27a – 27n exhibited enhanced microsome stability, while retaining their inhibition of T. cruzi in cell-based assays. It should be noted that all these compounds had low nM affinity for TcCYP51 (Table 4). Of particular interest is that the potency of 27i in the cell-based T. cruzi assay was increased 50-fold (EC50 = 0.014 µM) compared to that of LP10. Furthermore, 27i exhibited substantially diminished inhibition of human CYP enzymes (10/91/38/69% inhibition of human CYP1A2/2C9/2D6/3A4 at 1 µM). The microsome stability of analog 27k significantly increased (t1/2 = 34/125/83 min, for human, rat and mouse liver microsomes, respectively), and its inhibition of human CYP enzymes was also significantly decreased (% inhibition = 0/85/16/21% at 1 µM), while retaining sub-micromolar potency against T. cruzi in infected cells (EC50 = 0.23 µM).
Lastly, the potency, microsome stability, and CYP selectivity of several aminoaryl-containing analogs was assessed. The morpholinoaryl and sulfonylpiperazine derivatives 27q and 27r showed excellent anti-T. cruzi potency (EC50 = 0.057 and 0.018 µM, respectively), but were moderately stable only in rat liver microsomes (t1/2 = 19 min). Interestingly, the inhibition potency of amine salt 27s was slightly improved compared to that of LP10. As discussed previously, the amine salts 3c – 3f lost binding affinity and inhibition potency due to the conflict between the polar ammonium ion and the hydrophobic active site of TcCYP51. However, based on the co-crystal structure of 14t and TbCYP51 (Fig. 3), the polar ammonium ion of 27s can be oriented toward the solvent accessible area (Fig. 4). The microsome stability of 27s was also significantly increased, particularly in rat and mouse liver microsomes (t1/2 = 36 and 41 min respectively), and inhibition of human CYPs was notably decreased. Finally, the inhibition potency of 27t, which also possesses a basic amine, was ca. 20-fold increased (EC50 = 0.039 µM) compared to LP10; 27t was also fairly stable in rat and mouse liver microsomes (t1/2 = 26 and 20 min, respectively) and exhibited good selectivity toward human CYP enzymes (0/87/51/61% inhibition at 1 µM).
Figure 4. Predicted binding modes of inhibitors in Tc CYP51.

Binding modes of 27l (A), 27s (B), 27k (C), and 27r (D) resulting from molecular docking using Glide XP. Inhibitors are in stick mode colored by atom type: carbon in yellow, oxygen in red, nitrogen in blue, fluorine in cyan, hydrogen on the tertiary amino group of 27s is in gray. The protein is shown as a semi-transparent gray surface; heme is displayed as orange spheres.
Binding poses of inhibitors 27
All potent inhibitors 27 in Table 4 were docked in the 3D structure of TcCYP51, generated from the x-ray co-crystal structure of TbCYP51 complexed with 14t, by using Glide XP mode.20 In the results of docking studies, the terminal 3-fluoro-4-hydroxylphenyl ring of 27l (Fig 4A) and the protonated secondary amine of the piperazine unit of 27s (Fig 4B) are oriented towards the solvent accessible area. This analysis suggested that unfilled space near the phenolic hydroxyl group of 27l, could be filled by other substituents, as is the case with O-methyl (in 27i) and O-benzyl (in 27k) derivatives. This speculation is consistent with the excellent potency of benzenesulfonamide (27r) and benzylamine (27t) substituted inhibitors against T. cruzi, both of which have relatively large groups that may project into the unfilled region identified above (Fig. 4C, D).
CONCLUSION
The non-azole, indolylpyridinecarboxamide-based CYP51 inhibitor LP10, identified by HTS, was shown in prior studies to have moderate potency (EC50 = 0.65 µM) against T. cruzi in cell culture and to be effective in an acute mouse model of T. cruzi infection (60% cure rate).14 Accordingly, LP10 was selected as the starting point for hit-to-lead optimization, with the objective of improving activity against T. cruzi in cell culture, as well as improving microsome stability and enhancing selectivity against the human CYPs 1A2, 2C9, 2D6 and 3A4. A series of first-generation biaryl inhibitors (e.g., 14t) were synthesized and shown to have improved microsome stability and enhanced in vitro inhibitor potency (Table 2). The x-ray co-crystal structure of TbCYP51 with bound 14t was determined and employed in structure-based design of the next round of CYP51 inhibitors. Several potent inhibitors such as 27i, 27q, 27r, and 27t (EC50 = 14, 57, 18 and 39 nM against T. cruzi) were developed and had the same or better microsome stability compared to LP10, as well as enhanced selectivity against human CYPs. The microsome stability of many other inhibitors containing biaryl units, particularly 14t, 27k, 27l, 27p, and 27s, was improved, as was the selectivity of 27k and 27s when tested against the battery of human CYPs. However, 14t, 27k, 27l, and 27p were only moderately more potent against T. cruzi in cell culture than LP10 (EC50 = 190 to 470 nM).
Especially noteworthy is that the binding mode of 14t in the co-crystal structure with TbCYP51 is similar to that of posaconazole with the exception that the biaryl unit of 14t extends towards the solvent accessible area though a different hydrophobic tunnel than used by posaconazole (Fig. 2B,C). The indole ring of 14t occupies the same hydrophobic cavity as the 2,4-difluorophenyl moiety of posaconazole. The cavity extends beyond these groups along the heme macrocycle and has sufficient space to accommodate an alkoxy group attached to C5 of the indole nucleus (inhibitor 3k).
In summary, the SAR/SPR analysis presented here provides a basis to understand the structural features that lead to enhanced biochemical, cell-based activity and microsome stability of the LP10 series of CYP51 inhibitors. The 4-acylaminopyridine and indole rings of the new inhibitors contribute to their ability to bind tightly to both CYP51 and the human metabolic CYPs in liver microsomes. Introduction of a structurally rigid biaryl unit with appropriately placed substituents resulted in enhanced stability of inhibitors in liver microsomes and also contributed to decreased inhibition of human CYP enzymes, culminating in inhibitor 27k. In addition, our results show that the binding affinity and inhibition of TcCYP51 as well as other CYP enzymes is highly dependent on the position of amine substituents in the inhibitors due to the hydrophobic nature of the inhibitor binding sites of these enzymes (see data for 3c – 3f against 27s and 27t). On the other hand, appropriately placed amine substituents contribute to enhanced microsome stability and diminished inhibition of human CYP enzymes. The design of future generations of CYP51 inhibitors should take these considerations into account.
Experimental Section
Chemistry, General Methods
All reaction solvents were purified before use. Dichloromethane, tetrahydrofuran, dimethylformamide and toluene were purified by passing through a column of activated A-1 alumina. All other reagents purchased from commercial suppliers were used as received. All reactions sensitive to moisture or oxygen were conducted under an argon atmosphere using flame-dried (under vacuum) or oven-dried (overnight) glassware. Removal of solvents was accomplished by using a rotary evaporator under reduced pressure in a water bath below 35 °C, followed by exposure to high vacuum using a vacuum pump. Microwave assisted reactions were performed using a Biotage® Initiator microwave reactor.
Proton nuclear magnetic resonance (1H NMR) spectra and carbon (13C) NMR spectra were recorded on a commercially available NMR spectrometer at 400 MHz and 100 MHz, respectively. The proton signal for non-deuterated solvent (δ 7.26 for CHCl3 or δ 2.50 for DMSO) was used as an internal reference for 1H NMR chemical shifts. Coupling constants (J) are reported in Hertz (Hz). 13C chemical shifts are reported relative to the δ 77.16 resonance of CDCl3 or the δ 39.52 resonance of DMSO-d6.
Analytical thin layer chromatography (TLC) was performed using glass plates precoated with a 0.25-mm thickness of silica gel. The TLC plates were visualized with UV light. Column chromatography was performed using a Biotage® Isolera flash purification system using Biotage® SNAP HP-SIL cartridge (30 µm silica, 10 g to 100 g size). Unless noted otherwise, all compounds isolated by chromatography were sufficiently pure by 1H NMR analysis for use in subsequent reactions. Polar compounds were purified using preparative high performance liquid chromatography (HPLC) using SunFire column (30 mm × 250 mm) with a linear gradient elution at 60 mL/min.
The purity of all final compounds (typically ≥96%) was assayed at 254 nm wavelength by using analytical HPLC (Varian 1100 series) on a reverse phase ZORBAX Eclipse XDB-C18 column (4.6 × 150 mm, 5 µm). A linear gradient elution ranging from 2% to 98% CH3CN and H2O (containing 0.1% TFA and 1% CH3CN) at 1.5 mL/min was used. Compounds were lyophilized before dissolution in DMSO to give 10 mM stock solutions for use in biochemical and cell-based assays.
Synthesis of inhibitors 1, 2, 3, 14 and 27 by acylation of trytophan pyridinyl carboxamide (6)
General Procedure A. To a solution of a substituted benzoic or naphthoic acid (ca.1.2 eq), PyBOP (ca. 1.4 eq) and HOBt (ca. 10 mol%) in dry CH2Cl2 (5 mL) was slowly added triethylamine (ca. 4 eq.) at ambient temperature over 15 min. After the reaction mixture became homogenous, 6 was added, and the reaction mixture was stirred at room temperature for 1 h. After completion of the reaction as determined by TLC analysis, the solvent was removed under reduced pressure. The crude mixture was dissolved in ethyl acetate (10 mL) and was washed with saturated aqueous NaHCO3 (2 mL × 2) and brine (2 mL × 2). The organic layer was concentrated in vacuo and directly subjected to purification by flash chromatography to provide the amide products in ca. 80% yield.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)cyclohexanecarboxamide, (1)
To a solution of 6 (0.112 g, 0.353 mmol) in CH2Cl2 (10 mL) were added cyclohexanecarbonyl chloride (0.06 mL) and (iPr)2EtN (0.1 mL) at 0 °C After 10 min, the reaction mixture was warmed to ambient temperature and stirred for 1 h. After completion of the reaction monitored by TLC, ethyl acetate (40 mL) was added to the crude mixture, which was washed with saturated aqueous NaHCO3 (10 mL × 2) and brine (10 mL × 2). The organic layer was dried over magnesium sulfate, filtered, and concentrated under reduced pressure. Purification of the crude product by flash chromatography provided 1 as a light yellow solid (0.103 g, 0.263 mmol, 75%). 1H NMR (400 MHz, DMSO-d6) δ 10.81 (d, J = 2.5 Hz, 1H), 10.56 (s, 1H), 8.50 – 8.38 (m, 2H), 8.06 (d, J = 7.6 Hz, 1H), 7.73 – 7.55 (m, 3H), 7.31 (d, J = 8.1 Hz, 1H), 7.15 (d, J = 2.3 Hz, 1H), 7.04 (ddd, J = 8.1, 7.0, 1.2 Hz, 1H), 7.00 – 6.89 (m, 1H), 4.67 (td, J = 8.4, 5.7 Hz, 1H), 3.22 – 2.94 (m, 2H), 2.19 (ddd, J = 11.1, 7.7, 3.2 Hz, 1H), 1.61 (dt, J = 41.4, 10.3 Hz, 5H), 1.20 (dddd, J = 29.7, 15.4, 12.1, 5.8 Hz, 5H). 13C NMR (101 MHz, DMSO-d6) δ 175.36, 172.31, 149.72, 146.03, 135.97, 127.19, 123.64, 120.88, 118.50, 118.16, 113.42, 111.24, 109.67, 54.21, 43.45, 29.09, 28.98, 27.58, 25.42, 25.21, 25.16. MS (ESI) 391 m/z [M + H]+
(S)-N-(3-(1H-Indol-3-yl)-1-((2-methoxypyridin-4-yl)amino)-1-oxopropan-2-yl)cyclohexanecarboxamide (3a)
Compound 3a was obtained by following the procedure for the synthesis of 1 (69%) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.78 (s, 1H), 8.20 (s, 1H), 7.95 (d, J = 5.7 Hz, 1H), 7.64 (d, J = 7.9 Hz, 1H), 7.35 (d, J = 8.1 Hz, 1H), 7.23 – 7.15 (m, 1H), 7.14 – 7.05 (m, 1H), 7.02 (d, J = 2.3 Hz, 1H), 6.89 (d, J = 1.8 Hz, 1H), 6.78 (dd, J = 5.7, 1.9 Hz, 1H), 6.36 (d, J = 7.3 Hz, 1H), 4.93 (q, J = 7.0 Hz, 1H), 3.88 (s, 3H), 3.42 – 3.16 (m, 2H), 2.13 – 2.06 (m, 1H), 1.87 – 1.55 (m, 5H), 1.48 – 1.03 (m, 5H). 13C NMR (101 MHz, CDCl3) δ 177.24, 170.87, 165.37, 147.42, 146.95, 136.35, 127.39, 123.32, 122.63, 120.07, 118.83, 111.49, 110.41, 108.43, 99.77, 54.54, 53.73, 45.26, 29.61, 29.53, 27.76, 25.74, 25.68, 25.63. MS (ESI) 421 m/z [M + H]+.
(S)-5-Bromo-N-(1-((3,5-dimethylisoxazol-4-yl)amino)-3-(1H-Indol-3-yl)-1-oxopropan-2-yl)-2-fluorobenzamide (3b)
Compound 3b (75%) was obtained as a white solid by following the general procedure A with 5-bromo-2-fluorobenzoic acid. 1H NMR (400 MHz, DMSO-d6) δ 10.90 (d, J = 2.5 Hz, 1H), 9.48 (s, 1H), 8.71 (dd, J = 7.3, 2.3 Hz, 1H), 7.82 – 7.67 (m, 2H), 7.64 (d, J = 7.9 Hz, 1H), 7.35 (dd, J = 8.1, 0.9 Hz, 1H), 7.29 (dd, J = 10.0, 8.6 Hz, 1H), 7.24 (d, J = 2.4 Hz, 1H), 7.07 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 6.98 (ddd, J = 7.9, 6.9, 1.0 Hz, 1H), 4.80 (td, J = 7.8, 6.5 Hz, 1H), 3.39 – 3.09 (m, 2H), 2.12 (s, 3H), 1.95 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.77, 162.42, 162.36, 159.74, 157.49, 157.25, 136.09, 135.13, 132.41, 132.38, 127.26, 125.69, 125.53, 123.89, 120.97, 118.77, 118.53, 118.44, 118.31, 115.91, 115.88, 113.94, 111.34, 109.48, 54.59, 27.43, 10.57, 9.22. MS (ESI) 499/501 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)piperidine-4-carboxamide hydrochloride (3c)
To a solution of 9 (0.12 g, 0.25 mmol) in CH2Cl2 (10 mL) was added trifluoroacetic acid (0.5 mL), and the reaction mixture was stirred for 1 h at room temperature. After removing solvent under reduced pressure, the reaction mixture was directly subjected to HPLC, and the product 3c (58 mg, 0.15 mmol, 47%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 11.44 (s, 1H), 10.90 (d, J = 2.5 Hz, 1H), 8.80 (s, 1H), 8.68 – 8.61 (m, 2H), 8.49 (d, J = 7.3 Hz, 1H), 8.00 – 7.93 (m, 2H), 7.63 (d, J = 7.8 Hz, 1H), 7.36 – 7.23 (m, 2H), 7.21 (d, J = 1.9 Hz, 2H), 7.18 – 6.91 (m, 3H), 4.72 (ddd, J = 9.0, 7.3, 5.5 Hz, 1H), 3.30 – 3.15 (m, 2H), 3.09 (dd, J = 14.6, 9.0 Hz, 1H), 2.95 – 2.73 (m, 2H), 1.85 (dd, J = 14.4, 3.8 Hz, 1H), 1.79 – 1.51 (m, 3H). 13C NMR (101 MHz, DMSO-d6) δ 173.41, 173.12, 158.33, 158.02, 151.00, 144.33, 136.02, 127.09, 123.92, 120.95, 118.24, 115.59, 114.26, 111.33, 109.20, 54.90, 42.28, 38.28, 27.28, 24.98. MS (ESI) 392.4 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-1-methylpiperidine-4-carboxamide (3d)
Compound 3d (47%) as a yellow solid was obtained by following the procedure for the synthesis of 9 with 1-methylpiperidine-4-carboxylic acid. 1H NMR (400 MHz, DMSO-d6) δ 11.31 (s, 1H), 10.93 (d, J = 2.5 Hz, 1H), 8.67 – 8.44 (m, 3H), 7.84 (d, J = 6.0 Hz, 2H), 7.66 (d, J = 7.9 Hz, 1H), 7.31 (d, J = 8.0 Hz, 1H), 7.21 (d, J = 2.3 Hz, 1H), 7.08 – 6.99 (m, 1H), 6.94 (t, J = 7.3 Hz, 1H), 4.72 (q, J = 7.5 Hz, 1H), 3.44 – 3.27 (m, 2H), 3.27 – 3.02 (m, 2H), 2.99 – 2.76 (m, 2H), 2.66 (s, 3H), 2.04 – 1.57 (m, 5H). 13C NMR (101 MHz, DMSO-d6) δ 173.03, 172.60, 148.52, 147.02, 139.68, 136.01, 127.21, 123.87, 120.91, 118.57, 118.22, 113.87, 111.32, 109.45, 108.75, 54.91, 52.45, 42.47, 38.41, 27.54, 25.67. MS (ESI) 406 m/z [M + H]+.
N-((S)-3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-1-methylpiperidine-3-carboxamide (3e)
Compound 3e (light yellow, racemic mixture) was obtained by following the procedure for the synthesis of 9 with 1-methylpiperidine-3-carboxylic acid (58%). 1H NMR (400 MHz, DMSO-d6) δ 11.57 (d, J = 6.3 Hz, 1H), 10.95 (dd, J = 14.9, 2.5 Hz, 1H), 8.75 (dd, J = 19.9, 7.4 Hz, 1H), 8.61 (dd, J = 6.1, 3.7 Hz, 2H), 7.96 (d, J = 5.9 Hz, 2H), 7.67 (d, J = 7.9 Hz, 1H), 7.45 (s, 1H), 7.37 – 7.28 (m, 2H), 7.27 – 7.16 (m, 2H), 7.11 – 6.99 (m, 1H), 6.95 (dd, J = 7.6, 4.8 Hz, 1H), 4.73 (t, J = 7.5 Hz, 1H), 3.40 – 2.72 (m, 6H), 2.67 (d, J = 4.2 Hz, 3H), 2.05 – 1.68 ( m, 3H). 13C NMR (101 MHz, DMSO-d6) δ 172.71, 158.41, 158.11, 150.31, 145.03, 136.03, 136.00, 127.15, 124.02, 120.91, 118.69, 118.57, 118.28, 115.71, 114.18, 114.12, 111.35, 109.26, 109.20, 55.10, 53.74, 52.86, 42.79, 27.24, 25.46, 22.52, 21.87. MS (ESI) 406 m/z [M + H]+.
(S)-2-((2-Fluorobenzyl)amino)-3-(1H-indol-3-yl)-N-(pyridin-4-yl)propanamide hydrochloride (3f)
To a solution of 6 (0.107 g, 0.338 mmol) in CH2Cl2 were added Et3N (0.05 mL) and 2-fluorobenzylbromide (0.06 mL), and the reaction mixture was stirred at room temperature for 12 h. Solvent was removed by using rotary evaporator, and the product mixture was directly subjected to HPLC. The product 3f (38.7 mg, 0.0997 mmol, 30%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 10.86 (d, J = 2.5 Hz, 1H), 8.94 (d, J = 8.3 Hz, 1H), 8.26 (dd, J = 7.5, 1.8 Hz, 1H), 8.12 (dd, J = 7.3, 1.9 Hz, 1H), 7.90 – 7.77 (m, 1H), 7.63 (d, J = 7.7 Hz, 1H), 7.51 – 7.42 (m, 1H), 7.42 – 7.21 (m, 6H), 7.18 (d, J = 2.4 Hz, 1H), 7.08 – 6.92 (m, 3H), 6.73 (dd, J = 7.6, 2.9 Hz, 1H), 5.39 (s, 2H), 4.51 (td, J = 8.6, 5.1 Hz, 1H), 3.44 – 2.98 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.66, 158.96, 156.65, 143.80, 141.67, 136.00, 131.30, 130.44, 130.41, 127.04, 125.12, 125.08, 124.07, 122.52, 120.95, 118.37, 115.92, 115.72, 111.32, 111.11, 109.13, 105.57, 56.65, 54.15, 28.01. MS (ESI) 389 m/z [M + H]+.
(S)-N-(3-(Benzo[b]thiophen-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)cyclohexanecarboxamide (3g)
Compound 3g (68%) was obtained as a white solid by following the procedure for the synthesis of 1 with Boc-L-3-benzothienylalanine. 1H NMR (400 MHz, DMSO-d6) δ 10.58 (s, 1H), 8.54 – 8.37 (m, 2H), 8.23 (d, J = 7.9 Hz, 1H), 8.05 – 7.90 (m, 2H), 7.67 – 7.54 (m, 2H), 7.45 (s, 1H), 7.44 – 7.33 (m, 2H), 4.81 (ddd, J = 9.5, 7.9, 5.1 Hz, 1H), 3.46 – 3.05 (m, 2H), 2.17 (ddd, J = 10.9, 7.5, 3.5 Hz, 1H), 1.77 – 1.44 (m, 6H), 1.36 – 1.01 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 175.44, 171.62, 150.29, 145.48, 139.47, 138.62, 131.74, 124.27, 123.96, 123.89, 122.81, 121.99, 113.43, 52.91, 43.50, 30.44, 29.12, 28.89, 25.41, 25.22, 25.13. MS (ESI) 408.2 m/z [M + H]+.
(S)-N-(3-(Naphthalen-1-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)cyclohexanecarboxamide (3h)
Compound 3h (67%) was obtained as a white solid by following the procedure for the synthesis of 1 with Boc-L-1-naphthylalanine. 1H NMR (400 MHz, DMSO-d6) δ 10.47 (s, 1H), 8.49 – 8.37 (m, 2H), 8.24 (t, J = 8.0 Hz, 2H), 7.91 (dd, J = 7.9, 1.4 Hz, 1H), 7.78 (dd, J = 7.5, 1.9 Hz, 1H), 7.64 – 7.46 (m, 4H), 7.46 – 7.32 (m, 2H), 4.82 (td, J = 8.5, 5.8 Hz, 1H), 3.61 – 3.26 (m, 2H), 2.16 (dtd, J = 15.7, 8.4, 7.6, 4.1 Hz, 1H), 1.73 – 1.41 (m, 5H), 1.36 – 0.95 (m, 5H). 13C NMR (101 MHz, DMSO-d6) δ 175.39, 171.60, 150.31, 145.38, 133.33, 133.24, 131.67, 128.51, 127.29, 127.13, 126.07, 125.57, 125.22, 123.92, 113.48, 53.85, 43.47, 34.49, 29.14, 28.87, 25.41, 25.22, 25.12. MS (ESI) 402.3 m/z [M + H]+.
(S)-N-(3-(1-Methyl-1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)cyclohexanecarboxamide (3i)
Compound 3i (51%) was obtained as a yellow solid by following the procedures for the synthesis of 13 and 1 starting with 1-methyl-L-tryptophan. Rf = 0.57 (10% MeOH in ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.46 (s, 1H), 8.51 – 8.33 (m, 2H), 8.04 (d, J = 7.8 Hz, 1H), 7.64 (d, J = 7.9 Hz, 1H), 7.60 – 7.51 (m, 2H), 7.35 (d, J = 8.2 Hz, 1H), 7.17 – 7.07 (m, 2H), 6.99 (t, J = 7.4 Hz, 1H), 4.66 (td, J = 8.2, 5.6 Hz, 1H), 3.70 (s, 3H), 3.21 – 2.95 (m, 2H), 2.29 – 2.14 (m, 1H), 1.77 – 1.49 (m, 5H), 1.40 – 1.01 (m, 5H). 13C NMR (101 MHz, DMSO-d6) δ 175.37, 172.06, 150.30, 145.48, 136.45, 128.04, 127.53, 121.02, 118.80, 118.26, 113.35, 109.45, 109.13, 54.24, 43.45, 32.26, 29.05, 29.00, 27.50, 25.42, 25.20, 25.15. MS (ESI) 405 m/z [M + H]+.
(S)-N-(3-(5-Hydroxy-1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)cyclohexanecarboxamide (3j)
To a solution of 3k (0.328 g, 0.66 mmol) in methanol (10 mL) was added 10% Pd/C (ca. 30 mg) at room temperature. After air was removed from the flask using a vacuum pump, hydrogen gas was introduced using a balloon. The reaction mixture was stirred for 1h, and the flask was evacuated under vacuum and refilled with hydrogen gas. This procedure was repeated three times, and the reaction mixture was stirred overnight at ambient temperature. Palladium on carbon was removed by filtration through Celite pad. The filtrate was collected and evaporated to give the crude product, which was purified by flash chromatography to afford the product 3j as a brown solid (0.077 g, 0.19 mmol, 29%). 1H NMR (400 MHz, DMSO-d6) δ 10.47 (d, J = 3.6 Hz, 1H), 8.55 (s, 1H), 8.48 – 8.33 (m, 2H), 8.01 (d, J = 7.7 Hz, 1H), 7.66 – 7.49 (m, 2H), 7.09 (d, J = 8.5 Hz, 1H), 7.04 (d, J = 2.4 Hz, 1H), 6.93 (d, J = 2.2 Hz, 1H), 6.58 (dd, J = 8.6, 2.3 Hz, 1H), 4.63 (td, J = 8.2, 6.0 Hz, 1H), 3.15 – 2.83 (m, 2H), 2.19 (ddd, J = 14.4, 9.5, 3.5 Hz, 1H), 1.91 (s, 1H), 1.63 (dd, J = 32.3, 11.3 Hz, 5H), 1.38 – 1.00 (m, 5H). 13C NMR (101 MHz, DMSO-d6) δ 175.33, 172.27, 150.30, 150.20, 145.50, 130.54, 127.96, 123.98, 113.36, 111.46, 111.24, 108.72, 102.55, 54.02, 43.46, 29.13, 28.99, 27.67, 25.43, 25.23, 25.17, 21.07. MS (ESI) 407 m/z [M + H]+.
(S)-N-(3-(5-(Benzyloxy)-1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)cyclohexanecarboxamide (3k)
The procedure for the synthesis of 1 was followed using 13 to provide 3k as a light yellow solid (52%, over 3 steps). Rf = 0.21 (100% ethyl acetate), Rf = 0.63 (10% MeOH in ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.67 (d, J = 2.6 Hz, 1H), 10.52 (s, 1H), 8.41 (d, J = 5.5 Hz, 2H), 8.08 (d, J = 7.8 Hz, 1H), 7.58 (d, J = 5.5 Hz, 2H), 7.46 (d, J = 7.5 Hz, 2H), 7.39 (t, J = 7.5 Hz, 2H), 7.32 (t, J = 7.2 Hz, 1H), 7.24 (d, J = 2.2 Hz, 1H), 7.21 (d, J = 8.7 Hz, 1H), 7.12 (d, J = 2.3 Hz, 1H), 6.77 (dd, J = 8.6, 2.3 Hz, 1H), 5.16 – 4.90 (m, 2H), 4.66 (td, J = 8.3, 5.4 Hz, 1H), 3.17 – 2.90 (m, 2H), 2.27 – 2.13 (m, 1H), 1.61 (dt, J = 32.1, 11.7 Hz, 5H), 1.36 – 1.00 (m, 5H). 13C NMR (101 MHz, DMSO-d6) δ 175.32, 172.24, 152.06, 150.32, 145.51, 137.78, 131.34, 128.33, 127.65, 127.60, 124.45, 113.34, 111.80, 111.40, 109.58, 102.22, 69.90, 54.28, 43.51, 29.08, 29.00, 27.73, 25.43, 25.22, 25.18. MS (ESI) 497 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-4,4-difluorocyclohexanecarboxamide (3l)
The procedure for the synthesis of 9 was followed using 4,4-difluorocyclohexanecarboxylic acid to provide 3l as a light yellow solid (41%). Rf = 0.39 (100% ethyl acetate), 1H NMR (400 MHz, DMSO-d6) δ 10.81 (d, J = 2.4 Hz, 1H), 10.51 (s, 1H), 8.52 – 8.37 (m, 2H), 8.23 (d, J = 7.8 Hz, 1H), 7.63 (d, J = 7.8 Hz, 1H), 7.60 – 7.52 (m, 2H), 7.31 (dt, J = 8.1, 0.9 Hz, 1H), 7.15 (d, J = 2.3 Hz, 1H), 7.05 (ddd, J = 8.1, 7.0, 1.2 Hz, 1H), 6.96 (ddd, J = 8.0, 6.9, 1.1 Hz, 1H), 4.71 (td, J = 8.4, 5.5 Hz, 1H), 3.24 – 2.94 (m, 2H), 2.42 – 2.29 (m, 1H), 2.07 – 1.37 (m, 8H). 13C NMR (101 MHz, DMSO-d6) δ 173.93, 172.03, 150.33, 145.46, 135.99, 127.19, 123.65, 120.91, 118.52, 118.17, 113.36, 111.25, 109.63, 54.19, 40.44, 30.67, 27.65, 25.52, 25.43, 25.38, 25.29. MS (ESI) 427.1 m/z [M + H]+.
(S)-3-(1H-Indol-3-yl)-2-(2-phenylacetamido)-N-(pyridin-4-yl)propanamide (3m)
The procedure for the synthesis of 1 was followed using 2-phenylacetyl chloride to provide 3m as a yellow solid (61%). Rf = 0.30 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.94 – 10.73 (m, 1H), 10.54 (s, 1H), 8.50 (d, J = 7.6 Hz, 1H), 8.46 – 8.31 (m, 2H), 7.63 (d, J = 7.9 Hz, 1H), 7.59 – 7.49 (m, 2H), 7.32 (d, J = 8.1 Hz, 1H), 7.29 – 7.09 (m, 6H), 7.05 (t, J = 7.5 Hz, 1H), 6.95 (t, J = 7.4 Hz, 1H), 4.72 (td, J = 8.3, 5.6 Hz, 1H), 3.46 (d, J = 2.2 Hz, 2H), 3.25 – 2.97 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.93, 170.16, 150.33, 145.42, 136.20, 136.02, 128.96, 128.08, 127.17, 126.21, 123.74, 120.90, 118.51, 118.22, 113.37, 111.26, 109.47, 68.24, 54.48, 41.85. MS (ESI) 399 m/z [M + H]+.
(S)-3-(1H-Indol-3-yl)-2-(3-phenylpropanamido)-N-(pyridin-4-yl)propanamide (3n)
The procedure for the synthesis of 9 was followed using 3-phenylpropanoic acid to provide 3n as a yellow solid (73%). Rf = 0.33 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.81 (d, J = 2.5 Hz, 1H), 10.50 (s, 1H), 8.50 – 8.37 (m, 2H), 8.30 (d, J = 7.7 Hz, 1H), 7.63 (d, J = 7.9 Hz, 1H), 7.60 – 7.54 (m, 2H), 7.35 – 7.28 (m, 1H), 7.25 – 7.09 (m, 6H), 7.06 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 6.97 (ddd, J = 8.0, 6.9, 1.1 Hz, 1H), 4.72 (td, J = 8.2, 5.9 Hz, 1H), 3.25 – 2.92 (m, 2H), 2.74 (dd, J = 8.7, 6.8 Hz, 2H), 2.42 (dd, J = 8.8, 6.7 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 172.01, 171.53, 150.31, 145.46, 141.20, 136.02, 128.19, 128.14, 127.19, 125.79, 123.64, 120.91, 118.50, 118.21, 113.37, 111.27, 109.62, 54.38, 36.68, 30.96, 27.66. MS (ESI) 413 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-4-phenylbutanamide (3o)
The procedure for the synthesis of 9 was followed using 4-phenylbutanoic acid to provide 3o as a yellow solid (67%). Rf = 0.33 (100% ethyl acetate). 1H NMR (400 MHz, CDCl3) δ 9.28 (s, 1H), 8.37 – 8.26 (m, 2H), 7.57 (dd, J = 8.0, 1.0 Hz, 1H), 7.38 – 7.34 (m, 2H), 7.32 (dt, J = 8.1, 1.0 Hz, 1H), 7.26 (s, 3H), 7.20 – 7.14 (m, 2H), 7.09 – 7.04 (m, 2H), 7.04 – 7.00 (m, 2H), 6.42 (d, J = 7.3 Hz, 1H), 4.96 (q, J = 7.1 Hz, 1H), 3.29 (dd, J = 7.0, 1.9 Hz, 2H), 2.54 (t, J = 7.6 Hz, 2H), 2.18 (td, J = 7.4, 2.0 Hz, 2H), 1.87 (p, J = 7.9 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 174.04, 171.16, 149.27, 145.96, 141.20, 136.36, 128.60, 128.53, 127.32, 126.26, 123.40, 122.64, 120.09, 118.58, 114.05, 111.62, 110.09, 54.97, 35.73, 35.09, 27.78, 27.01. MS (ESI) 427 m/z [M + H]+.
(S)-3-(1H-Indol-3-yl)-2-(3-(4-methoxyphenyl)propanamido)-N-(pyridin-4-yl)propanamide (3p)
The procedure for the synthesis of 9 was followed using 3-(4-methoxyphenyl)propanoic acid to provide 3p as a yellow solid (34%). 1H NMR (400 MHz, CDCl3) δ 8.98 (s, 1H), 8.42 – 8.33 (m, 2H), 8.33 – 8.24 (m, 1H), 7.55 (d, J = 7.8 Hz, 1H), 7.38 – 7.32 (m, 1H), 7.32 – 7.27 (m, 2H), 7.18 (ddd, J = 8.2, 7.0, 1.1 Hz, 1H), 7.07 (ddd, J = 8.1, 7.0, 1.0 Hz, 1H), 7.02 – 6.94 (m, 2H), 6.90 (d, J = 2.4 Hz, 1H), 6.75 – 6.70 (m, 2H), 6.40 (d, J = 7.4 Hz, 1H), 4.90 (q, J = 7.0 Hz, 1H), 3.71 (s, 3H), 3.34 – 3.12 (m, 2H), 2.88 – 2.74 (m, 2H), 2.46 (t, J= 7.7 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 173.49, 170.91, 158.26, 149.75, 145.45, 136.33, 132.29, 129.35, 127.36, 123.40, 122.62, 120.08, 118.57, 114.14, 114.01, 111.59, 110.04, 55.38, 54.84, 38.40, 30.65, 27.40. MS (ESI) 443 m/z [M + H]+.
(S)-3-(1H-Indol-3-yl)-N-(pyridin-4-yl)-2-(3-(4-(trifluoromethyl)phenyl)propanamido)-propanamide (3q)
The procedure for the synthesis of 9 was followed using 3-(4-(trifluoromethyl)phenyl)propanoic acid to provide 3q as a yellow solid (17%). 1H NMR (400 MHz, CDCl3) δ 8.71 (s, 1H), 8.41 – 8.34 (m, 2H), 8.29 (s, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.44 (d, J = 8.1 Hz, 2H), 7.36 (dt, J = 8.2, 0.9 Hz, 1H), 7.29 – 7.16 (m, 5H), 7.09 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H), 6.97 (d, J = 2.4 Hz, 1H), 6.44 (d, J = 7.3 Hz, 1H), 4.90 (q, J = 7.2 Hz, 1H), 3.42 – 3.13 (m, 2H), 2.94 (t, J = 7.5 Hz, 2H), 2.51 (td, J = 7.5, 2.7 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 172.70, 170.71, 149.97, 145.17, 144.48, 136.38, 128.74, 127.26, 125.66, 125.62, 125.58, 123.41, 122.77, 120.22, 118.55, 113.93, 111.68, 110.03, 54.90, 37.53, 31.13, 27.62. MS (ESI) 481 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-4-(4-fluorophenyl)butanamide (3r)
The procedure for the synthesis of 9 was followed using 4-(4-fluorophenyl)butanoic acid to provide 3r as a solid (86%). Rf = 0.24 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.91 – 10.76 (m, 1H), 10.54 (s, 1H), 8.53 – 8.37 (m, 2H), 8.23 (d, J = 7.7 Hz, 1H), 7.65 (d, J = 7.8 Hz, 1H), 7.61 – 7.56 (m, 2H), 7.31 (d, J = 8.0 Hz, 1H), 7.17 (d, J = 2.3 Hz, 1H), 7.15 – 7.00 (m, 5H), 6.96 (t, J = 7.4 Hz, 1H), 4.73 (td, J = 8.5, 5.7 Hz, 1H), 3.23 – 2.93 (m, 2H), 2.44 (t, J = 7.6 Hz, 2H), 2.23 – 2.01 (m, 2H), 1.70 (p, J = 7.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 172.22, 172.05, 161.75, 159.35, 150.07, 145.72, 137.85, 137.82, 136.02, 130.01, 129.93, 127.17, 123.66, 120.91, 118.53, 118.19, 114.93, 114.72, 113.39, 111.27, 109.67, 54.36, 34.30, 33.54, 27.60, 26.99. MS (ESI) 445 m/z [M + H]+.
(S)-tert-Butyl (3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)carbamate, (5)
To a solution of N-Boc-L-tryptophan (1.0 g, 3.3 mmol), PyBOP (2.0 g, 3.9 mmol), and HOBt (0.29 g) in dry CH2Cl2 (20 mL) was slowly added triethylamine (1.5 mL, ca. 4 eq.) at 0 °C, and the reaction mixture was stirred and warmed to ambient temperature for 15 min. After the mixture was cooled to 0 °C, 4-aminopyridine (0.39 g, 4.1 mmol) was added, and the reaction mixture was stirred at room temperature for 1 h. After completion of the reaction monitored by TLC, ethyl acetate (80 mL) was added to the crude mixture, which was washed with saturated aqueous NaHCO3 (20 mL × 2) and brine (20 mL × 2). The organic layer was dried over magnesium sulfate, filtered, and concentrated in vacuo. Purification of the crude product by flash chromatography provided 0.98 g (94%) of 5 as a light yellow solid. Rf = 0.45 (100% ethyl acetate). 1H NMR (400 MHz, CDCl3) δ 9.07 (s, 1H), 8.86 (dd, J = 23.3, 11.0 Hz, 1H), 8.36 – 8.23 (m, 2H), 7.58 (d, J = 8.0 Hz, 1H), 7.31 (d, J = 8.3 Hz, 1H), 7.23 (d, J = 5.7 Hz, 2H), 7.14 (t, J = 7.7 Hz, 1H), 7.03 (t, J = 7.6 Hz, 1H), 6.98 (s, 1H), 5.57 (q, J = 7.5, 6.2 Hz, 1H), 4.67 (s, 1H), 3.40 – 3.15 (m, 2H), 1.39 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 171.75, 156.25, 150.26, 145.03, 136.47, 127.27, 123.51, 122.34, 119.79, 118.62, 113.90, 111.52, 109.94, 80.82, 56.04, 28.37, 22.61. MS (ESI) 381 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)benzamide (14a)
The procedure for the synthesis of 1 was followed using benzoyl chloride to provide 14a as a solid (99%). Rf = 0.39 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.82 (d, J = 2.5 Hz, 1H), 10.64 (s, 1H), 8.74 (d, J = 7.5 Hz, 1H), 8.56 – 8.37 (m, 2H), 7.92 – 7.80 (m, 2H), 7.75 (d, J = 7.7 Hz, 1H), 7.67 – 7.61 (m, 2H), 7.58 – 7.39 (m, 3H), 7.32 (d, J = 8.1 Hz, 1H), 7.28 (d, J = 2.3 Hz, 1H), 7.06 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 6.99 (ddd, J = 8.1, 6.9, 1.1 Hz, 1H), 4.89 (ddd, J = 9.1, 7.5, 5.6 Hz, 1H), 3.41 – 3.15 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 172.19, 166.50, 150.33, 145.55, 136.03, 133.75, 131.42, 128.19, 127.50, 127.15, 123.87, 120.95, 118.60, 118.22, 113.41, 111.32, 109.95, 55.28, 27.24. MS (ESI) 385 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2-fluorobenzamide (14b)
The procedure for the synthesis of 1 was followed using 2-fluorobenzoyl chloride to provide 14b as a light yellow solid (49%). Rf = 0.18 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.87 (d, J = 2.5 Hz, 1H), 10.63 (s, 1H), 8.50 (dd, J = 7.3, 3.6 Hz, 1H), 8.47 – 8.29 (m, 2H), 7.66 (d, J = 7.8 Hz, 1H), 7.60 (qd, J = 5.3, 1.8 Hz, 3H), 7.57 – 7.49 (m, 1H), 7.33 (d, J = 8.1 Hz, 1H), 7.31 – 7.21 (m, 3H), 7.06 (ddd, J = 8.0, 6.9, 1.2 Hz, 1H), 6.96 (ddd, J = 7.9, 6.9, 1.1 Hz, 1H), 4.89 (td, J = 8.1, 5.5 Hz, 1H), 3.40 – 3.11 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.52, 163.60, 160.61, 158.13, 150.37, 145.46, 136.06, 132.77, 130.33, 130.30, 127.17, 124.45, 124.41, 123.93, 123.09, 122.95, 120.97, 118.48, 118.25, 116.24, 116.02, 113.43, 111.33, 109.35, 55.08, 27.41. MS (ESI) 403 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-3-fluorobenzamide (14c)
The procedure for the synthesis of 1 was followed using 3-fluorobenzoyl chloride to provide 14c as a yellow solid (51%). Rf = 0.21 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.82 (d, J = 2.5 Hz, 1H), 10.66 (s, 1H), 8.88 (d, J = 7.5 Hz, 1H), 8.44 (d, J = 5.6 Hz, 2H), 7.74 (d, J = 7.8 Hz, 1H), 7.71 (dt, J = 7.7, 1.2 Hz, 1H), 7.69 – 7.63 (m, 1H), 7.63 – 7.59 (m, 2H), 7.51 (td, J = 8.0, 5.8 Hz, 1H), 7.38 (td, J = 8.3, 2.7 Hz, 1H), 7.31 (d, J = 8.0 Hz, 1H), 7.27 (d, J = 2.4 Hz, 1H), 7.06 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 7.02 – 6.94 (m, 1H), 4.89 (ddd, J = 9.4, 7.5, 5.4 Hz, 1H), 3.45 – 3.16 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.99, 165.18, 163.07, 150.26, 145.60, 136.03, 130.45, 127.13, 123.86, 123.72, 120.97, 118.59, 118.24, 113.45, 111.33, 109.86, 55.38, 27.22. MS (ESI) 403.2 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-3-phenoxybenzamide (14d)
The procedure for the synthesis of 9 was followed using 3-phenoxybenzoic acid to provide 14d as a white solid (84%). Rf = 0.60 (10% MeOH in ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.80 (d, J = 2.5 Hz, 1H), 10.63 (s, 1H), 8.82 (d, J = 7.6 Hz, 1H), 8.55 – 8.40 (m, 2H), 7.73 (d, J = 7.8 Hz, 1H), 7.65 (dt, J = 7.8, 1.2 Hz, 1H), 7.63 – 7.57 (m, 2H), 7.53 – 7.36 (m, 4H), 7.31 (d, J = 8.1 Hz, 1H), 7.24 (d, J = 2.4 Hz, 1H), 7.21 – 7.13 (m, 2H), 7.09 – 7.00 (m, 3H), 7.00 – 6.92 (m, 1H), 4.86 (ddd, J = 9.3, 7.5, 5.5 Hz, 1H), 3.32 – 3.11 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 172.06, 165.66, 156.62, 156.39, 150.31, 145.54, 136.01, 135.63, 130.15, 129.98, 127.12, 123.85, 123.73, 122.54, 121.68, 120.94, 118.73, 118.59, 118.22, 117.57, 113.42, 111.31, 109.91, 55.34, 27.18. MS (ESI) 477 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-4-(trifluoromethyl)benzamide (14e)
The procedure for the synthesis of 1 was followed using 4-trifluoromethylbenzoyl chloride to provide 14e as a white solid (50%). Rf = 0.27 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.81 (d, J = 2.5 Hz, 1H), 10.66 (s, 1H), 9.04 (d, J = 7.5 Hz, 1H), 8.58 – 8.27 (m, 2H), 8.05 (d, J = 8.1 Hz, 2H), 7.84 (d, J = 8.3 Hz, 2H), 7.74 (d, J = 7.6 Hz, 1H), 7.66 – 7.56 (m, 2H), 7.38 – 7.28 (m, 1H), 7.26 (d, J = 2.3 Hz, 1H), 7.06 (ddd, J = 8.2, 7.0, 1.3 Hz, 1H), 6.99 (ddd, J = 7.9, 6.8, 1.1 Hz, 1H), 4.92 (ddd, J = 9.3, 7.5, 5.4 Hz, 1H), 3.39 – 3.15 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.87, 165.37, 150.36, 145.48, 137.51, 136.03, 128.43, 127.14, 125.26, 123.82, 120.97, 118.58, 118.24, 113.44, 111.32, 109.84, 55.39, +27.25. MS (ESI) 453.3 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-4-methoxybenzamide (14f)
The procedure for the synthesis of 1 was followed using 4-methoxybenzoyl chloride to provide 14f as a yellow solid (90%). 1H NMR (400 MHz, DMSO-d6) δ 10.81 (d, J = 2.5 Hz, 1H), 10.62 (s, 1H), 8.58 (d, J = 7.6 Hz, 1H), 8.47 – 8.37 (m, 2H), 7.90 – 7.81 (m, 2H), 7.74 (d, J = 7.8 Hz, 1H), 7.66 – 7.58 (m, 2H), 7.31 (dt, J = 8.1, 1.0 Hz, 1H), 7.26 (d, J = 2.4 Hz, 1H), 7.14 – 6.88 (m, 4H), 4.86 (ddd, J = 9.1, 7.5, 5.7 Hz, 1H), 3.80 (d, J = 2.8 Hz, 3H), 3.39 – 3.18 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 172.39, 165.96, 161.71, 150.34, 145.57, 136.02, 129.38, 127.16, 125.94, 123.85, 120.93, 118.61, 118.22, 113.39, 111.31, 110.02, 55.34, 55.25, 27.24. MS (ESI) 415 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2-chlorobenzamide (14g)
The procedure for the synthesis of 1 was followed using 2-chlorobenzoyl chloride to provide 14g as a yellow solid (83%). Rf = 0.54 (10% MeOH in ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.95 – 10.79 (m, 1H), 10.59 (s, 1H), 8.85 (d, J = 7.6 Hz, 1H), 8.50 – 8.35 (m, 2H), 7.68 (d, J = 7.9 Hz, 1H), 7.62 – 7.56 (m, 2H), 7.50 – 7.40 (m, 2H), 7.40 – 7.28 (m, 3H), 7.24 (d, J = 2.3 Hz, 1H), 7.11 – 7.03 (m, 1H), 6.98 (t, J = 7.5 Hz, 1H), 4.90 (td, J = 8.5, 5.9 Hz, 1H), 3.42 – 3.08 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.53, 166.33, 150.35, 145.49, 136.16, 136.04, 130.90, 130.06, 129.59, 129.09, 127.19, 126.89, 123.84, 120.93, 118.53, 118.21, 113.40, 111.28, 109.57, 54.85, 27.39. MS (ESI) 419 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2,5-difluorobenzamide (14h)
The procedure for the synthesis of 1 was followed using 2,5-difluorobenzoyl chloride to provide 14h as a light yellow solid (50%). Rf = 0.39 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 12.31 (s, 1H), 11.00 (d, J = 2.5 Hz, 1H), 8.83 (dd, J = 7.0, 2.8 Hz, 1H), 8.72 (d, J = 7.0 Hz, 2H), 8.31 – 8.15 (m, 2H), 7.72 (d, J = 7.8 Hz, 1H), 7.47 – 7.27 (m, 5H), 7.10 – 6.98 (m, 1H), 6.93 (t, J = 7.4 Hz, 1H), 4.96 (ddd, J = 8.9, 7.0, 5.4 Hz, 1H), 3.50 – 3.18 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 172.85, 162.66, 158.86, 156.79, 156.46, 154.35, 152.93, 142.15, 136.07, 127.15, 124.34, 124.27, 124.18, 124.11, 120.98, 119.60, 119.51, 119.36, 119.27, 118.58, 118.30, 118.23, 118.06, 117.98, 116.53, 116.50, 116.28, 116.25, 114.62, 114.56, 111.38, 108.94, 55.95, 26.99. MS (ESI) 421 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2,4-difluorobenzamide (14i)
The procedure for the synthesis of 1 was followed using 2,4-difluorobenzoyl chloride to provide 14i as a light yellow solid (68%). Rf = 0.39 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.93 – 10.83 (m, 1H), 10.65 (s, 1H), 8.54 (dd, J = 7.4, 3.2 Hz, 1H), 8.49 – 8.39 (m, 2H), 7.66 (dd, J = 8.3, 6.9 Hz, 2H), 7.63 – 7.55 (m, 2H), 7.34 (t, J = 8.6 Hz, 2H), 7.24 (d, J = 2.3 Hz, 1H), 7.17 (td, J = 8.5, 2.4 Hz, 1H), 7.06 (t, J = 7.5 Hz, 1H), 6.97 (t, J = 7.4 Hz, 1H), 4.88 (td, J = 8.2, 5.7 Hz, 1H), 3.53 – 3.08 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.48, 162.83, 162.22, 150.37, 145.45, 136.05, 131.99, 127.15, 123.92, 120.97, 119.77, 119.73, 118.47, 118.25, 113.43, 111.65, 111.33, 109.33, 104.59, 55.13, 27.40. MS (ESI) 421 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-5-bromo-2-fluorobenzamide (14j)
The general procedure A was followed using 5-bromo-2-fluorobenzoic acid to provide 14j as a light yellow solid (67%). 1H NMR (400 MHz, DMSO-d6) δ 10.87 (d, J = 2.4 Hz, 1H), 10.63 (s, 1H), 8.74 (dd, J = 7.5, 2.3 Hz, 1H), 8.51 – 8.37 (m, 2H), 7.72 (ddd, J = 8.7, 4.4, 2.6 Hz, 1H), 7.69 – 7.63 (m, 2H), 7.62 – 7.56 (m, 2H), 7.33 (d, J = 8.1 Hz, 1H), 7.28 (dd, J = 10.0, 8.8 Hz, 1H), 7.22 (d, J = 2.4 Hz, 1H), 7.10 – 7.02 (m, 1H), 6.97 (t, J = 7.5 Hz, 1H), 4.88 (td, J = 8.3, 5.6 Hz, 1H), 3.39 – 3.07 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.33, 162.38, 150.37, 145.41, 136.03, 132.41, 127.17, 123.85, 120.98, 118.55, 118.48, 118.26, 115.94, 113.44, 111.33, 109.37, 55.16, 27.40. MS (ESI) 481/483 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-4-bromo-2-fluorobenzamide (14k)
The general procedure A was followed using 4-bromo-2-fluorobenzoic acid to provide 14k as a yellow solid (70%). Rf = 0.30 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.86 (d, J = 2.3 Hz, 1H), 10.67 (s, 1H), 8.62 (dd, J = 7.4, 2.8 Hz, 1H), 8.51 – 8.36 (m, 2H), 7.70 – 7.63 (m, 2H), 7.63 – 7.58 (m, 2H), 7.51 (d, J = 6.4 Hz, 2H), 7.33 (d, J = 8.0 Hz, 1H), 7.22 (d, J = 2.3 Hz, 1H), 7.06 (t, J = 7.6 Hz, 1H), 6.96 (t, J = 7.4 Hz, 1H), 4.88 (td, J = 8.2, 5.6 Hz, 1H), 3.50 – 3.04 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.44, 162.92, 157.93, 150.14, 145.64, 136.04, 131.77, 127.70, 127.14, 123.90, 120.97, 119.41, 118.47, 118.25, 113.46, 111.33, 109.29, 55.14, 27.39. MS (ESI) 481/483 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2-naphthamide (14l)
The general procedure A was followed using 2-naphthoic acid to provide 14k as a yellow solid (24%). Rf = 0.30 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.83 (s, 1H), 10.67 (s, 1H), 8.91 (d, J = 7.5 Hz, 1H), 8.48 (s, 1H), 8.44 (d, J = 5.4 Hz, 2H), 8.12 – 7.87 (m, 4H), 7.78 (d, J = 7.8 Hz, 1H), 7.70 – 7.52 (m, 4H), 7.32 (d, J = 7.9 Hz, 2H), 7.03 (dt, J = 23.4, 7.2 Hz, 2H), 4.96 (q, J = 7.5 Hz, 1H), 3.44 – 3.21 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 172.15, 166.52, 150.35, 145.54, 136.04, 134.20, 132.03, 131.08, 128.82, 127.88, 127.77, 127.69, 127.61, 127.22, 126.74, 124.31, 123.86, 120.96, 118.63, 118.25, 113.43, 111.33, 109.98, 55.42, 27.33. MS (ESI) 435 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-1-fluoro-2-naphthamide (14m)
The procedure for the synthesis of 1 was followed using 1-fluoro-2-naphthoic acid to provide 14m as a yellow solid (61%). 1H NMR (400 MHz, DMSO-d6) δ 10.97 – 10.83 (m, 1H), 10.67 (s, 1H), 8.67 (dd, J = 7.5, 3.5 Hz, 1H), 8.51 – 8.42 (m, 2H), 8.23 – 8.08 (m, 1H), 8.08 – 7.98 (m, 1H), 7.82 (d, J = 8.5 Hz, 1H), 7.75 – 7.56 (m, 6H), 7.34 (d, J = 8.0 Hz, 1H), 7.27 (d, J = 2.3 Hz, 1H), 7.06 (t, J = 7.5 Hz, 1H), 6.96 (t, J = 7.4 Hz, 1H), 4.97 (td, J = 8.0, 5.6 Hz, 1H), 3.48 – 3.15 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.51, 163.76, 150.31, 145.52, 136.06, 135.32, 128.53, 127.68, 127.45, 127.22, 125.52, 123.89, 123.76, 122.60, 122.43, 120.97, 118.49, 118.26, 113.45, 111.33, 109.39, 55.21, 27.49. MS (ESI) 453 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-6-fluoro-2-naphthamide (14n)
The general procedure A was followed using 6-fluoro-2-naphthoic acid to provide 14n as a light yellow solid (42%). Rf = 0.27 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.83 (d, J = 2.5 Hz, 1H), 10.67 (s, 1H), 8.91 (d, J = 7.6 Hz, 1H), 8.52 (d, J = 1.2 Hz, 1H), 8.48 – 8.38 (m, 2H), 8.10 (dd, J = 9.1, 5.8 Hz, 1H), 7.97 (d, J = 1.2 Hz, 2H), 7.84 – 7.73 (m, 2H), 7.69 – 7.60 (m, 2H), 7.51 (td, J = 8.9, 2.6 Hz, 1H), 7.40 – 7.26 (m, 2H), 7.06 (ddd, J = 8.2, 7.0, 1.3 Hz, 1H), 7.03 – 6.95 (m, 1H), 4.96 (ddd, J = 9.1, 7.5, 5.6 Hz, 1H), 3.44 – 3.18 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 172.12, 166.33, 159.74, 150.31, 145.58, 136.04, 135.22, 135.13, 131.96, 131.86, 130.65, 129.23, 128.01, 127.31, 127.21, 125.40, 123.85, 120.96, 118.62, 118.26, 116.89, 113.44, 111.34, 109.95, 55.41, 27.35. MS (ESI) 453 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-6-bromo-2-naphthamide (14o)
The general procedure A was followed using 6-bromo-2-naphthoic acid to provide 14o as a light yellow solid (76%). Rf = 0.36 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.92 – 10.74 (m, 2H), 8.98 (d, J = 7.5 Hz, 1H), 8.57 – 8.41 (m, 3H), 8.28 (d, J = 2.0 Hz, 1H), 7.98 (d, J = 6.1 Hz, 3H), 7.85 – 7.63 (m, 4H), 7.32 (d, J = 7.8 Hz, 2H), 7.03 (dt, J = 24.7, 7.2 Hz, 2H), 4.96 (td, J = 8.4, 5.7 Hz, 1H), 3.32 (qd, J = 14.6, 7.3 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 172.23, 166.27, 149.45, 146.35, 136.03, 135.29, 131.63, 131.03, 130.58, 129.80, 129.59, 127.95, 127.19, 127.07, 125.49, 123.88, 121.07, 120.96, 118.60, 118.25, 113.56, 111.34, 109.88, 55.54, 27.29. MS (ESI) 515 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-[1,1'-biphenyl]-4-carboxamide (14p)
The general procedure A was followed using [1,1'-biphenyl]-4-carboxylic acid to provide 14p as a light yellow solid (28%). Rf = 0.36 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.83 (d, J = 2.5 Hz, 1H), 10.66 (s, 1H), 8.81 (d, J = 7.6 Hz, 1H), 8.51 – 8.39 (m, 2H), 7.96 (d, J = 8.4 Hz, 2H), 7.81 – 7.70 (m, 5H), 7.67 – 7.60 (m, 2H), 7.56 – 7.45 (m, 2H), 7.44 – 7.37 (m, 1H), 7.32 (d, J = 8.1 Hz, 1H), 7.29 (d, J = 2.3 Hz, 1H), 7.11 – 7.03 (m, 1H), 7.00 (t, J = 7.4 Hz, 1H), 4.92 (td, J = 8.1, 7.7, 5.8 Hz, 1H), 3.45 – 3.21 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 172.21, 166.15, 150.36, 145.56, 142.92, 139.10, 136.04, 132.54, 129.02, 128.21, 128.07, 127.17, 126.86, 126.40, 123.86, 120.95, 118.61, 118.24, 113.42, 111.32, 109.98, 55.32, 27.25. MS (ESI) 461 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-[1,1'-biphenyl]-3-carboxamide (14q)
The general procedure A was followed using [1,1'-biphenyl]-3-carboxylic acid to provide 14q as a yellow solid (42%). Rf = 0.30 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.90 – 10.80 (m, 1H), 10.66 (s, 1H), 8.94 (d, J = 7.7 Hz, 1H), 8.50 – 8.37 (m, 2H), 8.14 (d, J = 1.8 Hz, 1H), 7.83 (dd, J = 7.8, 1.8 Hz, 2H), 7.78 (d, J = 7.8 Hz, 1H), 7.76 – 7.71 (m, 2H), 7.66 – 7.61 (m, 2H), 7.53 (dt, J = 11.7, 7.7 Hz, 3H), 7.41 (t, J = 7.3 Hz, 1H), 7.35 – 7.26 (m, 2H), 7.05 (t, J = 7.5 Hz, 1H), 6.99 (t, J = 7.4 Hz, 1H), 5.01 – 4.88 (m, 1H), 3.42 – 3.17 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 172.15, 166.41, 150.36, 145.53, 140.07, 139.47, 136.01, 134.42, 129.59, 128.98, 128.95, 127.78, 127.22, 126.84, 126.73, 125.65, 123.81, 120.94, 118.62, 118.25, 113.42, 111.32, 110.03, 55.44, 27.30. MS (ESI) 461 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2-fluoro-[1,1'-biphenyl]-4-carboxamide (14r)
The general procedure A was followed using 3-fluoro-[1,1'-biphenyl]-4-carboxylic acid to provide 14r as a light yellow solid (78%). Rf = 0.36 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.83 (d, J = 2.5 Hz, 1H), 10.66 (s, 1H), 8.93 (d, J = 7.6 Hz, 1H), 8.60 – 8.35 (m, 2H), 7.91 – 7.71 (m, 3H), 7.70 – 7.56 (m, 5H), 7.55 – 7.40 (m, 3H), 7.32 (d, J = 7.9 Hz, 1H), 7.28 (d, J = 2.3 Hz, 1H), 7.10 – 7.03 (m, 1H), 7.00 (t, J = 7.5 Hz, 1H), 4.92 (ddd, J = 9.3, 7.4, 5.4 Hz, 1H), 3.33 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.99, 164.88, 159.88, 157.44, 150.36, 145.53, 136.04, 134.24, 131.02, 130.71, 128.85, 128.83, 128.71, 128.40, 127.15, 124.04, 123.86, 120.98, 118.60, 118.26, 113.46, 111.35, 109.89, 55.38, 27.25. MS (ESI) 479 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-3-fluoro-[1,1'-biphenyl]-4-carboxamide (14s)
The general procedure A was followed using 16s to provide 14s as a yellow solid (37%). 1H NMR (400 MHz, DMSO-d6) δ 10.88 (d, J = 2.7 Hz, 1H), 10.64 (s, 1H), 8.58 – 8.37 (m, 3H), 7.79 – 7.57 (m, 8H), 7.54 – 7.46 (m, 2H), 7.47 – 7.40 (m, 1H), 7.36 – 7.31 (m, 1H), 7.25 (d, J = 2.4 Hz, 1H), 7.06 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 6.96 (ddd, J = 8.1, 6.9, 1.0 Hz, 1H), 4.91 (td, J = 7.9, 5.5 Hz, 1H), 3.32 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.49, 150.35, 145.44, 137.75, 136.05, 129.07, 127.16, 126.90, 123.91, 121.49, 121.35, 120.95, 118.46, 118.25, 113.92, 113.42, 109.31, 55.08, 27.42. MS (ESI) 479 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-3,3'-difluoro-[1,1'-biphenyl]-4-carboxamide (14t)
The general procedure A was followed using 16t to provide 14t as a light yellow solid (54%). Rf = 0.45 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.88 (d, J = 2.4 Hz, 1H), 10.65 (s, 1H), 8.53 (dd, J = 7.4, 3.8 Hz, 1H), 8.45 (d, J = 5.6 Hz, 2H), 7.88 – 7.58 (m, 8H), 7.58 – 7.47 (m, 1H), 7.33 (d, J = 8.1 Hz, 1H), 7.31 – 7.19 (m, 2H), 7.06 (t, J = 7.5 Hz, 1H), 6.97 (t, J = 7.5 Hz, 1H), 4.97 – 4.84 (m, 1H), 3.46 – 3.17 (m, 2H). 13C NMR (101 MHz, DMSO) δ 171.49, 163.89, 163.22, 161.47, 161.08, 158.60, 150.37, 145.46, 136.07, 131.09, 131.00, 127.18, 123.92, 123.03, 122.61, 122.17, 122.03, 120.98, 118.48, 118.26, 113.87, 113.65, 113.43, 111.34, 109.34, 55.11, 27.43. MS (ESI) 497 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-3,4'-difluoro-[1,1'-biphenyl]-4-carboxamide (27a)
The general procedure A was followed using 16a to provide 27a as a light yellow solid (64%). Rf = 0.42 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.88 (d, J = 2.6 Hz, 1H), 10.67 (s, 1H), 8.54 – 8.39 (m, 3H), 7.88 – 7.76 (m, 2H), 7.74 – 7.55 (m, 6H), 7.33 (ddd, J = 8.8, 6.7, 2.2 Hz, 3H), 7.25 (d, J = 2.4 Hz, 1H), 7.06 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 7.01 – 6.88 (m, 1H), 4.91 (td, J = 8.1, 5.6 Hz, 1H), 3.45 – 3.17 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.53, 163.25, 161.28, 161.12, 158.64, 150.21, 145.61, 143.65, 143.57, 136.07, 130.98, 129.15, 129.06, 127.18, 123.92, 122.42, 120.98, 118.47, 118.26, 116.03, 115.82, 113.94, 113.46, 111.34, 109.32, 55.12, 27.42. MS (ESI) 497 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2',3-difluoro-[1,1'-biphenyl]-4-carboxamide (27b)
The general procedure A was followed using 16b to provide 27b as a light yellow solid (74%). 1H NMR (400 MHz, DMSO-d6) δ 10.88 (d, J = 2.5 Hz, 1H), 10.67 (s, 1H), 8.58 (dd, J = 7.3, 3.5 Hz, 1H), 8.53 – 8.36 (m, 2H), 7.73 – 7.64 (m, 2H), 7.64 – 7.57 (m, 3H), 7.48 (ddt, J = 9.7, 6.4, 1.4 Hz, 3H), 7.40 – 7.30 (m, 3H), 7.25 (d, J = 2.3 Hz, 1H), 7.06 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 6.97 (ddd, J = 7.9, 6.9, 1.0 Hz, 1H), 4.91 (td, J = 8.2, 5.6 Hz, 1H), 3.44 – 3.15 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.53, 163.31, 160.49, 160.24, 157.79, 150.14, 145.68, 139.51, 136.07, 130.75, 130.49, 127.17, 125.14, 125.10, 123.94, 122.30, 122.16, 120.98, 118.48, 118.26, 116.40, 116.18, 113.49, 111.34, 109.33, 55.14, 27.41. MS (ESI) 497 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-3,3',5'-trifluoro-[1,1'-biphenyl]-4-carboxamide (27c)
The general procedure A was followed using 16c to provide 27c as a light yellow solid (73%). Rf = 0.30 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.88 (d, J = 2.5 Hz, 1H), 10.65 (s, 1H), 8.57 (dd, J = 7.4, 3.6 Hz, 1H), 8.51 – 8.34 (m, 2H), 7.83 – 7.72 (m, 1H), 7.73 – 7.64 (m, 3H), 7.63 – 7.55 (m, 4H), 7.37 – 7.27 (m, 2H), 7.25 (d, J = 2.3 Hz, 1H), 7.06 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 7.01 – 6.93 (m, 1H), 4.91 (td, J = 8.2, 5.6 Hz, 1H), 3.40 – 3.16 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.48, 164.17, 164.03, 163.18, 161.72, 161.59, 161.01, 150.37, 145.46, 136.07, 130.93, 127.18, 123.92, 122.81, 122.72, 122.68, 120.99, 118.49, 118.27, 116.00, 114.71, 114.48, 113.45, 111.34, 110.37, 110.11, 109.34, 55.13, 27.43. MS (ESI) 515 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2',3,5'-trifluoro-[1,1'-biphenyl]-4-carboxamide (27d)
The general procedure A was followed using 16d to provide 27d as a light yellow solid (70%). Rf = 0.42 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.88 (d, J = 2.5 Hz, 1H), 10.64 (s, 1H), 8.61 (dd, J = 7.3, 3.3 Hz, 1H), 8.45 (d, J = 5.5 Hz, 2H), 7.73 – 7.65 (m, 2H), 7.63 – 7.58 (m, 2H), 7.57 – 7.47 (m, 3H), 7.42 (td, J = 9.5, 4.7 Hz, 1H), 7.37 – 7.29 (m, 2H), 7.25 (d, J = 2.4 Hz, 1H), 7.06 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 6.97 (ddd, J = 8.0, 6.9, 1.0 Hz, 1H), 4.91 (td, J = 7.9, 5.5 Hz, 1H), 3.46 – 3.14 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.47, 163.26, 162.71, 160.45, 159.54, 157.97, 157.14, 156.42, 152.87, 150.62, 150.36, 145.48, 136.06, 130.54, 127.18, 123.93, 120.98, 118.48, 118.27, 117.12, 113.45, 111.34, 109.34, 55.13, 27.42. MS (ESI) 515 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-4'-chloro-3-fluoro-[1,1'-biphenyl]-4-carboxamide (27e)
The general procedure A was followed using 16e to provide 27e as a light yellow solid (60%). Rf = 0.32 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.88 (d, J = 2.6 Hz, 1H), 10.65 (s, 1H), 8.51 (dd, J = 7.3, 3.9 Hz, 1H), 8.47 – 8.36 (m, 2H), 7.84 – 7.76 (m, 2H), 7.75 – 7.65 (m, 3H), 7.62 (ddd, J = 10.6, 5.3, 1.8 Hz, 3H), 7.58 – 7.52 (m, 2H), 7.33 (d, J = 8.1 Hz, 1H), 7.25 (d, J = 2.4 Hz, 1H), 7.06 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 6.96 (ddd, J = 8.0, 7.0, 1.1 Hz, 1H), 4.91 (td, J = 8.0, 5.4 Hz, 1H), 3.45 – 3.15 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.50, 163.25, 161.12, 158.64, 154.72, 153.81, 150.35, 145.49, 136.58, 136.07, 133.57, 131.04, 129.04, 128.75, 127.19, 123.93, 122.46, 121.74, 120.99, 118.48, 118.27, 114.25, 114.01, 113.45, 111.35, 109.33, 55.12, 27.43. MS (ESI) 513 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-3'-chloro-3-fluoro-[1,1'-biphenyl]-4-carboxamide (27f)
The general procedure A was followed using 16f to provide 27f, which was further purified by HPLC (36%, a white solid). 1H NMR (400 MHz, DMSO-d6) δ 10.88 (d, J = 2.5 Hz, 1H), 10.66 (s, 1H), 8.54 (dd, J = 7.3, 3.8 Hz, 1H), 8.49 – 8.39 (m, 2H), 7.85 (t, J = 1.9 Hz, 1H), 7.78 – 7.63 (m, 5H), 7.63 – 7.59 (m, 2H), 7.56 – 7.46 (m, 2H), 7.37 – 7.31 (m, 1H), 7.25 (d, J = 2.4 Hz, 1H), 7.06 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 6.96 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H), 4.92 (td, J = 7.9, 5.5 Hz, 1H), 3.44 – 3.15 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.50, 163.24, 161.07, 158.59, 150.28, 145.55, 143.03, 142.95, 139.89, 136.06, 133.93, 130.97, 130.89, 128.45, 127.18, 126.71, 125.67, 123.92, 122.69, 122.21, 122.07, 120.99, 118.48, 118.27, 114.55, 114.31, 113.45, 111.34, 109.34, 55.13, 27.43. MS (ESI) 513 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2'-chloro-3-fluoro-[1,1'-biphenyl]-4-carboxamide (27g)
The general procedure A was followed using 16g to provide 27g as a white solid (80%). Rf = 0.51 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.88 (d, J = 2.4 Hz, 1H), 10.65 (s, 1H), 8.62 (dd, J = 7.3, 3.2 Hz, 1H), 8.51 – 8.41 (m, 2H), 7.75 – 7.64 (m, 2H), 7.61 (dd, J = 4.8, 1.6 Hz, 4H), 7.49 – 7.43 (m, 3H), 7.38 (dd, J = 11.4, 1.6 Hz, 1H), 7.34 (dd, J = 7.9, 1.8 Hz, 2H), 7.26 (d, J = 2.4 Hz, 1H), 7.06 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 6.97 (ddd, J = 8.0, 6.9, 1.0 Hz, 1H), 4.91 (td, J = 8.3, 5.6 Hz, 1H), 3.43 – 3.17 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 172.59, 171.53, 163.42, 160.13, 157.65, 155.83, 150.29, 145.55, 143.07, 137.78, 136.07, 131.62, 131.39, 131.11, 130.16, 130.03, 129.98, 127.67, 127.17, 125.44, 123.95, 122.42, 122.28, 120.99, 118.49, 118.26, 117.14, 116.90, 113.45, 111.34, 109.37, 55.15, 27.40. MS (ESI) 513.3 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-3,3',4'-trifluoro-[1,1'-biphenyl]-4-carboxamide (27h)
The general procedure A was followed using 16h to provide 27h as a white solid (78%). Rf = 0.45 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.88 (d, J = 2.6 Hz, 1H), 10.65 (s, 1H), 8.53 (dd, J = 7.3, 3.8 Hz, 1H), 8.49 – 8.38 (m, 2H), 7.92 (ddd, J = 12.1, 7.7, 2.3 Hz, 1H), 7.75 – 7.49 (m, 8H), 7.33 (d, J = 8.0 Hz, 1H), 7.25 (d, J = 2.3 Hz, 1H), 7.06 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 7.01 – 6.90 (m, 1H), 4.91 (td, J = 8.1, 5.6 Hz, 1H), 3.46 – 3.16 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.50, 163.22, 161.07, 158.59, 150.35, 145.49, 136.07, 130.97, 127.18, 123.93, 122.55, 122.08, 121.95, 120.99, 118.48, 118.27, 118.18, 118.00, 116.29, 116.11, 114.45, 114.21, 113.45, 111.35, 109.34, 55.14, 27.43. MS (ESI) 515.3 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-3,3'-difluoro-4'-methoxy-[1,1'-biphenyl]-4-carboxamide (27i)
The general procedure A was followed using 16i to provide 27i, which was further purified by HPLC (45%, a white solid). 1H NMR (400 MHz, DMSO-d6) δ 10.89 (d, J = 2.5 Hz, 1H), 10.81 (s, 1H), 8.48 (dd, J = 7.0, 4.2 Hz, 3H), 7.74 – 7.64 (m, 5H), 7.64 – 7.58 (m, 3H), 7.37 – 7.31 (m, 1H), 7.31 – 7.23 (m, 2H), 7.06 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 6.96 (ddd, J = 8.0, 6.9, 1.0 Hz, 1H), 4.90 (td, J = 7.8, 5.6 Hz, 1H), 3.89 (s, 3H), 3.53 – 3.16 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.77, 163.31, 158.73, 152.96, 150.54, 149.03, 147.66, 147.56, 146.71, 136.07, 130.89, 127.16, 123.96, 123.22, 121.00, 118.45, 118.28, 114.27, 114.22, 113.74, 113.65, 113.51, 111.36, 109.25, 56.14, 55.21, 27.34. MS (ESI) 527.2 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-3'-cyano-3-fluoro-[1,1'-biphenyl]-4-carboxamide (27j)
The general procedure A was followed using 16j to provide 27j as a white solid (47%). 1H NMR (400 MHz, DMSO-d6) δ 10.89 (d, J = 2.5 Hz, 1H), 10.80 (s, 1H), 8.60 (dd, J = 7.4, 3.6 Hz, 1H), 8.48 (d, J = 5.6 Hz, 2H), 8.29 (t, J = 1.8 Hz, 1H), 8.12 (dt, J = 8.1, 1.3 Hz, 1H), 7.90 (dt, J = 7.8, 1.4 Hz, 1H), 7.80 – 7.74 (m, 1H), 7.74 – 7.64 (m, 6H), 7.33 (d, J = 8.1 Hz, 1H), 7.26 (d, J = 2.4 Hz, 1H), 7.11 – 7.02 (m, 1H), 6.96 (t, J = 7.3 Hz, 1H), 4.92 (td, J = 8.1, 5.6 Hz, 1H), 3.29 (dqt, J = 23.3, 14.6, 9.3, 8.5 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.68, 163.27, 161.08, 158.60, 149.34, 142.31, 138.83, 136.06, 132.14, 131.71, 131.03, 130.63, 130.27, 127.17, 123.95, 122.75, 120.99, 118.59, 118.47, 118.28, 114.70, 114.46, 113.60, 112.28, 111.35, 109.30, 55.23, 27.36. MS (ESI) 504 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-4'-(benzyloxy)-3,3'-difluoro-[1,1'-biphenyl]-4-carboxamide (27k)
The general procedure A was followed using 16k to provide 27k as a light yellow solid (88%). Rf = 0.39 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.90 (d, J = 2.5 Hz, 1H), 10.69 (s, 1H), 8.48 (dd, J = 7.4, 4.2 Hz, 1H), 7.87 – 7.52 (m, 8H), 7.51 – 7.38 (m, 5H), 7.38 – 7.30 (m, 4H), 7.25 (d, J = 2.4 Hz, 1H), 7.11 – 7.02 (m, 1H), 6.96 (t, J = 7.4 Hz, 1H), 5.26 (s, 2H), 4.91 (td, J = 8.0, 5.6 Hz, 1H), 3.40 – 3.16 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.55, 163.25, 161.21, 158.73, 153.26, 150.84, 150.22, 146.62, 146.52, 145.55, 143.14, 143.06, 136.39, 136.07, 130.93, 130.90, 130.83, 130.78, 128.53, 128.11, 127.80, 127.19, 123.94, 123.17, 123.14, 122.00, 121.97, 121.19, 121.06, 120.99, 118.49, 118.27, 115.72, 114.67, 114.47, 113.78, 113.54, 111.35, 109.33, 70.24, 55.13, 29.60, 27.43. MS (ESI) 603 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-3,3'-difluoro-4'-hydroxy-[1,1'-biphenyl]-4-carboxamide (27l)
The procedure for the synthesis of 3j was followed using 27k to provide 27l as a brown solid (81%). 1H NMR (400 MHz, DMSO-d6) δ 10.88 (d, J = 2.5 Hz, 1H), 10.75 (s, 1H), 10.20 (s, 1H), 8.60 – 8.36 (m, 3H), 7.71 – 7.53 (m, 7H), 7.45 (dd, J = 8.4, 2.2 Hz, 1H), 7.33 (d, J = 8.1 Hz, 1H), 7.25 (d, J = 2.4 Hz, 1H), 7.10 – 7.00 (m, 2H), 6.96 (t, J = 7.4 Hz, 1H), 4.90 (td, J = 8.0, 5.6 Hz, 1H), 3.47 – 3.11 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.70, 158.76, 149.42, 136.07, 127.16, 126.73, 123.95, 123.20, 121.74, 120.99, 118.46, 118.27, 111.36, 109.27, 55.17, 27.37. MS (ESI) 513 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-4'-amino-3-fluoro-[1,1'-biphenyl]-4-carboxamide (27m)
The general procedure A was followed using 16l to provide 27m as a yellow solid (59%). 1H NMR (400 MHz, DMSO-d6) δ 10.88 (d, J = 2.5 Hz, 1H), 10.64 (s, 1H), 8.52 – 8.38 (m, 2H), 8.27 (dd, J = 7.2, 5.3 Hz, 1H), 7.64 (dd, J = 8.1, 6.2 Hz, 2H), 7.62 – 7.57 (m, 2H), 7.51 – 7.41 (m, 4H), 7.33 (d, J = 8.1 Hz, 1H), 7.24 (d, J = 2.4 Hz, 1H), 7.06 (ddd, J = 8.2, 6.9, 1.2 Hz, 1H), 6.99 – 6.92 (m, 1H), 6.64 (d, J = 8.6 Hz, 2H), 5.47 (s, 2H), 4.90 (td, J = 7.8, 5.5 Hz, 1H), 3.49 – 3.16 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.58, 163.30, 161.48, 150.32, 149.68, 145.52, 136.08, 127.59, 127.19, 124.41, 123.93, 120.99, 120.66, 118.92, 118.79, 118.47, 118.27, 114.04, 113.44, 112.05, 111.81, 111.35, 109.29, 55.06, 27.45. MS (ESI) 494 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2-fluoro-4-(phenylethynyl)benzamide (27n)
The general procedure A was followed using 21b to provide 27n as a light yellow solid (86%). 1H NMR (400 MHz, DMSO-d6) δ 10.89 (d, J = 2.5 Hz, 1H), 10.68 (s, 1H), 8.66 (dd, J = 7.4, 3.1 Hz, 1H), 8.52 – 8.36 (m, 2H), 7.68 (d, J = 7.9 Hz, 1H), 7.65 – 7.56 (m, 5H), 7.52 (dd, J = 11.1, 1.5 Hz, 1H), 7.45 (dd, J = 7.0, 2.8 Hz, 4H), 7.34 (d, J = 8.1 Hz, 1H), 7.25 (d, J = 2.4 Hz, 1H), 7.07 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 7.01 – 6.93 (m, 1H), 4.97 – 4.84 (m, 1H), 3.34 – 3.14 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.45, 163.05, 160.32, 157.83, 150.39, 145.45, 136.07, 131.59, 130.76, 130.72, 129.43, 128.86, 127.54, 127.51, 127.17, 126.43, 126.33, 123.93, 123.38, 123.24, 121.50, 121.00, 118.90, 118.65, 118.50, 118.27, 113.44, 111.35, 109.34, 91.92, 87.54, 87.51, 55.15, 27.42. MS (ESI) 503.3 m/z [M + H]+.
(S,E)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-4-(4-chlorostyryl)-2-fluorobenzamide (27o)
The general procedure A was followed using 17 to provide 27o as a light yellow solid (78%). Rf = 0.51 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.88 (d, J = 2.5 Hz, 1H), 10.67 (s, 1H), 8.55 – 8.33 (m, 3H), 7.72 – 7.57 (m, 6H), 7.54 (dd, J = 12.4, 1.5 Hz, 1H), 7.51 – 7.41 (m, 4H), 7.37 – 7.28 (m, 2H), 7.24 (d, J = 2.4 Hz, 1H), 7.06 (ddd, J = 8.1, 7.0, 1.2 Hz, 1H), 6.96 (ddd, J = 8.0, 6.9, 1.0 Hz, 1H), 4.90 (td, J = 8.0, 5.6 Hz, 1H), 3.44 – 3.12 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.56, 163.23, 158.65, 150.17, 145.66, 141.97, 141.89, 136.07, 135.40, 132.67, 130.79, 130.31, 128.82, 128.50, 127.18, 123.94, 122.66, 121.32, 120.99, 118.48, 118.26, 113.46, 113.27, 111.35, 109.31, 55.13, 27.43. MS (ESI) 539.3 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2-fluoro-4-(phenylamino)benzamide (27p)
The general procedure A was followed using 20 to provide 27p as a yellow solid (88%). Rf = 0.33 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 10.89 (d, J = 2.5 Hz, 1H), 10.66 (s, 1H), 8.83 (s, 1H), 8.53 – 8.38 (m, 2H), 8.18 (s, 1H), 7.81 (t, J = 7.4 Hz, 1H), 7.70 – 7.54 (m, 3H), 7.42 – 7.27 (m, 3H), 7.22 (d, J = 2.3 Hz, 1H), 7.17 (d, J = 7.8 Hz, 2H), 7.03 (dt, J = 19.1, 7.4 Hz, 2H), 6.94 (t, J = 7.4 Hz, 1H), 6.85 (dd, J = 8.7, 2.2 Hz, 1H), 6.74 (dd, J = 14.4, 2.2 Hz, 1H), 4.96 – 4.80 (m, 1H), 3.27 (qd, J = 14.7, 6.8 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.74, 162.93, 162.90, 162.54, 160.09, 150.20, 148.88, 148.76, 145.65, 140.97, 136.11, 132.11, 132.07, 129.42, 127.19, 123.94, 122.23, 121.01, 119.48, 118.46, 118.28, 113.45, 111.37, 111.04, 110.92, 110.71, 109.20, 100.72, 100.44, 54.93, 27.56. MS (ESI) 494.2 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2-fluoro-4-morpholinobenzamide (27q)
The general procedure A was followed using 16m to provide 27q as a white solid (49%). Rf = 0.48 (10% MeOH in ethyl acetate), Rf = 0.15 (100% ethyl acetate). 1H NMR (400 MHz, DMSO-d6) δ 11.29 (s, 1H), 10.91 (d, J = 2.5 Hz, 1H), 8.68 – 8.49 (m, 2H), 7.96 – 7.78 (m, 3H), 7.70 – 7.53 (m, 2H), 7.32 (d, J = 8.1 Hz, 1H), 7.24 (d, J = 2.4 Hz, 1H), 7.04 (ddd, J = 8.1, 6.9, 1.1 Hz, 1H), 6.93 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H), 6.86 – 6.69 (m, 2H), 4.88 (q, J 13 = 6.8 Hz, 1H), 3.71 (dd, J = 5.9, 3.9 Hz, 4H), 3.44 – 3.18 (m, 6H). 13C NMR (101 MHz, DMSO-d6) δ 172.48, 136.08, 127.12, 124.03, 118.40, 118.29, 114.00, 111.37, 110.19, 109.53, 109.01, 65.71, 46.89, 45.87, 25.93. MS (ESI) 488.3 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2-fluoro-4-(4-(phenylsulfonyl)piperazin-1-yl)benzamide (27r)
The general procedure A was followed using 25b to provide 27r as a white solid (56%). 1H NMR (400 MHz, DMSO-d6) δ 10.85 (d, J = 2.5 Hz, 1H), 10.61 (s, 1H), 8.43 (d, J = 5.5 Hz, 2H), 7.85 – 7.70 (m, 4H), 7.70 – 7.62 (m, 2H), 7.62 – 7.51 (m, 4H), 7.31 (d, J 1 = 8.1 Hz, 1H), 7.18 (d, J = 2.4 Hz, 1H), 7.04 (ddd, J = 8.0, 7.0, 1.1 Hz, 1H), 6.92 (ddd, J = 8.0, 7.0, 1.1 Hz, 1H), 6.79 – 6.67 (m, 2H), 4.92 – 4.79 (m, 1H), 3.39 (t, J = 5.1 Hz, 4H), 3.35 – 3.14 (m, 2H), 2.97 (t, J = 5.0 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 171.65, 162.87, 162.49, 160.03, 153.45, 150.22, 145.57, 136.06, 134.56, 133.45, 131.65, 131.60, 129.51, 127.58, 127.17, 123.89, 120.97, 118.41, 118.25, 113.42, 111.33, 110.86, 110.23, 109.18, 101.42, 54.91, 46.30, 45.34, 27.50. MS (ESI) 627 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2-fluoro-4-(piperazin-1-yl)benzamide hydrochloride (27s)
The general procedure A was followed using 4-(4-(tert-butoxycarbonyl)piperazin-1-yl)-2-fluorobenzoic acid obtained by hydrolysis of 23 (79%). 1H NMR (400 MHz, CDCl3) δ 9.37 (s, 1H), 8.60 (d, J = 2.5 Hz, 1H), 8.31 – 8.21 (m, 2H), 7.81 (t, J = 9.1 Hz, 1H), 7.56 (d, J = 7.9 Hz, 1H), 7.44 – 7.28 (m, 4H), 7.14 – 7.02 (m, 2H), 6.91 (t, J = 7.5 Hz, 1H), 6.61 (dd, J = 9.1, 2.4 Hz, 1H), 6.41 (dd, J = 16.0, 2.4 Hz, 1H), 5.07 (q, J = 6.6, 6.1 Hz, 1H), 3.54 (dd, J = 6.8, 3.9 Hz, 4H), 3.41 – 3.33 (m, 2H), 3.25 (dd, J = 6.6, 4.1 Hz, 4H), 1.47 (s, 9H). The procedure for the synthesis of 3c was followed to provide 27s, which was further purified by HPLC (83%, a brown solid). 1H NMR (400 MHz, DMSO-d6) δ 12.43 (s, 1H), 11.03 (d, J = 2.5 Hz, 1H), 9.63 (s, 2H), 8.72 (d, J = 6.9 Hz, 2H), 8.34 – 8.18 (m, 2H), 8.02 (t, J = 6.8 Hz, 1H), 7.69 (d, J = 7.9 Hz, 1H), 7.61 (t, J = 8.9 Hz, 1H), 7.37 – 7.28 (m, 2H), 7.03 (t, J = 7.5 Hz, 1H), 6.90 (t, J = 7.6 Hz, 1H), 6.87 – 6.75 (m, 2H), 4.95 (dt, J = 8.2, 6.2 Hz, 1H), 3.56 (t, J = 5.1 Hz, 4H), 3.37 (qd, J = 14.6, 6.9 Hz, 2H), 3.15 (p, J = 4.3 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 173.23, 163.08, 163.06, 162.64, 160.19, 153.49, 153.37, 153.11, 142.02, 136.12, 131.78, 131.74, 127.16, 124.23, 121.01, 118.58, 118.33, 114.57, 111.41, 111.01, 110.88, 110.27, 108.91, 101.64, 101.36, 55.80, 43.91, 42.01, 27.01. MS (ESI) 487.3 m/z [M + H]+.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2-fluoro-4-(4-(4-fluorobenzyl)piperazin-1-yl)benzamide hydrochloride (27t)
The general procedure A was followed using 26b to provide 27t, which was further purified by HPLC (26%, a white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.59 (s, 1H), 10.96 (d, J = 2.5 Hz, 1H), 8.69 (d, J = 6.8 Hz, 2H), 8.12 – 7.93 (m, 3H), 7.72 – 7.53 (m, 5H), 7.33 (ddd, J = 8.7, 6.3, 2.8 Hz, 3H), 7.26 (d, J = 2.4 Hz, 1H), 7.13 – 7.00 (m, 1H), 6.93 (t, J = 7.4 Hz, 1H), 6.90 – 6.77 (m, 2H), 4.87 (dt, J = 8.2, 6.0 Hz, 1H), 4.36 (s, 2H), 3.86 – 3.02 (m, 7H). 13C NMR (101 MHz, DMSO-d6) δ 172.94, 163.99, 163.11, 161.54, 160.12, 158.53, 158.21, 153.03, 152.92, 151.54, 143.88, 136.11, 133.64, 133.55, 131.73, 127.08, 126.05, 124.13, 121.05, 118.34, 115.94, 115.72, 115.50, 114.39, 111.43, 111.26, 111.13, 110.33, 108.90, 101.72, 101.44, 57.87, 55.52, 49.87, 44.16, 27.04. MS (ESI) 595.4 m/z [M + H]+, 298.3 m/z [M + 2H]2+
General procedure B—synthesis of substituted biphenyl carboxlic acids
A mixture of 2-fluoro-4-bromobenzoic acid (15) (ca. 0.10 g, 0.46 mmol), phenyl boronic acid (ca. 1.1 eq), Pd2(dba)3 (3 mol%), PCy3 (6 mol %), and K3PO4 (2 M, 1 mL) in dioxane (4 mL) was stirred under microwave heating (100 °C) for 1 h. The palladium catalyst was removed by filtration. The filtrate was acidified with 2N HCl (aq), and a white solid precipitated. The product mixture was diluted with ethyl acetate (30 mL) and washed with water (10 mL × 2) and brine (10 mL × 2). The organic layer was dried over magnesium sulfate, filtered, and concentrated in vacuo. The resulting product was purified by flash chromatography to afford 2-fluorobiphenylcarboxylic acid 16 in ca. 90% yield.
Hepatic microsomal stability
Microsome stability was evaluated by incubating 1 µM compound with 1 mg/mL hepatic microsomes (human, rat, or mouse) in 100 mM potassium phosphate buffer, pH 7.4 at 37 °C with continuous shaking. The reaction was initiated by adding NADPH, 1 mM final concentration. The final incubation volume was 300 µL and 40 µL aliquots were removed at 0, 5, 10, 20, 40, and 60 minutes. The aliquots were added to 160 µL acetonitrile to stop the reaction and precipitate the protein. NADPH dependence of the reaction is evaluated in parallel incubations without NADPH. At the end of the assay, the samples are centrifuged through a 0.45 micron filter plate (Millipore Solventer low binding hydrophilic plates, cat# MSRLN0450) and analyzed by LC-MS/MS. The data were log transformed and results are reported as half-life.
P450 inhibition
Cytochrome P450 inhibition was evaluated in human liver microsomes using four selective marker substrates (CYP1A2, phenaceten demethylation to acetaminophen; CYP2C9, tolbutamide hydroxylation to hydroxytolbutamide; CYP2D6, bufuralol hydroxylation to 4′-hydroxybufuralol; and CYP3A4, midazolam hydroxylation to 1′-hydroxymidazolam) in the presence or absence of 10 or 1 µM test compound. The reaction is initiated by the addition of 1 mM NADPH and stopped after ten minutes by the addition of 2-times volume of acetonitrile containing dextrorphan as an internal standard. The concentration of each marker substrate is approximately its Km.21 Furafylline, sulfaphenazole, quinidine, and ketoconazole were included in each run to validate that the assay could identify selective inhibitors of each isoform.
Molecular docking
The homology model of TcCYP51 was generated based on the x-ray co-crystal structure of TbCYP51 complexed with 14t (PDB ID code: 4BJK) by using the homology model module implemented in Molecular Operating Environment (MOE). The homology model was refined with Protein Preparation Wizard implemented in Maestro 9.3. A receptor grid was generated from the refined structure using default values except for positional constraint at the nitrogen of 4-acylaminopyridine (radius: 0.8). The structure of 14t was docked into the active site of TcCYP51 by using Glide5.5 in extra precision (XP) mode with the predefined positional constraint (ligand feature: neutral acceptor). The binding pose of 14t was the same as that in the original co-crystal structure with 1.5 Å RMSD of all atom pairs (maximum difference = 5.0 Å of fluoro atoms). The structures of 27k, 27l, 27r, and 27s were subsequently docked to the model structure of TcCYP51 by applying the same parameters to predict their binding poses in the TcCYP51 active site.
T. cruzi CYP51 expression, purification and UV-vis assay
Recombinant TcCYP51 was expressed and purified as described elsewhere.18–19, 22 TcCYP51 was used to monitor compound binding in UV-vis spectral assay as previously described.22 Binding affinity of hits was estimated from the titration curves using the quadratic tight-binding equation:23
| (1) |
where Aobs is the absorption shift determined at any ligand concentration; Amax is the maximal absorption shift obtained at saturation; KD is dissociation constant for the inhibitor-enzyme complex; S is the ligand concentration; Et is the total enzyme concentration.
As differences in affinity between tight-binding ligands are reflected in the sharpness of the titration curve, KD values recovered from fits to experimental data approximated by equation (1) are disproportionally sensitive to error in data points near inflection point, setting a limit to the method’s sensitivity. Thus, caution was exercised when comparing the tight binding constants. At 0.5 µM TcCYP51 working concentration, only the upper limit of KD at ≤5 nM (a hundredth of the target concentration) could be estimated for the tightest binding inhibitors, if a plateau in the titration curve was reached at the stoichiometric enzyme-inhibitor ratio.
T. cruzi cell-based assay
EC50 of compounds were determined in the automated cell-based assay adapted from Engel and co-authors24 and modified as previously described.22
X-ray Crystallography
Recombinant TbCYP51 mutant V34M/D249A/D250A/D251A modified by inserting a His8-tag at the C-terminus and replacing the first 31 residues upstream of P32 with the fragment MAKKTSSKGKL was used to obtain co-crystal structure with 14t. Concentrated purified protein stored at −80°C was diluted to 0.1 mM prior to crystallization by mixing with water supplemented with 14t to reach 1:1 protein:inhibitor ratio. Crystallization conditions were determined using commercial high-throughput screening kits available in deep-well format (Hampton Research), a nanoliter drop-setting Mosquito robot (TTP LabTech) operating with 96-well plates, and a hanging drop crystallization protocol. Crystals were further optimized in 96-well plates for diffraction data collection and harvested directly from the 200-nL drops. Prior to data collection, crystals were cryo-protected by plunging them into a drop of reservoir solution supplemented with 20% ethylene glycol, then flash frozen in liquid nitrogen.
Diffraction data were collected at 100–110 K at Beamline 8.3.1, Advanced Light Source, Lawrence Berkeley National Laboratory, USA. Data indexing, integration, and scaling were conducted using MOSFLM25 and the programs implemented in the ELVES software suite.26 The crystal structures were determined by molecular replacement using diffraction data processed in the C2 space group, with Rmerge of 8.6% and atomic coordinates of T. brucei CYP51 (PDB ID code: 2×2N) as a search model. The final model was built using COOT27 and refinement was performed by using REFMAC5 software28 until R and Rfree converged to 19.4% and 27.4%, respectively. Data collection and refinement statistics are shown in Table 5.
Table 5.
Data collection and refinement statistics
| Protein | T. brucei CYP51 |
|---|---|
| Inhibitor | 14t (Small molecule code 18I) |
| PDB ID | 4BJK |
| Data collection | |
| Space group | C2 |
| Cell dimensions | |
| a, b, c (Å) | 199.3, 114.7, 136.22 |
| α, β, γ (°) | 90.0, 135.5, 90.0 |
| Molecules in AU | 4 |
| Wavelength | 1.11587 |
| Resolution (Å) | 2.67 |
| Rsym or Rmerge (%) | 8.6 (88.8)d |
| I / σI | 7.7 (1.4) |
| Completeness (%) | 99.9 (99.9) |
| Redundancy | 4.1 (4.1) |
| Crystallization | 12% pentaerythritol propoxylate (5/4 PO/OH) |
| conditions | 50 mM HEPES, pH 7.5 |
| 50 mM KCl | |
| 10% Jeffamine 600 | |
| Refinement | |
| No. reflections | 60761 |
| Rwork / Rfree (%) | 19.4/27.4 |
| No. atoms | |
| Protein | 14083 |
| Heme | 172 |
| Ligand | 100 |
| Solvent | 136 |
| Mean B value | 66.1 |
| B-factors | |
| Protein | 67.1 |
| Heme | 54.8 |
| Ligand | 84.9 |
| Solvent | 54.8 |
| R.m.s deviations | |
| Bond lengths (Å) | 0.011 |
| Bond angles (°) | 1.515 |
Values in parentheses are for highest-resolution.
Only one of the four protein chains (chain A) constituting an asymmetric unit contained electron density corresponding to the whole molecule of 14t; 14t was assigned PDB code 18I. In three other chains, only the N-indolylpyridinyl portion of 14t could be unambiguously placed. Thus, coordinates for the disordered biaryl moiety in chains B, C and D were omitted from the PDB entry.
Supplementary Material
ACKNOWLEDGMENTS
We thank Mr. Potter Wickware for critical reading of the manuscript, the staff members of Beamline 8.3.1, James Holton, George Meigs and Jane Tanamachi, at the Advanced Light Source at Lawrence Berkeley National Laboratory, for assistance with data collection. This research was generously supported by NIH R01 grant AI095437, C.M.C was supported by Conselho Nacional de Desenvolvimento Cientifico e Tecnologico (CNPq) and FIOCRUZ, Brazil. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
ABBREVIATIONS USED
- T. cruzi
Trypanosoma cruzi
- CYP51
cytochrome P450 family 51
- CYP1A2, CYP2C9, CYP2D6, CYP3A4
cytochrome P450 isoform 1A2, 2C9, 2D6, and 3A4
- SAR
structure activity relationship
- SPR
structure property relationship
- EC50
half maximal effective concentration
Footnotes
ASSOCIATED CONTENT
Supporting Information. Synthetic procedures and tabulated spectroscopic data for synthetic intermediates. This material is available free of charge via the Internet at https//pubs.acs.org.
The atomic coordinates and structure factors (PDB ID code 4BJK) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
Reference
- 1.(a) Bern C. Antitrypanosomal therapy for chronic Chagas' disease. N. Engl. J. Med. 2011;364:2527–2534. doi: 10.1056/NEJMct1014204. [DOI] [PubMed] [Google Scholar]; (b) Bern C, Kjos S, Yabsley MJ, Montgomery SP. Trypanosoma cruzi and Chagas' Disease in the United States. Clin. Microbiol. Rev. 2011;24:655–681. doi: 10.1128/CMR.00005-11. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rassi A, Jr., Rassi A, Marin-Neto JA. Chagas disease. Lancet. 2010;375:1388–1402. doi: 10.1016/S0140-6736(10)60061-X. [DOI] [PubMed] [Google Scholar]; (d) Rossi MA, Tanowitz HB, Malvestio LM, Celes MR, Campos EC, Blefari V, Prado CM. Coronary microvascular disease in chronic Chagas cardiomyopathy including an overview on history, pathology, and other proposed pathogenic mechanisms. PLoS Negl. Trop. Dis. 2010;4:e674. doi: 10.1371/journal.pntd.0000674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chagas C. Nova trypanozomiaze humana: estudos sobre a morfolojia e o ciclo evolutivo do Schizotrypanum cruzi n. gen., n. sp., ajente etiolojico de nova entidade morbida do homem. Mem. Inst. Oswaldo Cruz. 1909;1:159–218. [Google Scholar]
- 3.(a) Apt W. Current and developing therapeutic agents in the treatment of Chagas disease. Drug. Des. Devel. Ther. 2010;4:243–253. doi: 10.2147/dddt.s8338. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Guedes PM, Silva GK, Gutierrez FR, Silva JS. Current status of Chagas disease chemotherapy. Expert. Rev. Anti. Infect. Ther. 2011;9:609–620. doi: 10.1586/eri.11.31. [DOI] [PubMed] [Google Scholar]
- 4.Rassi A, Jr., Dias JC, Marin-Neto JA, Rassi A. Challenges and opportunities for primary, secondary, and tertiary prevention of Chagas' disease. Heart. 2009;95:524–534. doi: 10.1136/hrt.2008.159624. [DOI] [PubMed] [Google Scholar]
- 5.(a) Buckner FS. Sterol 14-demethylase inhibitors for Trypanosoma cruzi infections. Adv. Exp. Med. Biol. 2008;625:61–80. doi: 10.1007/978-0-387-77570-8_6. [DOI] [PubMed] [Google Scholar]; (b) Lepesheva GI, Waterman MR. Sterol 14alpha-demethylase (CYP51) as a therapeutic target for human trypanosomiasis and leishmaniasis. Curr. Top. Med. Chem. 2011;11:2060–2071. doi: 10.2174/156802611796575902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kathiravan MK, Salake AB, Chothe AS, Dudhe PB, Watode RP, Mukta MS, Gadhwe S. The biology and chemistry of antifungal agents: a review. Bioorg. Med. Chem. 2012;20:5678–5698. doi: 10.1016/j.bmc.2012.04.045. [DOI] [PubMed] [Google Scholar]
- 7.(a) Lepesheva GI, Ott RD, Hargrove TY, Kleshchenko YY, Schuster I, Nes WD, Hill GC, Villalta F, Waterman MR. Sterol 14alpha-demethylase as a potential target for antitrypanosomal therapy: enzyme inhibition and parasite cell growth. Chem. Biol. 2007;14:1283–1293. doi: 10.1016/j.chembiol.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Urbina JA, Payares G, Sanoja C, Molina J, Lira R, Brener Z, Romanha AJ. Parasitological cure of acute and chronic experimental Chagas disease using the longacting experimental triazole TAK-187. Activity against drug-resistant Trypanosoma cruzi strains. Int. J. Antimicrob. Agents. 2003;21:39–48. doi: 10.1016/s0924-8579(02)00274-1. [DOI] [PubMed] [Google Scholar]
- 8.(a) Buckner FS, Urbina JA. Recent developments in sterol 14-demethylase inhibitors for Chagas Disease. Int. J. Parasitol. Drugs Drug. Resist. 2012;2:236–242. doi: 10.1016/j.ijpddr.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Clayton J. Chagas disease: pushing through the pipeline. Nature. 2010;465:S12–S15. doi: 10.1038/nature09224. [DOI] [PubMed] [Google Scholar]
- 9.(a) Kraus JM, Tatipaka HB, McGuffin SA, Chennamaneni NK, Karimi M, Arif J, Verlinde CL, Buckner FS, Gelb MH. Second generation analogues of the cancer drug clinical candidate tipifarnib for anti-Chagas disease drug discovery. J. Med. Chem. 2010;53:3887–3898. doi: 10.1021/jm9013136. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kraus JM, Verlinde CL, Karimi M, Lepesheva GI, Gelb MH, Buckner FS. Rational modification of a candidate cancer drug for use against Chagas disease. J. Med. Chem. 2009;52:1639–1647. doi: 10.1021/jm801313t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andriani G, Amata E, Beatty J, Clements Z, Coffey BJ, Courtemanche G, Devine W, Erath J, Juda CE, Wawrzak Z, Wood JT, Lepesheva GI, Rodriguez A, Pollastri MP. Antitrypanosomal Lead Discovery: Identification of a Ligand-Efficient Inhibitor of Trypanosoma cruzi CYP51 and Parasite Growth. J. Med. Chem. 2013;56:2556–2567. doi: 10.1021/jm400012e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Chen CK, Leung SS, Guilbert C, Jacobson MP, McKerrow JH, Podust LM. Structural characterization of CYP51 from Trypanosoma cruzi and Trypanosoma brucei bound to the antifungal drugs posaconazole and fluconazole. PLoS Negl. Trop. Dis. 2010;4:e651. doi: 10.1371/journal.pntd.0000651. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lepesheva GI, Hargrove TY, Anderson S, Kleshchenko Y, Furtak V, Wawrzak Z, Villalta F, Waterman MR. Structural insights into inhibition of sterol 14alpha-demethylase in the human pathogen Trypanosoma cruzi. J. Biochem. 2010;285:25582–25590. doi: 10.1074/jbc.M110.133215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Buckner FS, Wilson AJ, White TC, Van Voorhis WC. Induction of resistance to azole drugs in Trypanosoma cruzi. Antimicrob. Agents Chemother. 1998;42:3245–3250. doi: 10.1128/aac.42.12.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kanafani ZA, Perfect JR. Antimicrobial resistance: resistance to antifungal agents: mechanisms and clinical impact. Clin. Infect. Dis. 2008;46:120–128. doi: 10.1086/524071. [DOI] [PubMed] [Google Scholar]
- 13.(a) Chen CK, Doyle PS, Yermalitskaya LV, Mackey ZB, Ang KK, McKerrow JH, Podust LM. Trypanosoma cruzi CYP51 inhibitor derived from a Mycobacterium tuberculosis screen hit. PLoS Negl. Trop. Dis. 2009;3:e372. doi: 10.1371/journal.pntd.0000372. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Podust LM, von Kries JP, Eddine AN, Kim Y, Yermalitskaya LV, Kuehne R, Ouellet H, Warrier T, Altekoster M, Lee JS, Rademann J, Oschkinat H, Kaufmann SH, Waterman MR. Small-molecule scaffolds for CYP51 inhibitors identified by high-throughput screening and defined by X-ray crystallography. Antimicrob. Agents Chemother. 2007;51:3915–3923. doi: 10.1128/AAC.00311-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doyle PS, Chen CK, Johnston JB, Hopkins SD, Leung SS, Jacobson MP, Engel JC, McKerrow JH, Podust LM. A nonazole CYP51 inhibitor cures Chagas' disease in a mouse model of acute infection. Antimicrob. Agents Chemother. 2010;54:2480–2488. doi: 10.1128/AAC.00281-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Kumar S, Sharma R, Roychowdhury A. Modulation of cytochrome-P450 inhibition (CYP) in drug discovery: a medicinal chemistry perspective. Curr. Med. Chem. 2012;19:3605–3621. doi: 10.2174/092986712801323180. [DOI] [PubMed] [Google Scholar]; (b) Gubbins PO. Triazole antifungal agents drug-drug interactions involving hepatic cytochrome P450. Expert Opin. Drug Metab. Toxicol. 2011;7:1411–1429. doi: 10.1517/17425255.2011.627854. [DOI] [PubMed] [Google Scholar]
- 16.(a) Green M, Berman J. Preparation of pentafluorophenyl esters of Fmoc protected amino-acids with pentafluorophenyl trifluoroacetate. Tetrahedron Lett. 1990;31:5851–5852. [Google Scholar]; (b) Choi JY, Plummer MS, Starr J, Desbonnet CR, Soutter H, Chang J, Miller JR, Dillman K, Miller AA, Roush WR. Structure guided development of novel thymidine mimetics targeting Pseudomonas aeruginosa thymidylate kinase: from hit to lead generation. J. Med. Chem. 2012;55:852–870. doi: 10.1021/jm201349f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ross D. The role of metabolism and specific metabolites in benzene-induced toxicity: evidence and issues. J. Toxicol. Environ. Health A. 2000;61:357–372. doi: 10.1080/00984100050166361. [DOI] [PubMed] [Google Scholar]
- 18.Chen C-K, Doyle PS, Yermalitskaya LV, Mackey ZB, Ang KKH, McKerrow JH, Podust LM. Trypanosoma cruzi CYP51 inhibitor derived from a Mycobacterium tuberculosis screen hit. PLoS Negl. Trop. Dis. 2009;3:e372. doi: 10.1371/journal.pntd.0000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen C-K, Leung SSF, Guilbert C, Jacobson MP, McKerrow JH, Podust LM. Structural characterization of CYP51 from Trypanosoma cruzi and Trypanosoma brucei bound to the antifungal drugs posaconazole and fluconazole. PLoS Negl. Trop. Dis. 2010;4:e651. doi: 10.1371/journal.pntd.0000651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 21.Testino SA, Jr., Patonay G. High-throughput inhibition screening of major human cytochrome P450 enzymes using an in vitro cocktail and liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2003;30:1459–1467. doi: 10.1016/s0731-7085(02)00480-6. [DOI] [PubMed] [Google Scholar]
- 22.Gunatilleke SS, Calvet CM, Johnston JB, Chen CK, Erenburg G, Gut J, Engel JC, Ang KK, Mulvaney J, Chen S, Arkin MR, McKerrow JH, Podust LM. Diverse inhibitor chemotypes targeting Trypanosoma cruzi CYP51. PLoS Negl. Trop. Dis. 2012;6:e1736. doi: 10.1371/journal.pntd.0001736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morrison JF. Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim. Biophys. Acta. 1969;185:269–286. doi: 10.1016/0005-2744(69)90420-3. [DOI] [PubMed] [Google Scholar]
- 24.Engel JC, Ang KKH, Chen S, Arkin MR, McKerrow JH, Doyle PS. Image-based high-throughput drug screening targeting the intracellular stage of Trypanosoma cruzi, the agent of Chagas' Disease. Antimicrob. Agents Chemother. 2010;54:3326–3334. doi: 10.1128/AAC.01777-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 ESF-EAMCB Newslett. Protein Crystallogr. 1992:26. [Google Scholar]
- 26.Holton J, Alber T. Automated protein crystal structure determination using ELVES. Proc. Natl. Acad. Sci. U S A. 2004;101:1537–1542. doi: 10.1073/pnas.0306241101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 28.(a) Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]; (b) Collaborative Computational Project, Number 4. Acta Crysallogr. D. 1994;50:760–763. [Google Scholar]
- 29.DeLano WL. The PyMOL molecular graphics system. San Carlos, CA, USA: DeLano Scientific; 2002. [Google Scholar]
- 30.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.