

Abstract
Reactive electrophiles produced during oxidative stress, such as 4-hydroxynonenal (HNE), are increasingly recognized as contributing factors in a variety of degenerative and inflammatory diseases. Here we used the RNA-seq technology to characterize transcriptome responses in RKO cells induced by HNE at subcytotoxic and cytotoxic doses. RNA-seq analysis rediscovered most of the differentially expressed genes reported by microarray studies and also identified novel gene responses. Interestingly, differential expression detection at the coding DNA sequence (CDS) level helped to further improve the consistency between the two technologies, suggesting the utility and importance of the CDS level analysis. RNA-seq data analysis combining gene and CDS levels yielded an informative and comprehensive picture of gradually evolving response networks with increasing HNE doses, from cell protection against oxidative injury at low dose, initiation of cell apoptosis and DNA damage at middle dose and significant deregulation of cellular functions at high dose. These evolving dose-dependent pathway changes, which cannot be observed by the gene level analysis alone, clearly reveal the HNE cytotoxic effect and are supported by IC50 experiments. Additionally, differential expression at the CDS level provides new insights of isoform regulation mechanisms. Taken together, our data demonstrate the power of RNA-Seq to identify subtle transcriptome changes and to characterize effects induced by HNE through the generation of high-resolution data coupled with differential analysis at both gene and CDS levels.
Introduction
4-Hydroxynonenal (HNE), one of the major aldehydic products of lipid peroxidation, has been suggested to contribute to the development and progression of various diseases.1–6 HNE reacts with a number of cellular molecules, including DNA, RNA and proteins, and has been shown to trigger multistep signal transduction cascades for suppression of cellular functions in a dose- and time-dependent manner.7–14
In a previous study, we used the microarray technology to examine the effects of HNE on gene expression in the RKO cell line.15 Significant alterations were observed for genes involved in DNA damage and antioxidant, heat shock and ER stress responses. Integrating gene expression changes with protein adduction data further elucidated signaling and transcriptional regulatory mechanisms through which protein adduction triggers gene expression changes.16, 17 However, the datasets and integrative analysis represented only high micromolar treatment concentrations of HNE (i.e., 60 μM), as few gene expression changes were detected at lower concentrations by microarray technologies, making it difficult to study the dose-dependent response upon HNE treatment.
RNA-seq has become increasingly used to quantify expression of all genes with their alternative isoforms. Compared with microarray, the digital nature of RNA-seq enables a larger dynamic range, higher resolution and lower technical variance in measuring expression abundance, which makes RNA-seq more sensitive in capturing expression differences. 18–22 By properly assigning reads to each isoform, RNA-seq enables quantifying gene expression at individual transcript level. Moreover, gene expression can also be quantified by grouping isoforms into biologically meaningful units. For example, isoforms with the same transcription start site (TSS) can be grouped together (Fig. 1, isoforms A and B). These isoforms are derived from the same pre-mRNA and differential expression at the TSS level suggests differential regulation of the pre-mRNA. Similarly, isoforms with the same coding DNA sequences (CDS) can be grouped together because they encode the same protein product (Fig. 1, isoforms B and C), and differential expression at the CDS level indicates potentially different protein outputs. Expression quantification at the transcript level and the intermediate functional unit levels allows the detection of expression changes that may not be observable at the gene level. As shown in Figure 1, RNA-seq reveals expression changes at the transcript level (isoform A) and the CDS level (CDS 1), although no significant change can be observed at the gene level. However, as many isoforms share exons, some reads cannot be accurately assigned to individual isoform. This read assignment uncertainty 23 and noisy splicing23, 24 make differential expression at the transcript level hard to detect and introduce false positives. To our knowledge, which level (gene, CDS group, TSS group, and transcript) is best suited for detecting differential expression has not been well studied.25
Figure 1.

Significant changes detected at the high-resolution level but not at the low-resolution gene level by RNA-seq. (a)Gene produces three isoforms A, B and C at different abundances. TSS or CDS groups are formed by grouping isoforms sharing the same transcription start site (TSS) or coding the same protein sequences (CDS). For example, A and B are within the same TSS group, while B and C are within the same CDS group.(b) Analyzing expression difference at the transcript level, different isoform groups level and the gene level. At the transcript level, the expression of isoform A is significantly changed across conditions, while B and C are not. Adding expression values of A and B yields the expression value for TSS1 group. At the TSS level, TSS1 and TSS2 groups are not significant changed. Adding expression values of B and C yields the expression value for CDS2 group. At the CDS level, CDS1 group is significantly changed but CDS2 group is not. Adding expression values of three isoforms yields the expression value of the gene, which is not significantly changed across conditions.
Here we applied RNA-seq to study the transcriptome changes in RKO cells in response to low, middle and high micromolar doses of HNE treatment. We first compared results from RNA-seq and microarrays at high HNE dose. Then we investigated whether the ability of RNA-seq to quantify expression of isoforms or isoform groups (CDS and TSS groups) could provide novel insights. We found that combining gene- and CDS- level analyses improved the consistency between RNA-seq and microarray and helped identify novel genes closely related to HNE response, especially under low and middle HNE dose. It presented a clear picture of gradually evolving response networks with increasing HNE doses, from cell protection against oxidative injury, initiation of cell apoptosis and DNA damage to significant deregulation of cellular pathways. These dose-dependent pathway changes revealed the HNE cytotoxic effect and were supported by IC50 experiments. Additionally, we discussed the relative contribution of transcriptional noise and isoform switching to the obscured expression changes at the gene level. Our study demonstrates that RNA-seq is a powerful tool to study dose-response relationships of altered pathways. Expression summarized at the CDS level complements gene-level analysis and provides novel and valuable information for characterizing molecular effects induced by HNE.
Results
RNA-seq was conducted to explore transcriptional changes in RKO cells following treatment for 6h with 15, 30, or 45 μM HNE. Among the total of 1195 million reads, about 81% were aligned to the human genome and 75% were uniquely mapped. Although exons constitute less than 3% of the human genome, about 87% of reads were mapped to exons, suggesting that our ploy(A)+ -selected RNA samples were highly enriched for exonic sequences (Supplementary Table S1).
Improved consistency between microarray and RNA-seq
We compared the intraplatform and interplatform correlations of gene expression in RNA-seq and microarray (Figure 2) after 45 μM HNE treatment. Within each platform, a high reproducibility was observed among biological replicates (RNA-seq: Pearson correlation r=0.98~1; microarray: r=0.98~1). Both platforms detected the same correlation trend between samples: correlations between replicates of 45 μM HNE treated samples (r=0.99~1) were slightly higher than that between replicates of controls (no HNE treatment, r=0.98~1), which were higher than that between 45 μM HNE treated samples and controls (r=0.95~0.98). In contrast, expression correlations between platforms were much lower, with Pearson correlations ranging from 0.70 to 0.74. These results are in good agreement with previous works that reported high reproducibility among replicates within platforms and much lower correlation coefficients between platforms.19, 21, 22
Figure 2.

Interplatform and Intraplatform correlations of gene expression under control and 45 μM HNE treatment between microarray and RNA-seq.
To compare the capacities of two platforms to capture the expression changes, we also calculated the fold-change-based correlation. Interestingly, the cross-platform correlation was improved by using fold changes (Figure 3A, Pearson correlation r=0.76). If only differentially expressed genes identified by either RNA-seq or microarray using the criteria of abs(log2 FC)>1 and FDR<0.01 were considered (91 genes), we obtained an even higher correlation (Figure 3B, Pearson correlation r=0.89), suggesting that the two platforms were quite consistent in detecting differential expression. Among the 91 differentially expressed genes, 24 were identified by both platforms and 11 could not be detected by RNA-seq gene-level analysis. In contrast, differential expression of another 56 genes could not be captured by microarray analysis (Figure 3C, Supplementary Table S2).
Figure 3.

Correlation of RNA-seq and microarray at the level of fold changes at 45 μM HNE treatment. A) Correlation of fold change for all genes in microarray and RNA-seq. B) Correlation of fold change of differentially expressed genes detected either by microarray or RNA-seq using the criteria of abs(log2FC)>1 and FDR<0.01. C) A Venn diagram of the number of genes detected by microarray and RNA-seq. D) POG values between the microarray and RNA-seq when gene-level analysis was combined with higher resolution level analysis, CDS, TSS and transcript levels.
We used Cuffdiff to extend differential analysis from gene level to higher resolution levels (transcript, CDS and TSS levels).26, 27 Among the 11 genes detected by microarray but missed by RNA-seq gene-level analysis, two upregulated (GCLM, TXNRD1) and four downregulated genes (PPRC1, DOT1L, URB2, EGR3) could be rediscovered by the CDS level analysis (Figure 3B). In contrast, only two upregulated (GCLM, TXNRD1) and two downregulated genes (EGR3, PPRC1) could be rediscovered by transcript or TSS-level analyses.
We further investigated the consistency in gene ranking between microarray and RNA-seq analyses at different levels. Using a FDR cutoff of 0.01, genes identified by microarray and RNA-seq through combining results from different levels were ranked by their fold change values, respectively, and were used to calculate the POG (Percentage of Overlapping genes). As shown in Figure 3D, POG between RNA-seq and microarray was improved when differential analysis at CDS, TSS or transcript levels was added to gene-level analysis. However, integrating TSS-level and transcript-level data introduced noise into highly changed genes, which led to lower POGs for the top ranked genes.
The six genes rediscovered by the CDS level analysis, including GCLM, TXNRD1, PPRC1, DOT1L, URB2, and EGR3, are important anti-oxidant genes or genes involved in DNA damage and cell proliferation, indicating their close relationship with HNE treatment. GCLM (the modifier subunit of glutamate cysteine ligase) is the first and the rate-limiting enzyme in the synthesis of GSH, a major player in cellular defense against oxidative stress.28 TXNRD1 (thioredoxin reductase 1) reduces thioredoxin as well as other substrates and protects the cell from oxidative damage.29, 30 DOT1L (DOT1-like, histone H3 methyltransferase) has been reported to be involved in DNA damage response. 31 Furthermore, based on the assumption that functionally related genes have similar expression changes, we systematically evaluated the biological relevance of differentially expressed CDS using three protein-protein interaction (PPI) datasets (PPI HQ, PPI all and PrePPI, see Method section for description).32, 33 Using the criteria of FDR<0.05 & abs(log2FC)>0.5, 297 genes were identified at the gene level and additional 195 genes were detected at the CDS level after 45μM HNE treatment (Figure 4). The 195 genes detected only at the CDS level were more likely to interact with the 297 genes detected at the gene level than randomly selected genes (p=3.5E-07 for PPI HQ, p=2.2e-15 for PPI all, p=2.4e-05 for PrePPI) (Table 1). These results suggest that differentially expressed CDS are highly likely to be involved in biological processes induced by HNE and differential analysis at the CDS level is a useful and appropriate complement to the gene level analysis.
Figure 4.
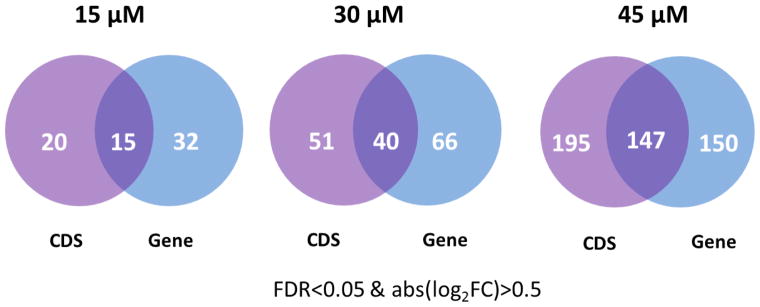
Differentially expressed genes detected at the CDS level and the gene level in HNE-treated RKO cells.
Table 1.
Relationships between differential expression detected at the gene level and that only at the CDS level based on three PPI datasets. The table lists the observed and the expected number of differential expression at the CDS level interacting with differentially expressed genes, and the probability to obtain at least the observed number by random
| Observed | Expected | P-value | |
|---|---|---|---|
| PPI HQ | 23 | 7.3 | 3.5e-07 |
| PPI all | 86 | 39.1 | 2.15e-15 |
| PrePPI | 69 | 45.8 | 2.4e-05 |
* PPI HQ (high quality protein-protein interaction dataset); PPI all(all protein-protein interaction dataset); PrePPI (protein-protein interaction dataset from PrePPI).
Gradually evolving response networks presented by the combined level
Differential expression at the CDS and gene levels were identified using Cuffdiff with FDR<0.05 & abs(log2FC)>0.5 after 15, 30 and 45 HNE treatment. CDS or genes were required to have FPKM>1 (Fragments Per Kilobase of transcript per Million fragments mapped) in at least one condition. The numbers of differentially expressed genes reported at CDS and gene levels at 15, 30, and 45 μM HNE are illustrated in Venn diagrams (Figure 4). In agreement with our previous study, the most pronounced changes in gene expression occurred in cells treated with the highest HNE concentrations.15
It should be noted that analysis at the CDS level detected a fraction of unique genes in each condition. Under 15 μM HNE treatment, 15 genes were detected at both CDS and gene levels, whereas 20 genes were only captured at the CDS level including GCLM, RRM2, SLC1A5 and TXNRD1. GCLM and TXNRD1, showing a 1.8-fold and 2.4-fold increase at the CDS level respectively, have been reported to play a vital role in protecting cells from oxidative stress.28–30 With 30 μM HNE treatment, 40 genes were detected at both CDS and gene levels, whereas 51 genes were only captured at the CDS level, including RRM1, RRM2, CCND1, DKC1, BUB1B, POLE3 and GADD45A, which are involved in cell cycle, DNA replication and glutathione metabolism. With 45μM HNE treatment, 147 genes were detected at both CDS and gene levels, whereas 195 genes were only captured at the CDS level, including many genes involved in cell cycle (e.g., EGFR, BUB1B, CCND1, and PPP5C), DNA replication (e.g., HMGB1, MCM10, MCM5, MCM8, RRM1, and DKC1) and glutathione metabolism (e.g., RRM1 and GCLM). Most of unique genes detected at the CDS level closely related to HNE response suggested that CDS level analysis is a useful complement to gene level analysis, which helps reveal important subtle biological changes.
Differentially expressed CDS and genes were further interpreted by functional enrichment analysis against Gene Ontology (GO) terms and KEGG pathways. Under 15μM HNE treatment, only MAPK signaling pathway and metabolic pathway were enriched in the differentially expressed genes. Besides these two pathways, glutathione metabolism was observed at the combined level (combining differentially expressed CDS and genes, FDR=0.0006) (Figure 5, Supplementary Table S3). Indeed, glutathione is a major intracellular antioxidant and glutathione synthesis is increased following HNE treatment to protect against oxidative injury.34, 35 Additionally, pyrimidine metabolism was also significantly represented at the combined level (FDR=0.015) and pyrimidines has been reported to be a rich source for the synthesis of new antioxidant compounds.36–38 With 30 μM HNE treatment, additional pathways, such as focal adhesion, endocytosis, spliceosome and cysteine and methionine metabolism, were detected at both gene level and the combined level. Interestingly, pathways associated with apoptosis and DNA repair were only revealed at the combined level, including programmed cell death (FDR=0.044), nucleotide excision repair (FDR=0.015), base excision repair (FDR=0.012), p53 signaling pathways (FDR=0.0006), DNA replication (FDR=0.038), etc (Figure 5, Supplementary Table S4). This is consistent with our previous studies, which reported that the IC50 values of HNE in RKO cells is 20 μM39 and a concentration equal to or greater than 30 μM begins to induce apoptosis and cell cycle deregulation.12 With 45 μM HNE treatment, a number of additional pathways, such as ubiquitin mediated proteolysis, DNA repair, microtubule-based process, and RNA transport were affected, while most of these pathways showed a higher level of enrichment in combined level analysis compared to gene level analysis (Figure 5, Supplementary Table S5). For example, the FDR value for “DNA repair” is 3.63e-07 at the combined level compared to 1e-04 at the gene level.
Figure 5.

Over represented pathways detected at the gene level and the combined level in HNE-treated RKO cells. Pathways observed only at the combined level are denoted by *.
Taken together, the HNE cytotoxic effect was clearly shown by the dose-dependent pathway changes at the combined gene and CDS levels. At a low HNE concentration (15 μM), adaptive changes that protect cells against oxidative injury (e.g., glutathione and pyrimidine metabolism) occurred. At the 30 μM HNE concentration, repair of DNA damage was introduced along with an increase in the apoptotic response, which is consistent with IC50 experiments. Notably, cell protection against oxidative injury occurring at low dose and cell apoptosis initiated at middle dose was not identified by gene level analysis alone. At the 45 μM concentration, HNE triggered many changes in signal transduction pathways that suppress cellular functions, which may lead to cell cycle arrest and apoptosis. Compared with gene level analysis combining gene and CDS levels helped reveal a gradual and continual involvement of biological pathways after low to high HNE dose treatment, which present an informative and comprehensive picture of the dose-dependent cellular function changes.
Discussion
RNA-seq provides the highest resolution of transcriptome information at the transcript level and the lowest resolution at the gene level. Our study is the first to estimate which level(s) are best suited to identify differential expression across conditions in terms of maximizing overlap with microarray data and providing biological relevance. At the gene level, differential expression identified from RNA-seq and microarrays were quite consistent, with more genes identified by RNA-seq. At higher resolution, differential expression identified at the CDS level seemed to be a useful complement to gene-level analysis. Differential expression detected by the combined level(CDS and gene) achieved a higher overlap with microarray results and provided higher sensitivity in revealing biological insights into HNE dose-dependent responses than from gene-level analysis alone. The combined level analysis helped reveal gradually evolving response network with increasing HNE dose, from cell protection against oxidative stress (e.g., glutathione metabolism) at 15 μM HNE treatment, initiation of apoptosis and the DNA damage response at 30 μM HNE treatment, and significant deregulation of cellular pathways at 45 μM HNE treatment.
Detection of differentially expressed CDS is technically more difficult than differentially expressed genes, due to greater uncertainty of read assignment and more stringent multiple test correction to account for a larger number of comparisons. There are two main possible explanations for differential expression detected at the CDS level but not at the gene level: transcriptional noise obscuring gene-level signal and isoform switching inducing differential splice variants without gene-level expression changes. To evaluate the relative contributions of these two factors to obscured gene expression changes, we compared the “CDS-only” group (differential expression detected only at the CDS level, 195 genes, Figure 4) with the “both CDS and gene” group (differential expression detected at both CDS and gene levels, 147 genes, Figure 4) after 45 μM HNE treatment. These two groups both have differentially expressed CDS but differ in gene-level expression changes. With the potential to change protein output, differentially expressed CDS is likely to function in HNE response and thus more informative. This informative CDS in the “CDS-only” group contributed less to the overall gene expression than those in the “both CDS and gene” group (Figure 6A), suggesting higher background noise or splicing complexity in the “CDS-only” group. Furthermore, compared with genes in “both CDS and gene group”, which exhibited similarity in both fold changes and expression variability (calculated by Cuffdiff26, including biological and technical variance) with their corresponding CDS, genes in the “CDS-only” group showed similar fold change with but higher expression variances than their corresponding CDS (Figures 6B and 6C). The high gene expression variability, resulting from transcriptional noise, obscures the gene level signal in the “CDS-only” group. Additionally, we only found one instance (SEPT6) out of 195 genes where isoform switching led to differentially expressed CDS (log2FC=−1.58, FDR=0.004) without detectable changes at the gene-level (log2FC=−0.24, FDR=1) (Supplementary Text and Figures S1–S6). Thus transcriptional noise instead of isoform switching might be the main reason for the insignificant gene-level expression changes.
Figure 6.
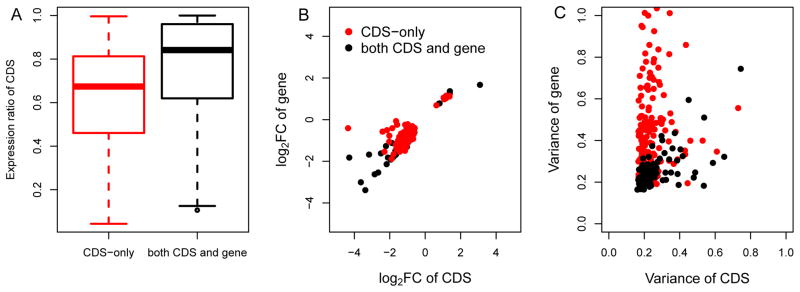
A) Cumulative distribution of the relative contributions of differentially expressed CDS to the genes in the “CDS-only” group and the “both CDS and gene” group. B) Fold change of differentially expressed CDS vs. fold change of the corresponding genes in the “CDS-only” group and the “both CDS and gene” group at 45 μM HNE treatment. C) Variances for differentially expressed CDS vs. variances of the corresponding genes in the “CDS-only” group and the “both CDS and gene” group under 45 μM HNE treatment.
Transcriptional noise mainly stems from noncoding isoforms. Among 248675 transcripts detected in the HNE transcriptome, 154780 (62%) are noncoding isoforms. Noncoding isoforms, classified as retained intron or processed transcript, lack protein-coding capacity and do not contribute to protein output and thus may not be as functionally important as protein coding isoforms 40. They are generally subject to less functional constraints on isoform abundance and have larger expression variances, which obscure gene-level signal. For example, NEDD4 was identified to undergo significant expression changes at the CDS level (FDR<0.05), but not at the gene level (FDR=0.99) with 45 μM HNE treatment (Figure 7A). NEDD4 had two highly expressed transcripts, ENST00000435532 and ENST0000508075 (Figure 7B). ENST00000435532 codes for a protein product and its expression was significantly changed (FDR=0.027), whereas ENST0000508075 is a noncoding transcript whose expression varies a lot in two conditions and was not changed after 45 μM HNE treatment (FDR=1). Another possible source of transcriptional noise is from those coding isoforms lacking strong transcriptional control. Their expressions, to a large extent reflecting background transcription, make gene-level signal hard to detect. For example, HNRNPR underwent significant expression changes at the CDS level (FDR<0.05), but not at the gene level (FDR=0.29) with 45 μM HNE treatment (Figure 8A). Besides the differentially expressed CDS (containing two isoforms, ENST00000302271 and ENST00000374612), HNRNPR had another CDS without significant expression change (ENST00000426846, FDR=1), which obscured the gene-level signal (Figures 7A). Comparing the transcript structure of these two CDS, we found that the significantly changed CDS has one more exon than the non-significant CDS (Figure 8B). This exon encodes RNA recognition motif domain 1 (RRM1)(Figure 8C), which is predicted to interact with many differentially expressed genes or CDS by PrePPI, including HNRPDL, SRSF1, RNPS1, HNRNPF, HNRNPL, etc. (Figure 7D). The CDS containing this important exon might be subjected to strong constraints on its expression, showing a higher transcriptional signal-to-noise ratio. In contrast, the non-informative CDS lacking the exon, subject to less functional constraints on isoform abundance, might undergo noisy splicing by erroneous splice site choice24 and results in lower signal-to-noise ratio. This agrees with a previous study demonstrating that noise in gene expression is a biologically important variable and subject to natural selection.24
Figure 7.

A) Gene and transcript expression changes of NEDD4 in response to 45 μM HNE treatment. B) Transcript structure of differentially expressed CDS and non-differentially expressed transcripts. ENST00000435532 encodes differentially expressed CDS, while ENST00000508075 is a processed transcript.
Figure 8.

A) Gene and CDS expression changes of HNRNPR in response to 45 μM HNE treatment. B) Transcript structure of differentially expressed CDS and non-differentially expressed CDS. Two transcripts, ENST00000302271 and ENST00000374612 encode differentially expressed CDS, while ENST00000426846 encodes non-differentially expressed CDS, which lacks an exon situated in RRM1 domain. C) Comparison of protein sequences between differentially expressed and non-differentially expressed CDS.D) The RRM1 domain of HNRNPR is predicted to interact with many genes by PrePPI, whose expressions are significantly changed. The number on the edge denotes the likelihood ratio score based on three-dimensional structural interaction. A score greater than 50 suggests the high probability of interaction between two proteins. The number beside the node shows the domain information. For example, RRM1 domain of HNRNPR is predicted to interact with the domain 17–87 of SRSF1 with the score of 574.54.
Additionally, differential expression observed at the CDS level but not at the gene level may present an opportunity for exploring potential post-transcriptional regulatory mechanisms to gain insights into isoform specific regulation. For example, the small expression variation of the functional transcripts within biological replicates suggest that their expression might be controlled by the coupling of transcription and splicing since RNA binding proteins usually have a low degree of transcriptional noise.41 As another example, post-transcriptional regulation might be involved if only functional transcripts changed their abundance across conditions (e.g., miRNA targeting of specific isoforms to induce mRNA decay). Analyzing the 3′ UTR of genes with differentially expressed CDS is one way to find the miRNA involved in the process. For example, NEDD4 was found to be the target of several miRNAs from MSigDB (c3.mir.v3.1.symbols.gmt)42, including miR-30, miR-27, miR-9 and miR-144. The binding sites are evolutionarily conserved and the miRNA-target relationships are also supported by other prediction algorithms (Supplementary Table S6). Consistently, miR-144 targets were highly enriched in differentially expressed gene sets, not only in those detected at the CDS level but also at the combined level (CDS and gene levels)(FDR<1e-06, Table 2). Previous studies have found that the RKO cell line exhibits low expression levels of miR-144 and down regulation of miR-144 leads to colorectal cancer progression via activation of the mTOR signaling pathway.43 Thus, miR-144 might be upregulated by HNE treatment, which leads to the down regulation of transcripts or genes and the inhibition of cell proliferation.
Table 2.
miRNA targets enrichment analysis on differential expression at the CDS level and the combined level (CDS or gene)
| DEGs at CDS-level | DEGs at combined level | |||
|---|---|---|---|---|
| miRNA | Num. of targets | FDR | Num. of targets | FDR |
| miR-144 | 13 | 1.25e-07 | 15 | 1.26e-07 |
| miR-524 | 14 | 6.07e-05 | 19 | 2.20e-06 |
| miR-518a-2 | 10 | 6.07e-05 | 13 | 2.23e-06 |
| miR-101 | 10 | 2e-04 | 14 | 3.89e-06 |
| miR-519a, b, c | 13 | 2e-04 | 16 | 6.01e-05 |
| miR-522 | 8 | 2e-04 | 11 | 6.82e-06 |
| miR-204, miR-211 | 9 | 3e-04 | 10 | 3e-04 |
| miR-181a, b, c, d | 13 | 4e-04 | 18 | 1.55e-05 |
| miR-324-3p | 6 | 5e-04 | 6 | 0.001 |
| miR-30a-5p, 30c, 30d, 30b, 30e-5p | 14 | 5e-04 | 24 | 1.26e-07 |
Differential CDS analysis can identify significant CDS abundance changes no matter gene expression changes or not, but this method is quite different from methods aimed to detect differential spliced genes or differential exon usage, e.g., MISO44, ALEXA-Seq45, DEXseq46 and DSGseq47. The major difference is that if the gene’s overall expression changes but the relative abundances of the different transcripts stay the same, the genes will be called significant by differential CDS analysis but not be called by methods focusing on differential splicing. Among 35 significantly changed genes detected by microarray at 45 μM HNE treatment, 30 were called by differential gene and CDS analysis from RNA-seq, but none of them were identified by DEXSeq. In addition, differential CDS analysis has several advantages. CDS is an important function unit, thus differential CDS analysis is more biologically meaningful and easier to interpret than differential exon usage. Furthermore, although exons are more sensitive and easier to calculate than CDS, the results based on exon level are more prone to noise and will be less robust and less stable. A large portion of differentially expressed/spliced genes at low dose is expected to be also significantly changed at high dose since HNE response networks will gradually evolve with the increasing dose. This expectation is better supported by differential CDS analysis than alternative splicing methods. 77% (23) of 30 significant CDS at 15 μM were found to be still significantly changed at 30 μM, and 86% (78) of 91 significant CDS at 30 μM were supported by 45 μM. In contrast, only one (50%) of two exons detected by DEXSeq at 15 μM found evidence of differential usage at 30 μM, and 78% (18) of 23 exons detected at 30 μM were re-identified at 45 μM HNE. Even worse, MISO identified 29 exon skipping and 11 exon inclusion events at 15 μM, but only one exon skipping and 2 inclusion events (8%) reappeared at 30 μM. Among 23 exon skipping and 10 exon inclusion events detected at 30 μM, only 4 skipping and 2 inclusion events (18%) were re-identified at 45 μM (Supplementary Figures S7).
Although RNA-seq offers high resolution transcriptome information, read assignment uncertainty remains a major challenge, especially for low abundance genes with many isoforms. Differential expression at the transcript level is the most difficult to detect, due to the largest read assignment uncertainty and the highest statistical significance required to account for the largest number of comparisons. Additionally, noisy splicing leads to false positives, especially when the number of replicates is small. Therefore, transcript level analysis did not help find more biologically relevant results in our experiments with only 3 replicates in each condition. Standard RNA-seq methods are not suited to annotate the 5′ start site, which may explain why differential expression detection at the TSS level was not as useful as expected. In contrast, each CDS group encompasses all transcripts coding for the same protein product, which reduces the read assignment ambiguity and the noise due to erroneous splice site choice. Thus, differential expression analysis at the CDS level is a useful complement to gene-level analysis. Combining CDS and gene levels revealed more subtle biological responses triggered by HNE treatment. In the future, adding more replicates, increasing sequencing depth, and using long pair-end reads will facilitate differential expression detection at the transcript level, which will create opportunities for regulation analysis with unprecedented scope and scale and allow researchers to better disentangle the complex interplay between transcriptional and post-transcriptional regulation.
Materials and Methods
Cell culture and treatment
RKO human colorectal carcinoma cells were grown in McCoy’s 5A medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and antibiotics at 37 °C and 5% CO2. HNE was obtained from Cayman Chemical and was dissolved in MeOH as a 1000 × stock solution. RKO cells were seeded and were treated with vehicle or 15, 30, or 45 μM HNE for 6h. Cell treatments were conducted three times for each condition. Experimental details have been described previously15.
RNA sequencing
The twelve RNA samples were sequenced following the protocols recommended by the manufacturer (Illumina). Briefly, poly-A was purified and then fragmented into small pieces. Using reverse transcriptase and random primers, RNA fragments were used to synthesize the first and second strand cDNAs. Following end repair, addition of an “A” base, adapter ligation, size selection and amplification of cDNA templates, samples were sequenced in 5 lanes on the Illumina HiSeq 2000, generating about 70~110 million of 100 pair-end reads per sample (Supplementary Table S1).
RNA-seq and microarray analysis
Reads were mapped to human genome hg19 using TopHat version 1.4.0 with the reference annotation file (Homo_sapiens.GRCh37.65.gtf).26, 27, 48 Each sample obtained similar mapping quality, about 81% of the reads mapped to genome, of which 87% overlapping exons. The mapping results were summarized in Supplementary Table S1. The aligned reads was assembled and transcript expression was quantified using FPKM (Fragments Per Kilobase of transcript per Million fragments mapped) by Cufflinks version2.0.2, which uses a linear statistical model to compute the likelihood that the number of fragments would be observed given the proposed abundances on the transcripts26. Differential expression between four groups, HNE15 vs. HNE0, HNE 30 vs. HNE 0, and HNE 45 vs. HNE0 was detected by Cuffdiff.26, 27, 49 Genes, CDS, TSS or transcripts with FPKM>1 in any of four conditions were selected for further analysis.
Affymetrix cel files were normalized using the Robust MultiChip Analysis (RMA) algorithm 50 as implemented in Bioconductor.51 Probe set identifiers (IDs) were mapped to gene symbols. Probe sets that mapped to multiple genes were eliminated. When multiple probe sets were mapped to the same gene, the probe set with the maximal IQR was used to represent the gene expression level. Differential expression analysis between HNE 45 and HNE 0 was performed using limma.52
A common set of genes shared by RNA-seq and microarray was used to compare gene expression between these two platforms. If genes had FDR<0.01 at both gene-level and other levels (CDS, TSS or transcript), fold change values at gene level were used. A fold change ranking with FDR cutoff of 0.01 was applied separately to RNA-seq and microarray to calculate the percentage of overlapping genes (POG) using the equation POG = 100*(DD+UU)/2L, where DD and UU are the number of down- or up-regulated genes common in RNA-seq and microarray, respectively, and L is the number of selected genes ranked by fold change. Directionality of gene regulation is considered in POG calculations, that is, genes selected by two platforms but with different regulation directionalities are considered as discordant.53
Functional interpretation
Three protein-protein interaction datasest, PPI HQ, PPI all and PrePPI, were downloaded from PrePPI webserver (http://bhapp.c2b2.columbia.edu/PrePPI/).32, 33 PPI HQ contains 7,409 interactions of at least two publication supports, involving 2976 proteins. PPI all includes 82,551 interactions between 12,104 proteins from HPRD, DIP, IntAct, BioGRID, and MINT. PrePPI comprises 317,813 high confidence interactions (LR>600) for 11219 proteins. For 492 genes whose differential expression was detected at the gene or the CDS level after 45 μM HNE treatment (Figure 4), 154 was contained in PPI HQ, 405 were included in PPI all, and 217 was involved in PrePPI. Hypergeometric test was used to calculate the probability of differentially expressed CDS randomly connected to differentially expressed genes in the protein-protein network.
GO, KEGG and miRNA targets enrichment analysis were performed using WebGestalt54. Functional categories or pathways containing no less than two differentially expressed CDS or genes with FDR<0.05 were selected. Potential miRNAs targeting NEDD4 were obtained from MSigDB (c3.mir.v3.1.symbols.gmt)4242, which were further validated by evolutionary conservation and other miRNA target prediction algorithms, including TargetScan, DIANAmT, miRanda, miRDB, miRWalk, RNAhybrid, PICTAR4, PICTAR5, PITA, and RNA22.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health grants P01 ES013125, P30 ES000267, U54CA126479and R01GM088822. QL was partially supported by the State Key Program of National Natural Science of China (31230058) and National Natural Science Foundation of China (31070746).
References
- 1.Hennig B, Chow CK. Free Radic Biol Med. 1988;4:99–106. doi: 10.1016/0891-5849(88)90070-6. [DOI] [PubMed] [Google Scholar]
- 2.Jurgens G, Chen Q, Esterbauer H, Mair S, Ledinski G, Dinges HP. Arterioscler Thromb. 1993;13:1689–1699. doi: 10.1161/01.atv.13.11.1689. [DOI] [PubMed] [Google Scholar]
- 3.Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y. Proc Natl Acad Sci U S A. 1996;93:2696–2701. doi: 10.1073/pnas.93.7.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Traverso N, Menini S, Cosso L, Odetti P, Albano E, Pronzato MA, Marinari UM. Diabetologia. 1998;41:265–270. doi: 10.1007/s001250050902. [DOI] [PubMed] [Google Scholar]
- 5.Dou X, Li S, Wang Z, Gu D, Shen C, Yao T, Song Z. Am J Pathol. 2012;181:1702–1710. doi: 10.1016/j.ajpath.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee WC, Wong HY, Chai YY, Shi CW, Amino N, Kikuchi S, Huang SH. Biochem Biophys Res Commun. 2012;425:842–847. doi: 10.1016/j.bbrc.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 7.Nakashima I, Liu W, Akhand AA, Takeda K, Kawamoto Y, Kato M, Suzuki H. Mol Aspects Med. 2003;24:231–238. doi: 10.1016/s0098-2997(03)00018-9. [DOI] [PubMed] [Google Scholar]
- 8.Liu W, Kato M, Akhand AA, Hayakawa A, Suzuki H, Miyata T, Kurokawa K, Hotta Y, Ishikawa N, Nakashima I. J Cell Sci. 2000;113(Pt 4):635–641. doi: 10.1242/jcs.113.4.635. [DOI] [PubMed] [Google Scholar]
- 9.Biswas D, Sen G, Biswas T. Toxicol Appl Pharmacol. 2010;244:315–327. doi: 10.1016/j.taap.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 10.Abarikwu SO, Pant AB, Farombi EO. Basic Clin Pharmacol Toxicol. 2012;110:441–448. doi: 10.1111/j.1742-7843.2011.00834.x. [DOI] [PubMed] [Google Scholar]
- 11.Sharma A, Sharma R, Chaudhary P, Vatsyayan R, Pearce V, Jeyabal PV, Zimniak P, Awasthi S, Awasthi YC. Arch Biochem Biophys. 2008;480:85–94. doi: 10.1016/j.abb.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ji C, Amarnath V, Pietenpol JA, Marnett LJ. Chem Res Toxicol. 2001;14:1090–1096. doi: 10.1021/tx000186f. [DOI] [PubMed] [Google Scholar]
- 13.Ruef J, Moser M, Bode C, Kubler W, Runge MS. Basic Res Cardiol. 2001;96:143–150. doi: 10.1007/s003950170064. [DOI] [PubMed] [Google Scholar]
- 14.Herbst U, Toborek M, Kaiser S, Mattson MP, Hennig B. J Cell Physiol. 1999;181:295–303. doi: 10.1002/(SICI)1097-4652(199911)181:2<295::AID-JCP11>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 15.West JD, Marnett LJ. Chem Res Toxicol. 2005;18:1642–1653. doi: 10.1021/tx050211n. [DOI] [PubMed] [Google Scholar]
- 16.Jacobs AT, Marnett LJ. Acc Chem Res. 2010;43:673–683. doi: 10.1021/ar900286y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang B, Shi Z, Duncan DT, Prodduturi N, Marnett LJ, Liebler DC. Mol Biosyst. 2011;7:2118–2127. doi: 10.1039/c1mb05014a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Z, Gerstein M, Snyder M. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nookaew I, Papini M, Pornputtapong N, Scalcinati G, Fagerberg L, Uhlen M, Nielsen J. Nucleic Acids Res. 2012;40:10084–10097. doi: 10.1093/nar/gks804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiong Y, Chen X, Chen Z, Wang X, Shi S, Zhang J, He X. Nat Genet. 2010;42:1043–1047. doi: 10.1038/ng.711. [DOI] [PubMed] [Google Scholar]
- 21.Su Z, Li Z, Chen T, Li QZ, Fang H, Ding D, Ge W, Ning B, Hong H, Perkins RG, Tong W, Shi L. Chem Res Toxicol. 2011;24:1486–1493. doi: 10.1021/tx200103b. [DOI] [PubMed] [Google Scholar]
- 22.van Delft J, Gaj S, Lienhard M, Albrecht MW, Kirpiy A, Brauers K, Claessen S, Lizarraga D, Lehrach H, Herwig R, Kleinjans J. Toxicol Sci. 2012;130:427–439. doi: 10.1093/toxsci/kfs250. [DOI] [PubMed] [Google Scholar]
- 23.Garber M, Grabherr MG, Guttman M, Trapnell C. Nat Methods. 2011;8:469–477. doi: 10.1038/nmeth.1613. [DOI] [PubMed] [Google Scholar]
- 24.Pickrell JK, Pai AA, Gilad Y, Pritchard JK. PLoS Genet. 2010;6:e1001236. doi: 10.1371/journal.pgen.1001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oshlack A, Robinson MD, Young MD. Genome Biol. 2010;11:220. doi: 10.1186/gb-2010-11-12-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Nat Biotechnol. 2012;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cole TB, Giordano G, Co AL, Mohar I, Kavanagh TJ, Costa LG. J Toxicol. 2011;2011:157687. doi: 10.1155/2011/157687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karimpour S, Lou J, Lin LL, Rene LM, Lagunas L, Ma X, Karra S, Bradbury CM, Markovina S, Goswami PC, Spitz DR, Hirota K, Kalvakolanu DV, Yodoi J, Gius D. Oncogene. 2002;21:6317–6327. doi: 10.1038/sj.onc.1205749. [DOI] [PubMed] [Google Scholar]
- 30.Smart DK, Ortiz KL, Mattson D, Bradbury CM, Bisht KS, Sieck LK, Brechbiel MW, Gius D. Cancer Res. 2004;64:6716–6724. doi: 10.1158/0008-5472.CAN-03-3990. [DOI] [PubMed] [Google Scholar]
- 31.FitzGerald J, Moureau S, Drogaris P, O’Connell E, Abshiru N, Verreault A, Thibault P, Grenon M, Lowndes NF. PLoS One. 2011;6:e14714. doi: 10.1371/journal.pone.0014714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang QC, Petrey D, Garzon JI, Deng L, Honig B. Nucleic Acids Res. 2013;41:D828–833. doi: 10.1093/nar/gks1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang QC, Petrey D, Deng L, Qiang L, Shi Y, Thu CA, Bisikirska B, Lefebvre C, Accili D, Hunter T, Maniatis T, Califano A, Honig B. Nature. 2012;490:556–560. doi: 10.1038/nature11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schulz JB, Lindenau J, Seyfried J, Dichgans J. Eur J Biochem. 2000;267:4904–4911. doi: 10.1046/j.1432-1327.2000.01595.x. [DOI] [PubMed] [Google Scholar]
- 35.Kruman I, Bruce-Keller AJ, Bredesen D, Waeg G, Mattson MP. J Neurosci. 1997;17:5089–5100. doi: 10.1523/JNEUROSCI.17-13-05089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kotaiah Y, Harikrishna N, Nagaraju K, Venkata Rao C. Eur J Med Chem. 2012;58:340–345. doi: 10.1016/j.ejmech.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 37.Panda SS, Chowdary PV. Indian J Pharm Sci. 2008;70:208–215. doi: 10.4103/0250-474X.41457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bano T, Kumar N, Dudhe R. Org Med Chem Lett. 2012;2:34. doi: 10.1186/2191-2858-2-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGrath CE, Tallman KA, Porter NA, Marnett LJ. Chem Res Toxicol. 2011;24:357–370. doi: 10.1021/tx100323m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gonzalez-Porta M, Frankish A, Rung J, Harrow J, Brazma A. Genome Biol. 2013;14:R70. doi: 10.1186/gb-2013-14-7-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kornblihtt AR. Adv Exp Med Biol. 2007;623:175–189. doi: 10.1007/978-0-387-77374-2_11. [DOI] [PubMed] [Google Scholar]
- 42.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iwaya T, Yokobori T, Nishida N, Kogo R, Sudo T, Tanaka F, Shibata K, Sawada G, Takahashi Y, Ishibashi M, Wakabayashi G, Mori M, Mimori K. Carcinogenesis. 2012;33:2391–2397. doi: 10.1093/carcin/bgs288. [DOI] [PubMed] [Google Scholar]
- 44.Katz Y, Wang ET, Airoldi EM, Burge CB. Nat Methods. 2010;7:1009–1015. doi: 10.1038/nmeth.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Griffith M, Griffith OL, Mwenifumbo J, Goya R, Morrissy AS, Morin RD, Corbett R, Tang MJ, Hou YC, Pugh TJ, Robertson G, Chittaranjan S, Ally A, Asano JK, Chan SY, Li HI, McDonald H, Teague K, Zhao Y, Zeng T, Delaney A, Hirst M, Morin GB, Jones SJ, Tai IT, Marra MA. Nat Methods. 2010;7:843–847. doi: 10.1038/nmeth.1503. [DOI] [PubMed] [Google Scholar]
- 46.Anders S, Reyes A, Huber W. Genome Res. 2012;22:2008–2017. doi: 10.1101/gr.133744.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang W, Qin Z, Feng Z, Wang X, Zhang X. Gene. 2013;518:164–170. doi: 10.1016/j.gene.2012.11.045. [DOI] [PubMed] [Google Scholar]
- 48.Trapnell C, Pachter L, Salzberg SL. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 51.Reimers M, Carey VJ. Methods Enzymol. 2006;411:119–134. doi: 10.1016/S0076-6879(06)11008-3. [DOI] [PubMed] [Google Scholar]
- 52.Smyth GK. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 53.Shi L, Jones WD, Jensen RV, Harris SC, Perkins RG, Goodsaid FM, Guo L, Croner LJ, Boysen C, Fang H, Qian F, Amur S, Bao W, Barbacioru CC, Bertholet V, Cao XM, Chu TM, Collins PJ, Fan XH, Frueh FW, Fuscoe JC, Guo X, Han J, Herman D, Hong H, Kawasaki ES, Li QZ, Luo Y, Ma Y, Mei N, Peterson RL, Puri RK, Shippy R, Su Z, Sun YA, Sun H, Thorn B, Turpaz Y, Wang C, Wang SJ, Warrington JA, Willey JC, Wu J, Xie Q, Zhang L, Zhong S, Wolfinger RD, Tong W. BMC Bioinformatics. 2008;9(Suppl 9):S10. doi: 10.1186/1471-2105-9-S9-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang B, Kirov S, Snoddy J. Nucleic Acids Res. 2005;33:W741–748. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.