

Abstract
B cell central tolerance is a process through which self-reactive B cells are removed from the B cell repertoire. Self-reactive B cells are generally removed by receptor editing in the bone marrow and by anergy induction in the periphery. Interferon regulatory factor 8 (IRF8) is a critical transcriptional regulator of immune system development and function. A recent study has shown that marginal zone B cells and B1 B cells population are dramatically increased in the IRF8 deficient mice, indicating that there are B cell developmental defects in the absence of IRF8. Here, we report that mice deficient for IRF8 produced anti-dsDNA antibodies. Using hen egg lysozyme double transgenic model, we further demonstrate that B cell anergy was breached in the IRF8 deficient mice. While anergic B cells in the IRF8 proficient background were blocked at the transitional stage of development, anergic B cells in the IRF8 deficient background were able to further mature which allow them to regain responses to antigen stimulation. Interestingly, our results show that IRF8 deficient B cells were more sensitive to antigen stimulation and were resistant to antigen induced cell death. Moreover, our results show that IRF8 was expressed at a high level in the anergic B cells and elevated level of IRF8 promoted apoptosis in the transitional B cells. Thus, our findings presented here reveal a previously unrecognized function of IRF8 in B cell anergy induction.
Keywords: IRF8, anergy, HEL, transcription factor, SLE
Introduction
It has been estimated that almost 70% newly generated immature B cells are self-reactive (1). B cell central tolerance is a process through which self-reactive B cells are rendered non-self-reactive (2). Central tolerance consists of receptor editing, anergy induction and clonal deletion. In the bone marrow, self-reactive B cells are initially removed from the repertoires through receptor editing and deletion (3). The self-reactive B cells that somehow escape deletion in the bone marrow are functionally inactivated by anergy in the periphery. Anergy refers to a persistent state of unresponsiveness to antigen stimulation and anergic B cells are arrested in their development at the transitional stage (2). The majority of self-reactive B cells are inactivated by anergy and breach in B cell anergy has been linked to development of autoimmune diseases (2).
There are three groups of proteins that are critical for B cell anergy induction. The first group, are the molecules involved in BCR signaling. It has been shown that chronic antigen stimulation leads to monophosphorylation of Igα/β resulting in biased activation of phosphatase SHIP-1 and adaptor Dok-1 (4). In anergic B cells, SHIP-1 and Dok-1 are constitutively phosphorylated leading to activation of an inhibitory circuit that is critical for B cell anergy induction. Indeed, mice deficient in SHIP-1 in B cells alone is sufficient to cause lupus-like autoimmune disease (4). Besides SHIP-1, signaling molecules such as Cbl, PKCδ and PTEN are also critical for establishment of B cell anergy (5–7).
The second group, are the molecules that promote the survival and differentiation of B cells. BAFF is a cytokine that is critical for survival and differentiation of B2 cells (8). Anergic B cells have a shorter lifespan mainly because of their highly dependence on BAFF for survival than their normal counterparts (9). The reliance on BAFF for survival and differentiation helps reinforce B cell anergy as anergic B cells fail to compete with normal B cells for limited amounts of BAFF. However, when BAFF is overexpressed anergic B cells survive and mature, eventually leading to breakdown of anergy (10). Bim is a proapoptotic Bcl-2-related protein and Bim deficiency by itself is sufficient to breach B cell anergy (11). Interestingly, lack of Bim renders B cell independent of BAFF for survival, which allows anergic B cell in the Bim deficient mice to survive and mature (11).
The third group, are transcription factors that mediate B cell response to antigen stimulation. Transcription factors integrate the signals from BCR and its co-stimulatory receptors, in doing so they dictate B cell response to antigen via regulation of genes that control B cell survival and activation. NFAT1 is one such transcription factor which functions in a calcium-dependent manner and lack of NFAT1 is sufficient to break B cell anergy (12). NFAT1 deficient B cells are hyperresponsive to antigen stimulation, indicating that NFAT1 is a negative regulator of antigen response in B cells (13, 14). Similarly, c-Myc is rapidly induced upon antigen engagement and is critical for antigen-induced B cell expansion (15). However, anergic B cells produce low levels of c-Myc and fail to enter cell cycle upon antigen stimulation (16). Interestingly, overexpression of c-Myc in anergic B cells can restore B cell response to antigen stimulation and by itself is sufficient to breach B cell anergy (17). Although defective distal BCR signaling in anergic B cell renders them unresponsive to antigen stimulation, elevated expression of key transcription factors in the anergic B cells, such as c-Myc, could lower their activation threshold thereby restoring response of anergic B cells to antigen stimulation.
Interferon regulatory factor 8 (IRF8), also known as IFN consensus binding protein (ICSBP), is a critical transcriptional regulator that controls the development and differentiation of multiple lineages of immune cells, including macrophages, dendritic cells, B cells and T cells (18). It has been demonstrated that IRF8 is required for macrophage development and in its absence the granulocyte population is dramatically increased (19). IRF8 has also been shown to induce the expression of PU.1 and EBF1 in hematopoietic progenitor cells to directly promote B cell commitment and specification (20). Previous studies from us have shown that IRF8, together with its closely related family member IRF4, orchestrate the transition from large pre-B to small pre-B cells (21–23). Additionally, IRF8 has been shown to induce Bcl-6 in germinal center reaction (24). A recent study has shown that marginal zone B cells (MZ B) and B1 B cells are significantly increased in the IRF8 deficient mice. The defects were observed in IRF8 germline as well as conditional mutant mice, indicating that the defects are B cell intrinsic. Moreover, it has been shown that spleens of IRF8 conditional mutant mice contain twice as many mature B cells as their counterparts in wild type mice (25). MZ and B1 B cells are known to harbor B cells with self-reactive BCRs, and expansion of MZ and B1 B cells are often observed in mice with defects in B cell tolerance induction (26). In this report, we show that IRF8 deficient mice, both germline and conditional knockout, produced anti-dsDNA antibodies. Using a well-established mouse model of B cell anergy, we further demonstrate that B cell anergy was breached in the IRF8 deficient mice. Thus, our findings presented here identify IRF8 as a novel regulator of B cell anergy.
Materials and Methods
Mice
IRF8 germline (IRF8−/−) and conditional mutant mice have been previously described (25, 27). The BCR transgenic mice recognizing hen egg lysozyme (HEL-Tg, MD4) as well as the transgenic mice expressing secreted HEL were obtained from Jackson lab. IRF8 conditional mutant mice were bred with CD19Cre mice to generate IRF8 conditional mutant mice that are hemizygous for CD19Cre (IRF8fl/fl). All mice were in C57BL/6 genetic background and were maintained under specific pathogen-free conditions. Experiments were performed according to guidelines from the National Institutes of Health and with an approved protocol from the Institutional Animal Care and Use Committee of University of Nebraska Medical Center. The mice aged from 6 to 30 weeks were used for this study.
ELISA
Anti-HEL ELISA was performed with a previously described method (28). Sera at 1:200 dilutions were added to a 96-well plate pre-coated with HEL antigen (10μg/ml). After washing, the plate was subsequently incubated with biotinylated anti-IgMa antibody. The bound Ig was visualized by addition of alkaline phosphatase and pNPP substrate. The raw ODs were converted to Units/ml using a control serum from HEL-Tg mice. This would allow inter-plate comparison. Anti-dsDNA IgM and IgG ELISA were conducted with kits from Alpha Diagnostic (Texas) according to manufacturer’s recommendation.
Cell proliferation assay with CFSE tracking dye
B cells were isolated from mice spleen through negative selection. Briefly, splenocytes were incubated with cocktail of biotinylated antibodies against CD3, CD4, CD8, Gr-1, Ter119 and Dx5. After incubation with streptavidin conjugated microbeads (Miltenyi Biotech), B cells were isolated with a negative selection column. The isolated cells were incubated with 1 μM of CFSE tracking dye (Molecular probes) in PBS at 37 °C for 10 minutes. The stained cells were plated in a 96-well plate in triplicate at 5x 105 cells per well. The cells were stimulated with different mitogens including HEL, LPS and IgM and analyzed by FACS after four days.
Survival assay
The isolated B cells were plated into each well of a 96-well plate in triplicate, incubated with different concentrations of BAFF, HEL or both, and analyzed at different time points. To distinguish dead and alive cells, the treated cells were incubated with Propidium Iodide (PI) prior to FACS analysis.
FACS analysis
Cells were isolated from mice, pre-incubated with either 2% rat serum or Fc-Block (2.4G2), and stained with optimal amounts of specific antibodies, either biotinylated or directly fluorochrome-conjugated. The antibodies against B220, IgM, CD93 (AA4.1), CD21, IgMb were purchased from BD Pharmingen. The stained cells were analyzed with a FACS Calibur flow cytometer.
Measurement of Calcium Influx
Splenocytes were isolated from mice and stained with antibodies against B220. After washing, the stained cells (5x106 cells/ml) were incubated with 1 μM of Indo-1 AM (Molecular Probes) for 30 minutes at 37 °C in RPMI 1640 media containing 3 % FBS. The Calcium influx of loaded cells was analyzed with a LSR II flow cytometer. The baseline emission of fluorescence ratio (405:525 nm) was collected for 1 min, followed by stimulation by HEL at 100 ng/ml. The fluorescence ratio of 405 nm vs 525 nm was recorded for another 4 minutes. The increase in the fluorescence ratio was used to reflect the intensity of calcium mobilization upon BCR activation. The data were analyzed with a Flowjo software.
Western Blot analysis
Splenic B cells were isolated via negative selection. The negatively selected B cells were stimulated with different concentrations of HEL at 37°C for indicated times. The cells were lysed and proteins were separated by SDS-PAGE gel. The membranes were probed with indicated antibodies and the signals were revealed with ECL detection system (Pierce). The antibodies against IRF4, IRF8, c-Myc and cyclin D2 were obtained from Santa Cruz Biotech.
Generation of inducible IRF8 transgenic mice in B cells
Previously, we have generated an inducible IRF4 transgenic mice where the expression of IRF4 can be induced in B cell by Doxycycline (Dox) (29). Here, we used the same approach and generated an inducible IRF8 transgenic mice. Briefly, a full-length IRF8 cDNA was inserted into the pTet-Splice vector to generate the transgenic construct (IRF8Tg). Expression of the IRF8 transgene is driven by a minimal CMV promoter linked with a tetracycline response element (TRE). Three different founder lines were established. To induce transgene expression in B cells, we bred IRF8Tg mice with transgenic mice expressing a reverse tetracycline transactivator (rtTA) (30). The rtTA transgenic construct, which co-express GFP, was inserted into ROSA26 locus (ROSA-rtTA). The ROSA-rtTA was inactive due to an internal stop codon which can be cleaved by Cre recombinase. We bred ROSA-rtTA mice with CD19-cre mice to generate B cell specific expression of rtTA (CD19 rtTA). CD19rtTA mice were subsequently mated with IRF8Tg mice to generate mice where expression of IRF8 transgene can be induced exclusively in B cells (CD19creIRF8Tg). In this system, the expression of IRF8 transgene is induced only in presence of Dox. The founder line expressing the highest level of IRF8 transgene was chosen for subsequent studies. The mice of 8 to 12 weeks of age were used for this study.
TUNEL assay
The apoptosis status of splenic B cells in mice was examined with terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay. The TUNEL assay was conducted as we have previously described (31, 32). CD19creIRF8Tg and CD19cre mice were fed with Dox water for four weeks. Splenic B cells were isolated from the mice and stained with surface antibodies against CD93, B220 and IgM. The TUNEL positive cells were revealed with an APO-direct kit (BD Pharmingen).
Results
IRF8 deficient mice, both germline and B cell specific, produce anti-dsDNA antibodies
To determine whether deficiency in IRF8 deficient mice leads to breakdown of B cell tolerance, we measured titers of serum anti-dsDNA antibodies. As shown in Fig 1A, IRF8−/− mice produced higher titers of anti-dsDNA IgM and IgG antibodies than IRF8+/+ mice. Importantly, the titers of anti-dsDNA IgM and IgG were also found to be higher in IRF8fl/fl mice than in IRF8 heterozygous mutant mice (IRF8fl/+) (Fig. 1B). The anti-dsDNA antibodies can be detected in the IRF8−/− mice of three-months of age. In contrast, the anti-dsDNA antibodies can only be detected in the IRF8fl/fl mice of at least 5 months of age. Together, our results show that IRF8 deficient B cells produce anti-dsDNA antibodies, indicating that IRF8 plays a role in B cell tolerance induction.
Fig 1. IRF8 deficient mice, both germline and B cell specific, produce anti-dsDNA antibodies.
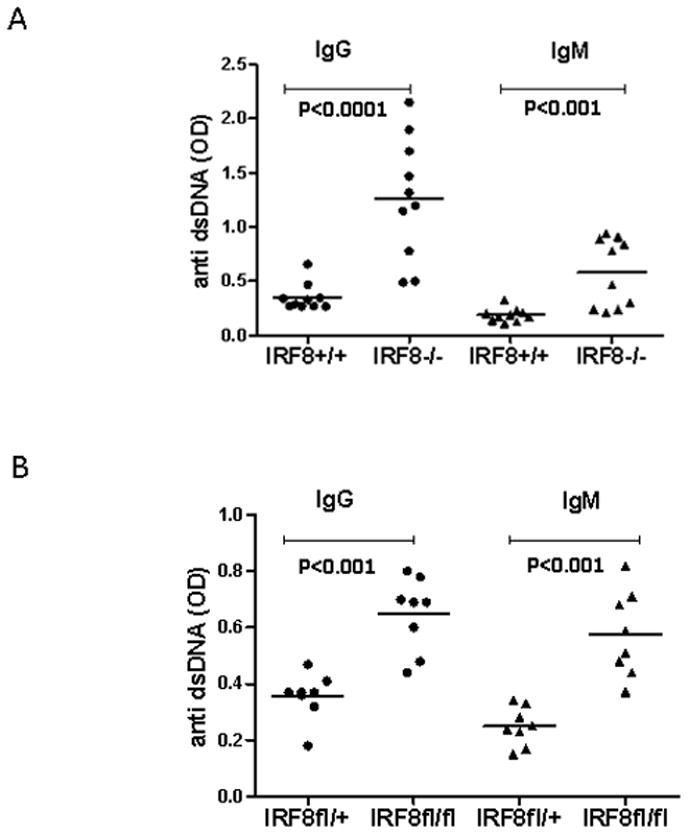
A) Sera were collected from IRF8+/+ and IRF8−/− mice three to four months of age. Ten mice from each group were used. The titers of anti-dsDNA IgM and IgG antibodies were measured by ELISA with kits from Alpha Diagnostic. B) Sera were collected from IRF8fl/+ and IRF8fl/fl mice five to eight months of age. Eight mice from each group were used. The titers of anti-dsDNA IgM and IgG antibodies were measured by ELISA.
B cell anergy is breached in the IRF8 deficient mice
The majority of self-reactive B cells are removed via anergy induction in the periphery. However, the role of IRF8 in B cell anergy induction is unclear. The most-widely used mouse model of B cell anergy is a double transgenic mouse which consists of a transgenic BCR recognizing hen egg lysozyme (HEL-Tg) and a second transgene expressing secreted HEL (33). B cells in the double transgenic mice (sHEL-Tg) are anergic and are unable to produce anti-HEL antibodies. To determine the role of IRF8 in B cell anergy induction, we bred IRF8−/− mice with sHEL-Tg mice to generate sHEL-Tg mice deficient for IRF8 (IRF8−/−sHEL-Tg). Blood was collected from mice and ELISA was performed to measure titers of anti-HEL antibody. As shown in Fig. 2A, anti-HEL antibodies titers were very low in IRF8+/+ and IRF8−/− mice, indicating that these mice do not make anti-HEL antibody. As expected, both IRF8+/+HEL-Tg and IRF8−/−HEL-Tg mice produced high levels of anti-HEL antibodies. Interestingly, the titers of anti-HEL antibodies were significantly higher in the IRF8−/−HEL-Tg mice than in the IRF8+/+HEL-Tg mice. In contrast, the titers of anti-HEL antibodies in the IRF8+/+sHEL-Tg mice were as low as those found in the IRF8+/+ mice. This finding is consistent with the fact that B cells in the IRF8+/+sHEL-Tg mice were anergic. However, anti-HEL antibodies were detected in the IRF8−/−sHEL-Tg mice at levels that are comparable to those produced by IRF8+/+HEL-Tg mice, indicating that B cells in the IRF8−/−sHEL-Tg mice are not anergic. We further examined and compared the numbers of splenic B cells in IRF8+/+HEL-Tg, IRF8−/−HEL-Tg, IRF8+/+sHEL-Tg and IRF8−/−sHEL-Tg mice. The numbers of splenic B cells were significantly higher in IRF8−/−HEL-Tg mice than in IRF8+/+HEL-Tg mice. Anergic B cells have a shortened lifespan which leads to their reduced number in mice. Indeed, there was only about 6 million anergic B cells in spleens of IRF8+/+sHEL-Tg mice, however, the number increased to 22 million in the IRF8−/−sHEL-Tg mice (Fig. 2B). Together, our results show that there is a defect in B cell anergy induction in the IRF8 deficient mice.
Fig 2. Breach in B cell anergy in the IRF8 deficient mice.

A) Sera were collected from mice of following genotypes: IRF8+/+, IRF8−/−, IRF8+/+HEL-Tg, IRF8−/−HEL-Tg, IRF8+/+sHEL-Tg and IRF8−/−sHEL-Tg. The mice aged from three to five months were used for this study. Eight mice from each group were used. The titers of serum anti-IgMa antibody were measured by ELISA. B) Splenic B cell populations were enumerated in mice of following genotypes: IRF8+/+HEL-Tg, IRF8−/−HEL-Tg, IRF8+/+sHEL-Tg and IRF8−/−sHEL-Tg. Six mice from each group were used. The numbers represent averages and standard deviations.
B cells in the IRF8−/−sHEL-Tg mice are more mature than B cells in the IRF8+/+ sHEL-Tg mice
Anergic B cells fail to mature in periphery and are arrested in their development at the transitional stage (34). To determine whether B cells in the IRF8−/−sHEL-Tg mice can overcome this developmental block, we analyzed and compared immunophenotypes of splenic B cells in the IRF8+/+sHEL-Tg and IRF8−/−sHEL-Tg mice. IRF8+/+HEL-Tg and IRF8−/−HEL-Tg mice were also analyzed. Transitional B cells stain positive for CD93 but CD93 staining is lost once they become mature B2 cells. Majority of the splenic B cells in IRF8+/+HEL-Tg and IRF8−/−HEL-Tg mice are mature B2 cells, thus stained negative for CD93 (Fig. 3A left). Since anergic B cells are blocked at the transitional stage, they should be CD93+. Indeed, anergic B cells in the IRF8+/+sHEL-Tg mice were stained positive for CD93 (Fig. 3A right). However, the intensity of CD93 staining reduced dramatically in the splenic B cells of IRF8−/−sHEL-Tg mice (Fig. 3A right), indicating that B cells in the IRF8−/−sHEL-Tg mice matured beyond the transitional stage. One of the key features of anergic B cells is the reduced level of surface IgM. Indeed, the expression level of IgM is much lower in the IRF8+/+sHEL-Tg mice than in the IRF8+/+HEL-Tg mice (Fig. 3B). However, B cells in the IRF8−/−sHEL-Tg mice expressed higher level of surface IgM than the anergic B cells in the IRF8+/+sHEL-Tg mice. It has been shown that anergic B cells in sHEL-Tg mice express low level of CD21 and upregulation of CD21 expression has been linked to breach in B cell anergy (11). Consistent with this previous report, CD21 was expressed at a low level in the anergic B cells of IRF8+/+sHEL-Tg mice (Fig. 3B). However, expression level of CD21 on B cells was much higher in the IRF8−/−sHEL-Tg mice than in the IRF8+/+sHEL-Tg mice. Self-reactive B cells can evade tolerance induction and mature in periphery via co-opting an additional BCR (35). To determine whether B cells in the IRF8−/−sHEL-Tg mice use dual BCRs to subvert anergy induction, we stained splenic B cells with anti-IgMb allotype antibody. The transgenic BCR is of IgMa allotype whereas the endogenous Ig locus is of IgMb allotype. As shown in Fig. 3B, 98% of splenic B cells in the IRF8−/−sHEL-Tg mice were IgMb negative, indicating that they only express transgenic BCR on their surface. Together, our results show that B cells in the IRF8−/−sHEL-Tg mice are more mature than their counterparts in the IRF8+/+sHEL-Tg mice.
Fig 3. B cells in the IRF8−/−sHEL-Tg mice are more mature than B cells in the IRF8+/+ sHEL-Tg mice.

Splenocytes were isolated from IRF8+/+HEL-Tg, IRF8−/−HEL-Tg, IRF8+/+sHEL-Tg and IRF8−/−sHEL-Tg mice of three to four months of age. The cells were stained with biotinylated HEL or antibodies against B220, CD93, IgM, CD21 and IgMa. A) The intensity of CD93 staining on isolated splenocytes was examined under a B220 gate. B) Expression of IgM, CD21, and IgMb was examined in isolated splenocytes. The binding to biotinylated HEL was also examined in isolated splenocytes. The analysis was done under a lymphocyte gate. The numbers represent the percentages of the cells that fall into each quadrant. The result is a representative of at least three independent experiments.
B cells in the IRF8−/−sHEL-Tg mice are responsive to antigen stimulation
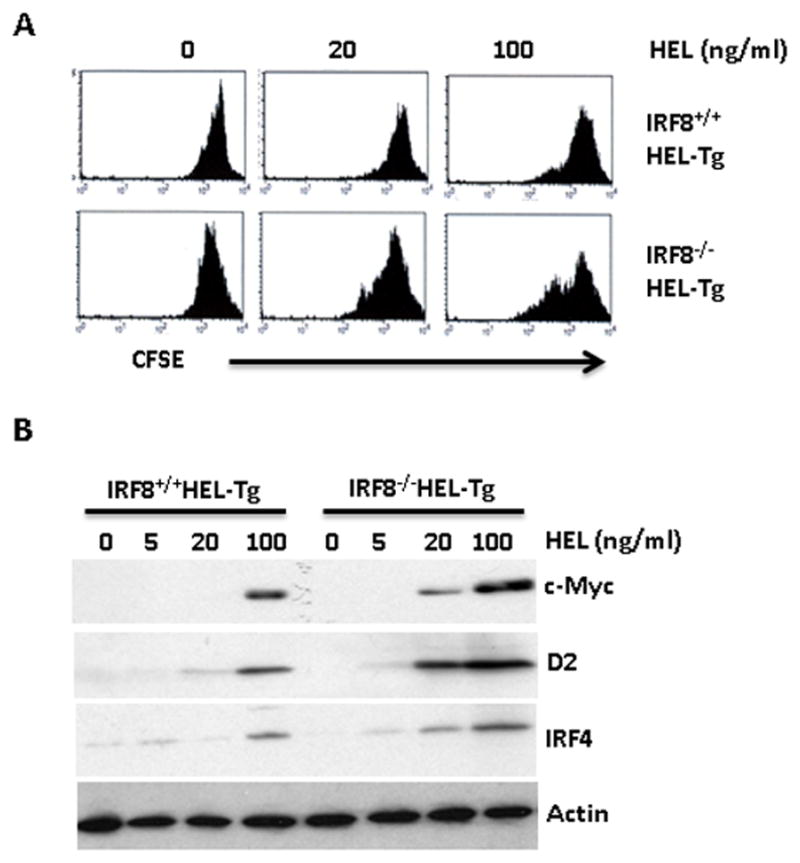
Anergic B cells exhibit defects in BCR signaling and are unresponsive to antigen stimulation. To further characterize splenic B cells in the IRF8−/−sHEL-Tg mice, we examined antigen-induced calcium influx and proliferation in the IRF8−/−sHEL-Tg mice. Splenocytes were isolated from IRF8−/−sHEL-Tg and IRF8+/+sHEL-Tg mice and loaded with Indo-1 Am dye to track calcium influx. Splenocytes from IRF8+/+HEL-Tg and IRF8−/−HEL-Tg were also analyzed. Upon addition of HEL antigen (100 ng/ml), calcium influx was detected in B cells isolated from IRF8+/+HEL-Tg mice and IRF8−/−HEL-Tg mice (Fig. 4A top). The amplitudes of calcium influx triggered by BCR signaling were comparable between IRF8−/−HEL-Tg and IRF8+/+HEL-Tg mice, although the amplitude of calcium influx appears to be slightly higher in B cells isolated from IRF8−/−HEL-Tg mice than B cells in the IRF8+/+sHEL-Tg mice. This result indicates that there is no gross defect in BCR signaling in the IRF8 deficient mice. Calcium influx was impaired in anergic B cells isolated from IRF8+/+sHEL-Tg mice (Fig. 4A bottom). In contrast, calcium influx, albeit at low amplitude, could still be detected in B cells isolated from IRF8−/−sHEL-Tg mice (Fig. 4A bottom), indicating that B cells were not anergic in those mice. We further examined antigen induced proliferation in B cells isolated from IRF8+/+sHEL-Tg and IRF8−/−sHEL-Tg mice. The isolated B cells were stained with CFSE tracking dye and stimulated with different concentrations of HEL. As shown in Fig. 4B, anergic B cells from IRF8+/+sHEL-Tg mice failed to respond to antigen stimulation as intensities of CFSE dye remained relatively unchanged between control and HEL treated cells. In contrast, HEL treatment led to dilution of CFSE dye in B cells isolated from the IRF8−/−sHEL-Tg mice, indicating that B cells in the IRF8−/−sHEL-Tg mice are responsive to antigen stimulation. The induction of genes that are critical for B cell proliferation, such as c-Myc, cyclin D2 and IRF4, are defective in anergic B cells (16). However, expression of c-Myc, cyclin D2 and IRF4 were found to be rapidly induced by HEL in B cells isolated from the IRF8−/−sHEL-Tg mice but not in the anergic B cells isolated from the IRF8+/+sHEL-Tg mice (Fig 4C). Together, our results confirm that B cells in the IRF8−/−sHEL-Tg mice lose their anergic state.
Fig 4. B cells in the IRF8−/−sHEL-Tg mice are responsive to antigen stimulation.

A) Splenocytes were isolated from IRF8+/+HEL-Tg, IRF8−/−HEL-Tg, IRF8+/+sHEL-Tg, IRF8−/−sHEL-Tg mice, stained with anti-B220 antibody and loaded with Indo-1 Am dye to track calcium influx. The Calcium influx of loaded cells was analyzed after HEL stimulation (100 ng/ml) with a LSR II flow cytometer. The calcium influx of B cells isolated from IRF8+/+HEL-Tg and IRF8−/−HEL-Tg mice were overlaid (Fig 4A top). The calcium influx between B cells of IRF8+/+sHEL-Tg and IRF8−/−sHEL-Tg mice were also overlaid (Fig 4A bottom). B) Splenic B cells in IRF8+/+sHEL-Tg, IRF8−/−sHEL-Tg mice were isolated through negative selection columns, stained with CFSE dye and plated into 96-well plate at 5x105 cells per well in presence of indicated concentrations of HEL antigens. Four days later, the cells were analyzed by FACS. C) Isolated B cells from IRF8+/+sHEL-Tg, IRF8−/−sHEL-Tg mice were stimulated with HEL (100 ng/ml) and lysed at different time points. Western Blot analysis was carried out to detect the expression of c-Myc, cyclin D2, IRF4, IRF8 and β-actin. The results are representatives of at least three independent experiments.
B cells in the IRF8−/−HEL-Tg are more resistant to antigen stimulation induced cell death than their counterparts in the IRF8+/+HEL-Tg mice
Our result suggests that IRF8 deficient B cells may have survival advantage as the numbers of splenic B cells significantly increased in the IRF8−/−HEL-Tg mice and the IRF8−/−sHEL-Tg mice (Fig 2B). BAFF is critical for survival and differentiation of B cells and overexpression of BAFF is sufficient to breach B cell tolerance (10). However, B cells isolated from IRF8+/+HEL-Tg and IRF8−/−HEL-Tg mice responded similarly to BAFF stimulation in vitro (Fig. 5A). We further incubated splenic B cells isolated from IRF8+/+HEL-Tg and IRF8−/−HEL-Tg mice with different concentrations of HEL antigen. The effect of antigen stimulation on cell survival was examined after 60h. Our result show that while antigen stimulation induced cell death was evident in B cells isolated from IRF8+/+HEL-Tg mice, this effect was attenuated in B cells isolated from IRF8−/−HEL-Tg mice (Fig. 5B). We further examined the effect of BAFF on antigen (200 ng/ml) induced death. As shown in Fig. 5C, BAFF, in a dose-dependent fashion, rescued HEL triggered cell death in B cells isolated from IRF8+/+HEL-Tg mice and IRF8−/−HEL-Tg mice. However, BAFF’s rescue effect was more pronounced in B cells isolated from IRF8−/−HEL-Tg mice than B cells from the IRF8+/+HEL-Tg mice. In absence of BAFF, 20% of IRF8−/−sHEL-Tg B cells were alive, however, in presence of BAFF at 100 ng/ml, 58% were still alive, a 2.9-fold increase in survival. In contrast, 10% of IRF8+/+HEL-Tg B cells were alive without BAFF, the percentage of living cells increased to 16% in presence of 100 ng/ml BAFF, an increase of only 1.6-fold. Thus, in presence of HEL, B cells from the IRF8−/−HEL-Tg mice are more sensitive to BAFF than their counterparts in the IRF8+/+HEL-Tg mice.
Fig 5. B cells in the IRF8−/−HEL-Tg mice are more resistant to antigen stimulation induced apoptosis than their counterparts in the IRF8+/+HEL-Tg mice.
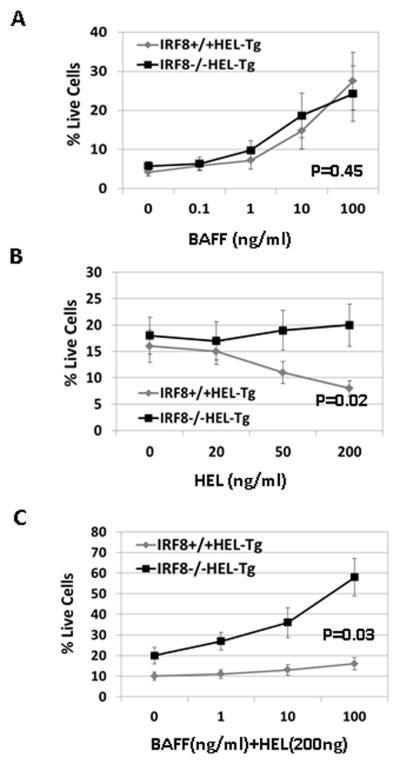
Splenocytes were isolated from IRF8−/−HEL-Tg and IRF8+/+HEL-Tg mice through negative selection. The isolated cells were plated into 96-well plate in triplicate at 5x105 cells per well in presence of increasing concentrations of BAFF (ng/ml). After 72h, the percentages of live cells were analyzed via PI staining. P=0.45 (Linear regression analysis). B) The isolated splenocytes were plated into 96-well plate in triplicate in presence of increasing concentrations of HEL (ng/ml). After 48h, the percentages of live cells were analyzed by FACS. P= 0.02 (linear regression analysis). C) The isolated splenocytes were incubated with HEL (200 ng /ml) in presence of increasing concentrations of BAFF (ng/ml). Two days later, the percentages of live cells were analyzed by FACS. P=0.03(linear regression analysis).
B cell in IRF8 deficient mice are hypersensitive to antigen stimulation
To further characterize the role of IRF8 in B cell activation and anergy induction, we examined cell response to antigen stimulation in IRF8 proficient and deficient B cells. Splenic B cells were isolated from IRF8+/+HEL-Tg and IRF8−/−HEL-Tg mice, stained with CFSE tracking dye and stimulated with different concentrations of HEL for four days. Our result show that B cells from the IRF8−/−HEL-Tg mice were more sensitive to antigen stimulation than their counterparts in the IRF8+/+HEL-Tg mice (Fig. 6A). At low HEL concentration (20 ng/ml), CFSE dilution was observed only in B cells isolated from the IRF8−/−HEL-Tg mice. Surprisingly, B cell proliferations induced by anti-IgM cross-linking and LPS were comparable between B cells isolated from IRF8+/+HEL-Tg and IRF8−/−HEL-Tg mice (Supplemental data 1). These results indicate that B cells in the IRF8−/−HEL-Tg mice are hyperproliferative only to stimulation by the cognate antigen. We further examined the induction of c-Myc, cyclin D2 and IRF4 by HEL (Fig. 6B). Our result shows that induction of c-Myc, cyclin D2 and IRF4 in B cells isolated from the IRF8−/−HEL-Tg mice were detected at HEL concentrations as low as 20 ng/ml. In contrast, their inductions in B cells isolated from IRF8+/+HEL-Tg mice was detected only at HEL concentration of 100 ng/ml. Together, these results show that B cells in the IRF8−/−HEL-Tg mice are more sensitive to HEL stimulation.
Fig 6. B cells in the IRF8 deficient mice are hypersensitive to antigen stimulation.
A) B cells were isolated from spleens of IRF8+/+HEL-Tg and IRF8−/−HEL-Tg mice, stained with CFSE dye and incubated with increasing concentrations of HEL. Four days later, the cells were analyzed by FACS. B) The isolated B cells from IRF8+/+HEL-Tg and IRF8−/−HEL-Tg mice were treated with increasing concentrations of HEL. The treated cells were lysed after 12h. Western Blot analysis was used to detect the expression of c-Myc, cyclin D2 and IRF4. The results are representatives of at least three independent experiments.
B cells in IRF8fl/fl mice are hypersensitive to antigen stimulation and are resistant to antigen stimulation induced cell death
Our results show that B cells in the IRF8 germline deficient mice (IRF8−/−) were more resistant to antigen induced cell death and were hypersensitive to antigen stimulation. However, IRF8−/− mice have defects in multiple lineages including T cells and Myeloid cells and it is possible that the B cell defects observed in the IRF8−/− mice are B cell extrinsic caused by the pleiotropic effects of IRF8. To address this, we bred IRF8fl/fl mice with HEL-Tg mice to generate B cell specific IRF8 knockout mice expressing HEL-BCR (IRF8fl/fl HEL-Tg). Compared to IRF8fl/+HEL-Tg control mice, the number of B cells was significantly increased in the IRF8fl/fl HEL-Tg mice (Fig. 7A). We next examined their response to BAFF and to antigen induced cell death. Consistent with results obtained from IRF8−/− mice, B cells in the IRF8fl/flHEL-Tg mice responded normally to BAFF stimulation alone (Fig. 7B), but were more resistant to HEL induced cell death than B cells in the IRF8fl/+HEL-Tg mice (Fig. 7C). Additionally, B cells in IRF8fl/flHEL-Tg mice were found to be more sensitive to BAFF in presence of antigen stimulation than their counterparts in the IRF8fl/+HEL-Tg mice (Fig. 7D). Finally, antigen induced cell proliferation was also found to be more pronounced in B cells isolated from IRF8fl/flHEL-Tg mice than B cells from IRF8fl/+HEL-Tg control mice (Fig. 7E). Together, our results indicate that IRF8 functions in a B cell intrinsic fashion to regulate cell survival and response to antigen stimulation.
Fig 7. B cells in IRF8fl/fl mice are hypersensitive to antigen stimulation and are resistant to antigen stimulation induced cell death.
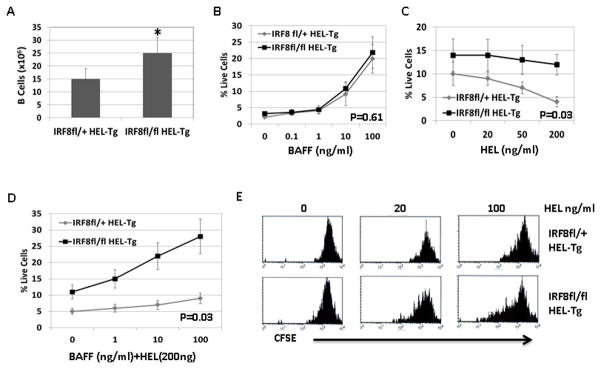
Splenic B cells were enumerated in IRF8fl/fl HEL-Tg and IRF8fl/+ HEL-Tg mice of 8 to 12 weeks of age. The results were means ± SD of five mice from each group. *p<0.05. B) Splenocytes were isolated from IRF8fl/fl HEL-Tg and IRF8fl/+ HEL-Tg mice through negative selection. The isolated cells were plated into 96-well plate in triplicate at 5x105 cells per well in presence of increasing concentrations of BAFF (ng/ml). After 72h, the percentages of live cells were analyzed via PI staining. P=0.61 (Linear regression analysis). C) The isolated splenocytes were plated into 96-well plate in triplicate in presence of increasing concentrations of HEL (ng/ml). After 48h, the percentages of live cells were analyzed by FACS. P= 0.03 (linear regression analysis). D) The isolated splenocytes were incubated with HEL (200 ng /ml) in presence of increasing concentrations of BAFF (ng/ml). Two days later, the percentages of live cells were analyzed by FACS. P=0.03 (linear regression analysis). E) The isolated splenocytes were stained with CFSE dye and incubated with increasing concentrations of HEL as indicated. Four days later, the cells were analyzed by FACS.
IRF8 is expressed at high level in anergic B cell and Induction of IRF8 transgene triggers apoptosis in the transitional B cells
IRF8 is expressed throughout B cell development; however, its expression level in the anergic B cells is unclear. We measured the expression level of IRF8 in B cells isolated from IRF8+/+HEL-Tg mice and IRF8+/+sHEL-Tg mice. Interestingly, IRF8 was expressed at a level that was twofold higher in the anergic B cells than normal B cells (Fig. 8A). This result is consistent with our findings that IRF8 is important for anergy induction. We wanted to determine the effect of a high level of IRF8 on B cells. To this end, we generated inducible IRF8 transgenic mice where the expression of IRF8 can be induced by Dox in B cells. The CD19CreIRF8Tg mice and CD19cre control mice were fed with Dox for four weeks and GFP+ splenic B cells were isolated by sorting. Our results show that expression levels of IRF8 were comparable between CD19creIRF8Tg and CD19cre control mice fed with regular water (Fig. 8B. Untreated). However, in presence of Dox water, the expression of IRF8 in B cells is found to be 2.5 fold higher in the CD19creIRF8Tg mice than in CD19Cre control mice, indicating that Dox treatment induced the expression of IRF8 transgene (Fig. 8B. Dox). To assess the effect of IRF8 transgene expression on B cell development in spleen, the number of transitional T1, T2, T3 and mature B cells (CD93−) were counted (Fig. 8C). Compared to CD19Cre mice, the number of CD93− mature B cells moderately decreased in the CD19creIRF8Tg mice. In contrast, the numbers of transitional B cells, particularly T2 and T3, were significantly decreased in the CD19creIRF8Tg mice. TUNEL assay further shows that percentages of apoptotic transitional B cells were significantly increased in the CD19creIRF8Tg mice (Fig. 8D). Together, our results show that induction of IRF8 transgene in B cells promotes apoptosis in the transitional B cells.
Fig 8. IRF8 is expressed at high level in the anergic B cells and induction of IRF8 transgene triggers apoptosis in the transitional B cells.
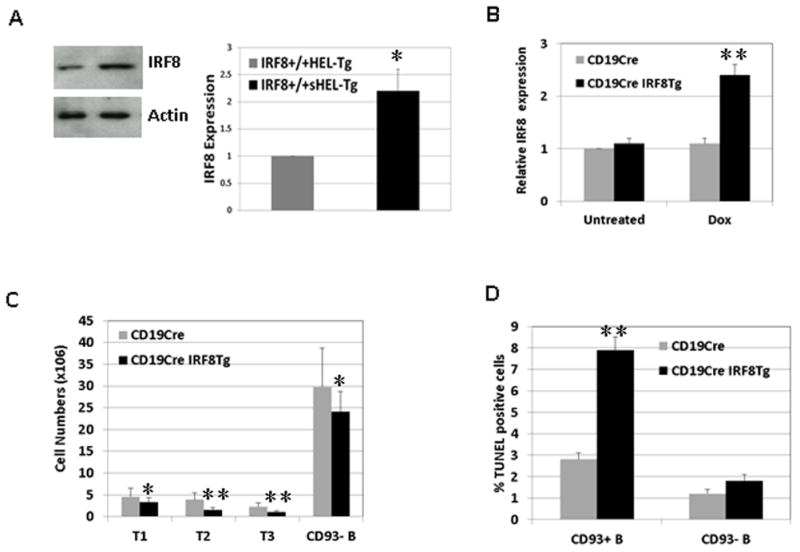
A) B cells were isolated from spleens of IRF8+/+HEL-Tg and IRF8+/+sHEL-Tg mice. The cells were lysed for Western Blot analysis to detect the expression of IRF8 and β-actin. The intensity of IRF8 signal in B cells of IRF8+/+sHEL-Tg mice was scanned, normalized to the signal of β-actin, and expressed as fold changes relative to its expression in the IRF8+/+HEL-Tg mice. B) To induce IRF8 transgene expression, CD19CreIRF8Tg mice and CD19Cre control mice were fed with Dox water for four weeks. Splenic B cells were subsequently analyzed. A) Splenic B cells (GFP+) were isolated by sorting from CD19CreIRF8Tg mice and CD19Cre control mice fed with Dox water (Dox). Additionally, B cells were also isolated from CD19CreIRF8Tg mice and CD19Cre control mice fed with regular water (Untreated). Real time PCR analysis was used to measure the expression of IRF8. The data was expressed as fold changes relative to the expression level of IRF8 in the untreated CD19Cre B cells. C) Splenic B cells in CD19CreIRF8Tg mice and CD19Cre control mice were stained with antibodies against CD93, CD23, IgM, and B220 and analyzed by FACS. The number of transitional T1 (CD93+IgMhiCD23−), T2 (CD93+IgMhiCD23+), T3 (CD93+IgMlowCD23+) and mature B cells (CD93-IgM+) were enumerated. A total of five mice were used from each group. D) Splenic B cells were stained with antibodies against B220, IgM and CD93. The TUNEL positive B cells were determined with a kit from BD Biosciences. The B220+IgM+CD93− mature B cells and B220+IgM+CD93+ transitional B cells were analyzed separately. The results were means ± SD of three independent experiments. *p<0.05, **p<0.01.
Discussion
Engagement of cognate antigen by mature B cells leads to B cell activation and antibody secretion, however, antigen encounter by developing B cells causes developmental arrest and anergy. Anergic B cells do not respond to antigen stimulation because they are transitional B cells with defective BCR signaling (2). In this report, we demonstrate that B cell anergy is breached in the IRF8 deficient mice. In contrast to anergic B cell in the wild type background, B cells in the IRF8−/−sHEL-Tg mice exhibited downregulated CD93 and elevated levels of surface IgM and CD21; these findings indicate that B cells in the IRF8−/−sHEL-Tg mice are able to mature beyond transitional stage which eventually allows them to regain responses to antigen stimulation and the ability to secrete antibodies.
Our findings indicate that IRF8 is an important regulator of B cell survival at the transitional stage. We show that expression of IRF8 was elevated in anergic B cells and induction of IRF8 transgene in B cells promoted apoptosis at the transitional stage. Additionally, antigen stimulation induced cell death was attenuated in the IRF8−/−HEL-Tg mice, indicating that IRF8 also regulates the survival of mature B cells. BAFF is a critical regulator of B cell survival and maturation. Although the response to BAFF stimulation alone was normal in B cells of IRF8−/−HEL-Tg mice, the response by B cells to a combined stimulation of BAFF and HEL was more potent in the IRF8−/−HEL-Tg mice than their counterparts in the IRF8+/+HEL-Tg mice. It appears that in presence of antigen stimulation, B cells in IRF8−/−HEL-Tg mice become hypersensitive to BAFF, a property that could lead to breach of anergy by promoting survival and maturation of anergic B cells. How IRF8 regulates B cell survival remains to be determined. Previous studies have linked the pro-apoptotic effect of IRF8 to several putative targets including Bax, Bcl2 and acid ceramidase (36–38).
Our results show that B cells in the IRF8−/−HEL-Tg mice are more sensitive to antigen stimulation than their counterparts in the IRF8+/+HEL-Tg mice. The hypersensitivity to HEL stimulation is not due to defect in BCR signaling as calcium influxes triggered by HEL stimulation were comparable between B cells isolated from IRF8−/−HEL-Tg and IRF8+/+HEL-Tg mice. Surpringsly, our results also show that B cells in the IRF8−/−HEL-Tg mice were hyperresponsive only to HEL stimulation, but not to anti-IgM crosslinking or LPS. Since IRF8 expression is elevated in the anergic B cells, IRF8 may function as a negative regulator of B cell activation in the anergic B cells. It is conceivable that the hypersensitivity to HEL stimulation could contribute to anergy breakdown by allowing B cells in the IRF8−/−sHEL-Tg mice to proliferate and secrete antibodies.
Our results indicate that IRF8 functions in a B cell intrinsic fashion to regulate B cell survival and response to antigen stimulation. IRF8 is critical for development of several lineages of blood cells and IRF8 germline deficient mice exhibit defects in multiple lineages including T, B and myeloid cells (39). Because of this, the B cell defects observed in the IRF8 germline mutant mice could be B cell extrinsic defects that are caused indirectly by the pleiotropic effects of IRF8. However, our study shows that B cell survival and response to antigen stimulation were similarly affected in the IRF8 conditional mutant mice, indicating that those defects are B cell specific. Since B cell tolerance can be influenced by defects in other immune cells, such as T cells and myeloid cells, it is possible that abnormalities in other lineage cells also contribute to the tolerance breakdown in the IRF8 germline deficient mice. Our findings that B cells in the IRF8 conditional mutant mice produced lower titers of anti-dsDNA antibody and did so at later time points than B cells in the IRF8 germline mutant mice are consistent with this scenario.
Two recent genome-wide SNP association studies have identified IRF8 as a major susceptibility gene for Systemic Lupus Erythematosus (SLE)(40, 41). One of the studies further shows that the SNPs cause a high level of IRF8 in lupus patients (40). The involvement of IRF8 in SLE has been linked to its function in interferon signaling and production (40, 41). Consistent with this assertion, a recent study demonstrates that lupus development was attenuated in New Zealand Black mice deficient for IRF8 (42). This study further shows that IRF8 deficient mice are lack of plasmacytoid dendritic cells (pDC), which are the major producers of type 1 interferon in vivo (42). Together, those studies establish the requirement for IRF8 in lupus development. However, our results presented here show that lack of IRF8 in B cells leads to breackdown of B cell tolerance which could also contribute to development of autoimmune diseases. The paradoxical role IRF8 plays in the development of autoimmune diseases mirrors that of TLRs. It has been well-documented that TLR signaling is critical for interferon production and class-switching and is absolutely required for Lupus development (43–45). However, it has also been shown that lack of TLR signaling during B cell development leads to breakdown of B cell tolerance (46). Like TLRs, the role of IRF8 in autoimmunity is complex and stage specific. Given the contradicting roles IRF8 plays in SLE, our findings would caution against targeting IRF8 as a therapeutic target for lupus.
Supplementary Material
Acknowledgments
This work was supported by a grant from NIH AI67891 (R.L). Simanta Pathak was supported by a predoctoral fellowship from the University of Nebraska Medical Center.
We thank Vicki Smith at the UNMC cell analysis core for help with data analysis.
Abbreviations
- IRF8
interferon regulatory factor 8
- HEL
hen egg lysozyme
- HEL-Tg
BCR transgenic mice recognizing HEL
- sHEL-Tg
HEL-Tg mice expressing secreted HEL antigen
- IRF8−/−
IRF8 germline mutant mice
- IRF8fl/fl
IRF8 B cell specific conditional mutant mice
- SLE
Systemic Lupus Erythematosus
References
- 1.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–1377. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 2.Yarkoni Y, Getahun A, Cambier JC. Molecular underpinning of B-cell anergy. Immunol Rev. 2010;237:249–263. doi: 10.1111/j.1600-065X.2010.00936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nemazee D. Receptor editing in lymphocyte development and central tolerance. Nat Rev Immunol. 2006;6:728–740. doi: 10.1038/nri1939. [DOI] [PubMed] [Google Scholar]
- 4.O’Neill SK, Getahun A, Gauld SB, Merrell KT, Tamir I, Smith MJ, Dal Porto JM, Li QZ, Cambier JC. Monophosphorylation of CD79a and CD79b ITAM motifs initiates a SHIP-1 phosphatase-mediated inhibitory signaling cascade required for B cell anergy. Immunity. 2011;35:746–756. doi: 10.1016/j.immuni.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Browne CD, Del Nagro CJ, Cato MH, Dengler HS, Rickert RC. Suppression of phosphatidylinositol 3,4,5-trisphosphate production is a key determinant of B cell anergy. Immunity. 2009;31:749–760. doi: 10.1016/j.immuni.2009.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kitaura Y, I, Jang K, Wang Y, Han YC, Inazu T, Cadera EJ, Schlissel M, Hardy RR, Gu H. Control of the B cell-intrinsic tolerance programs by ubiquitin ligases Cbl and Cbl-b. Immunity. 2007;26:567–578. doi: 10.1016/j.immuni.2007.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mecklenbrauker I, Saijo K, Zheng NY, Leitges M, Tarakhovsky A. Protein kinase Cdelta controls self-antigen-induced B-cell tolerance. Nature. 2002;416:860–865. doi: 10.1038/416860a. [DOI] [PubMed] [Google Scholar]
- 8.Mackay F, Schneider P, Rennert P, Browning J. BAFF AND APRIL: a tutorial on B cell survival. Annu Rev Immunol. 2003;21:231–264. doi: 10.1146/annurev.immunol.21.120601.141152. [DOI] [PubMed] [Google Scholar]
- 9.Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu HB, Cyster JG. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity. 2004;20:441–453. doi: 10.1016/s1074-7613(04)00079-2. [DOI] [PubMed] [Google Scholar]
- 10.Thien M, Phan TG, Gardam S, Amesbury M, Basten A, Mackay F, Brink R. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity. 2004;20:785–798. doi: 10.1016/j.immuni.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 11.Oliver PM, Vass T, Kappler J, Marrack P. Loss of the proapoptotic protein, Bim, breaks B cell anergy. J Exp Med. 2006;203:731–741. doi: 10.1084/jem.20051407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barrington RA, Borde M, Rao A, Carroll MC. Involvement of NFAT1 in B cell self-tolerance. J Immunol. 2006;177:1510–1515. doi: 10.4049/jimmunol.177.3.1510. [DOI] [PubMed] [Google Scholar]
- 13.Borde M, Barrington RA, Heissmeyer V, Carroll MC, Rao A. Transcriptional basis of lymphocyte tolerance. Immunol Rev. 2006;210:105–119. doi: 10.1111/j.0105-2896.2006.00370.x. [DOI] [PubMed] [Google Scholar]
- 14.Peng SL, Gerth AJ, Ranger AM, Glimcher LH. NFATc1 and NFATc2 together control both T and B cell activation and differentiation. Immunity. 2001;14:13–20. doi: 10.1016/s1074-7613(01)00085-1. [DOI] [PubMed] [Google Scholar]
- 15.de Alboran IM, O’Hagan RC, Gartner F, Malynn B, Davidson L, Rickert R, Rajewsky K, DePinho RA, Alt FW. Analysis of C-MYC function in normal cells via conditional gene-targeted mutation. Immunity. 2001;14:45–55. doi: 10.1016/s1074-7613(01)00088-7. [DOI] [PubMed] [Google Scholar]
- 16.Glynne R, Ghandour G, Rayner J, Mack DH, Goodnow CC. B-lymphocyte quiescence, tolerance and activation as viewed by global gene expression profiling on microarrays. Immunol Rev. 2000;176:216–246. doi: 10.1034/j.1600-065x.2000.00614.x. [DOI] [PubMed] [Google Scholar]
- 17.Refaeli Y, Field KA, Turner BC, Trumpp A, Bishop JM. The protooncogene MYC can break B cell tolerance. Proc Natl Acad Sci U S A. 2005;102:4097–4102. doi: 10.1073/pnas.0409832102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu R. Interferon regulatory factor 4 and 8 in B-cell development. Trends Immunol. 2008;29:487–492. doi: 10.1016/j.it.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tamura T, Nagamura-Inoue T, Shmeltzer Z, Kuwata T, Ozato K. ICSBP directs bipotential myeloid progenitor cells to differentiate into mature macrophages. Immunity. 2000;13:155–165. doi: 10.1016/s1074-7613(00)00016-9. [DOI] [PubMed] [Google Scholar]
- 20.Wang H, Lee CH, Qi C, Tailor P, Feng J, Abbasi S, Atsumi T, Morse HC., 3rd IRF8 regulates B-cell lineage specification, commitment, and differentiation. Blood. 2008;112:4028–4038. doi: 10.1182/blood-2008-01-129049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma S, Pathak S, Mandal M, Clark RM, Trinh L, Lu R. Ikaros and Aiolos inhibit pre-B cells proliferation by directly suppresssing c-Myc expression. Mol Cell Biol. 2010;30:4149–4158. doi: 10.1128/MCB.00224-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma S, Pathak S, Trinh L, Lu R. Interferon regulatory factors 4 and 8 induce the expression of Ikaros and Aiolos to down-regulate pre-B-cell receptor and promote cell-cycle withdrawal in pre-B-cell development. Blood. 2008;111:1396–1403. doi: 10.1182/blood-2007-08-110106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma S, Turetsky A, Trinh L, Lu R. IFN regulatory factor 4 and 8 promote Ig light chain kappa locus activation in pre-B cell development. J Immunol. 2006;177:7898–7904. doi: 10.4049/jimmunol.177.11.7898. [DOI] [PubMed] [Google Scholar]
- 24.Lee CH, Melchers M, Wang H, Torrey TA, Slota R, Qi CF, Kim JY, Lugar P, Kong HJ, Farrington L, van der Zouwen B, Zhou JX, Lougaris V, Lipsky PE, Grammer AC, Morse HC., 3rd Regulation of the germinal center gene program by interferon (IFN) regulatory factor 8/IFN consensus sequence-binding protein. J Exp Med. 2006;203:63–72. doi: 10.1084/jem.20051450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng J, Wang H, Shin DM, Masiuk M, Qi CF, Morse HC., 3rd IFN regulatory factor 8 restricts the size of the marginal zone and follicular B cell pools. J Immunol. 2011;186:1458–1466. doi: 10.4049/jimmunol.1001950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hardy RR. B-1 B cells: development, selection, natural autoantibody and leukemia. Curr Opin Immunol. 2006;18:547–555. doi: 10.1016/j.coi.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 27.Lu R, Medina KL, Lancki DW, Singh H. IRF-4,8 orchestrate the pre-B-to-B transition in lymphocyte development. Genes Dev. 2003;17:1703–1708. doi: 10.1101/gad.1104803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar KR, Li L, Yan M, Bhaskarabhatla M, Mobley AB, Nguyen C, Mooney JM, Schatzle JD, Wakeland EK, Mohan C. Regulation of B cell tolerance by the lupus susceptibility gene Ly108. Science. 2006;312:1665–1669. doi: 10.1126/science.1125893. [DOI] [PubMed] [Google Scholar]
- 29.Pathak S, Ma S, Trinh L, Eudy JD, Wagner K, Joshi SS, Lu R. IRF4 is a suppressor of c-Myc induced leukemia. PLoS ONE. 2011 doi: 10.1371/journal.pone.0022628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belteki G, Haigh J, Kabacs N, Haigh K, Sison K, Costantini F, Whitsett J, Quaggin SE, Nagy A. Conditional and inducible transgene expression in mice through the combinatorial use of Cre-mediated recombination and tetracycline induction. Nucleic Acids Res. 2005;33:e51. doi: 10.1093/nar/gni051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma S, Shukla V, Leilei F, Gould KA, Joshi SS, Lu R. Accelerated development of chronic lymphocytic leukemia in New Zealand Black mice expressing low level of interferon regulatory factor 4. J Biol Chem. 2013;288:26430–26440. doi: 10.1074/jbc.M113.475913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shukla V, Ma S, Hardy RR, Joshi SS, Lu R. A role for IRF4 in the development of CLL. Blood. 2013 doi: 10.1182/blood-2013-03-492769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, Pritchard-Briscoe H, Wotherspoon JS, Loblay RH, Raphael K, et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. 1988;334:676–682. doi: 10.1038/334676a0. [DOI] [PubMed] [Google Scholar]
- 34.Merrell KT, Benschop RJ, Gauld SB, Aviszus K, Decote-Ricardo D, Wysocki LJ, Cambier JC. Identification of anergic B cells within a wild-type repertoire. Immunity. 2006;25:953–962. doi: 10.1016/j.immuni.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 35.Li Y, Li H, Weigert M. Autoreactive B cells in the marginal zone that express dual receptors. J Exp Med. 2002;195:181–188. doi: 10.1084/jem.20011453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burchert A, Cai D, Hofbauer LC, Samuelsson MK, Slater EP, Duyster J, Ritter M, Hochhaus A, Muller R, Eilers M, Schmidt M, Neubauer A. Interferon consensus sequence binding protein (ICSBP; IRF-8) antagonizes BCR/ABL and down-regulates bcl-2. Blood. 2004;103:3480–3489. doi: 10.1182/blood-2003-08-2970. [DOI] [PubMed] [Google Scholar]
- 37.Hu X, Yang D, Zimmerman M, Liu F, Yang J, Kannan S, Burchert A, Szulc Z, Bielawska A, Ozato K, Bhalla K, Liu K. IRF8 regulates acid ceramidase expression to mediate apoptosis and suppresses myelogeneous leukemia. Cancer Res. 2011;71:2882–2891. doi: 10.1158/0008-5472.CAN-10-2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang J, Hu X, Zimmerman M, Torres CM, Yang D, Smith SB, Liu K. Cutting edge: IRF8 regulates Bax transcription in vivo in primary myeloid cells. J Immunol. 2011;187:4426–4430. doi: 10.4049/jimmunol.1101034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. 2008;26:535–584. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- 40.Cunninghame Graham DS, Morris DL, Bhangale TR, Criswell LA, Syvanen AC, Ronnblom L, Behrens TW, Graham RR, Vyse TJ. Association of NCF2, IKZF1, IRF8, IFIH1, and TYK2 with systemic lupus erythematosus. PLoS Genet. 2011;7:e1002341. doi: 10.1371/journal.pgen.1002341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lessard CJ, Adrianto I, Ice JA, Wiley GB, Kelly JA, Glenn SB, Adler AJ, Li H, Rasmussen A, Williams AH, Ziegler J, Comeau ME, Marion M, Wakeland BE, Liang C, Ramos PS, Grundahl KM, Gallant CJ, Alarcon-Riquelme ME, Alarcon GS, Anaya JM, Bae SC, Boackle SA, Brown EE, Chang DM, Cho SK, Criswell LA, Edberg JC, Freedman BI, Gilkeson GS, Jacob CO, James JA, Kamen DL, Kimberly RP, Kim JH, Martin J, Merrill JT, Niewold TB, Park SY, Petri MA, Pons-Estel BA, Ramsey-Goldman R, Reveille JD, Scofield RH, Song YW, Stevens AM, Tsao BP, Vila LM, Vyse TJ, Yu CY, Guthridge JM, Kaufman KM, Harley JB, Wakeland EK, Langefeld CD, Gaffney PM, Montgomery CG, Moser KL B Network and G Network. Identification of IRF8, TMEM39A, and IKZF3-ZPBP2 as susceptibility loci for systemic lupus erythematosus in a large-scale multiracial replication study. American journal of human genetics. 2012;90:648–660. doi: 10.1016/j.ajhg.2012.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baccala R, Gonzalez-Quintial R, Blasius AL, Rimann I, Ozato K, Kono DH, Beutler B, Theofilopoulos AN. Essential requirement for IRF8 and SLC15A4 implicates plasmacytoid dendritic cells in the pathogenesis of lupus. Proc Natl Acad Sci U S A. 2013;110:2940–2945. doi: 10.1073/pnas.1222798110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 44.Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K, Shlomchik MJ. TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol. 2010;184:1840–1848. doi: 10.4049/jimmunol.0902592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Subramanian S, Tus K, Li QZ, Wang A, Tian XH, Zhou J, Liang C, Bartov G, McDaniel LD, Zhou XJ, Schultz RA, Wakeland EK. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci U S A. 2006;103:9970–9975. doi: 10.1073/pnas.0603912103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Isnardi I, Ng YS, Srdanovic I, Motaghedi R, Rudchenko S, von Bernuth H, Zhang SY, Puel A, Jouanguy E, Picard C, Garty BZ, Camcioglu Y, Doffinger R, Kumararatne D, Davies G, Gallin JI, Haraguchi S, Day NK, Casanova JL, Meffre E. IRAK-4- and MyD88-dependent pathways are essential for the removal of developing autoreactive B cells in humans. Immunity. 2008;29:746–757. doi: 10.1016/j.immuni.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.