

Abstract
Objectives
Lafora disease is a rare yet invariably fatal form of progressive neurodegenerative epilepsy resulting from mutations in the phosphatase laforin. Several therapeutic options for Lafora disease patients are currently being explored, and these therapies would benefit from a biochemical means of assessing functional laforin activity following treatment. To date, only clinical outcomes such as decreases in seizure frequency and severity have been used to indicate success of epilepsy treatment. However, these qualitative measures exhibit variability and must be assessed over long periods of time. In this work, we detail a simple and sensitive bioassay that can be used for the detection of functional endogenous laforin from human and mouse tissue.
Design and methods
We generated antibodies capable of detecting and immunoprecipitating endogenous laforin. Following laforin immunoprecipitation, laforin activity was assessed via phosphatase assays using para-nitrophenylphosphate (pNPP) and a malachite green-based assay specific for glucan phosphatase activity.
Results
We found that antibody binding to laforin does not impede laforin activity. Furthermore, the malachite green-based glucan phosphatase assay used in conjunction with a rabbit polyclonal laforin antibody was capable of detecting endogenous laforin activity from human and mouse tissue. Importantly, this assay discriminated between laforin activity and other phosphatases.
Conclusions
The bioassay that we have developed utilizing laforin antibodies and an assay specific for glucan phosphatase activity could prove valuable in the rapid detection of functional laforin in patients to which novel Lafora disease therapies have been administered.
Keywords: Lafora disease, laforin, glucan phosphatase, bioassay, malachite green, epilepsy
1. Introduction
Lafora disease (LD, OMIM #254780) is a fatal autosomal recessive form of neurodegenerative epilepsy in which patients exhibit myoclonic, tonic clonic, and focal occipital seizures [1, 2]. Patients typically present with an initial seizure during early adolescence, and these seizures increase in frequency and severity prior to death within ten years from complications arising from neurodegeneration [1]. Concurrent with increasing seizure frequency and progressive neurodegeneration is the accumulation of insoluble glucan deposits termed Lafora bodies (LBs) in the cytoplasm of many cell types [1, 3]. LBs are sparsely-branched and hyperphosphorylated relative to normal cellular glycogen, resembling instead insoluble glucans such as plant starch [2–5]. LBs are a distinguishing characteristic of LD [6], and are caused by inactivating mutations in either the gene encoding a glucan phosphatase known as laforin [5, 7–10] or an E3 ubiquitin ligase called malin [11, 12]. LBs increase in size and number during the life of patients, and are hypothesized to be responsible for neuronal cell death [2, 3].
Laforin, encoded by the EPM2A gene, consists of an N-terminal carbohydrate-binding module (CBM) and a C-terminal dual-specificity phosphatase (DSP) domain [2, 7, 10, 13–15]. Laforin binds to and dephosphorylates glucans/glycogen, and is the only known phosphatase that exhibits glucan phosphatase activity in humans [5, 8, 9, 16]. Most LD-causing point mutations in the gene encoding laforin disrupt either glucan binding or phosphatase activity [5, 17]. Ganesh and colleagues generated a laforin knock-out mouse model and demonstrated that these mice exhibit LB accumulation [18]. Subsequent analysis of the LBs from this mouse model by the Roach and Minassian labs revealed that the LBs are hyperphosphorylated and poorly-branched and that laforin can remove phosphate from the LBs [4, 8]. Recent data demonstrated that phosphate may be misincorporated into glycogen due to an error by glycogen synthase, and in the absence of laforin activity this increased phosphate inhibits proper branching necessary for glycogen solubility [19]. However, a subsequent report questioned this mechanism and suggested that the phosphate is not introduce by glycogen synthase [20]. Regardless of how the phosphate is incorporated into LBs, multiple lines of evidence support the hypothesis that laforin’s role in the cell is to remove phosphate incorporated into cellular glycogen. Without functional laforin, LBs form and LD results.
To date, LD can only be managed for a short period using palliative therapeutics designed to limit the severity and frequency of epileptic episodes [6, 21]. More permanent therapeutic options, including EPM2A gene replacement using neutral pegylated immunoliposomes [22] and readthrough of nonsense mutations using aminoglycosides and functionally-related compounds [21, 23], are currently being explored. Additionally, results from mouse models suggest that downregulation of glycogen synthase is an additional treatment option [24, 25]. As therapeutic options for LD become available and enter clinical trials, it will be necessary to assess efficacy of these therapies. Currently, the quantitated neurological and electrophysiological states of LD patients are the only means by which therapeutics may be assessed for efficacy, and these methods are subject to varied response and must be assessed long-term [1, 21, 26]. We have developed a simple and sensitive bioassay for laforin activity that provides both rapid results and could be utilized at any time after treatment administration. This bioassay is suitable for both the detection of endogenous laforin protein concentrations and, more importantly, the assessment of enzymatic activity. Furthermore, this bioassay is specific for laforin, as no other human enzyme is known to possess glucan phosphatase activity [5, 27]. Additionally, we demonstrate that this bioassay can measure the activity of endogenous laforin from either human or mouse tissue.
2. Materials and Methods
2.1. Laforin antibody production
We collaborated with the NIH NeuroMab Facility (Davis, CA) to generate and characterize mouse monoclonal IgG1 antibodies raised against full-length wild-type human laforin-HIS6. We also generated rabbit IgG polyclonal antibodies (Cocalico Biologicals Inc, Reamstown, PA) against full-length wild-type human laforin-HIS6.
Laforin cloned into the pET21a vector (EMD Chemicals, Darmstadt, Germany) [13] was expressed in Escherichia coli BL21 (DE3) CodonPlus RIL cells (Stratagene, Santa Clara, CA) and then purified from soluble bacterial extracts using Ni2+-agarose (Qiagen, Hilden, Germany) affinity chromatography as previously described [11, 28]. Eluted laforin (1.2 mg/mL) was supplemented with 20% glycerol and used for antibody production.
Mouse monoclonal antibodies against laforin (NeuroMab N84/1 and N84/37.1; 30 μg/mL) were purified from tissue culture supernatant collected from cultured mouse hybridomas. The supernatant was filtered through a 0.22 μm filter (Millipore, Billerica, MA) and then affinity purified using either a HiTrap Protein A HP or HiTrap Protein G HP column in conjunction with an antibody purification kit (GE Healthcare, Little Chalfont, UK). Rabbit polyclonal antibodies against laforin (rabbits #113 and #139; 0.6 mg/mL) were obtained from exsanguination serum filtered through a 0.22μm filter and affinity purified with protein A using the Montage Antibody Purification Kit with PROSEP-A Media (Millipore). We also created a rabbit polyclonal antibody against a laforin peptide (amino acids 18–32; 21st Century Biochemicals Inc, Marlboro, MA) and obtained peptide affinity-purified α-laforin antibody (#4860; 50 μg/mL).
2.2. Assessment of antibodies for the immunoprecipitation of overexpressed laforin
Full-length human wild-type laforin cloned into a modified pcDNA3.1 vector (Invitrogen, Carlsbad, CA) featuring an N-terminal FLAG tag (pcDNA3.1 FLAG) [13] was first expressed in human embryonic kidney 293 (HEK293) cells grown to 80% confluence. 24 hours post-transfection, cells were rinsed twice with 5 mL Tris buffered saline (TBS) and lysates collected in 300 μl ice-cold modified RIPA buffer (mRIPA) supplemented with protease inhibitors [11, 28]. After centrifugation to clear the lysates (10 min, 10K, 4°C), 10 μl of each laforin antibody was then incubated while rocking with supernatants (1 hour, 4°C) prior to the addition of 35 μl Protein A Sepharose slurry (Sigma-Aldrich, St. Louis, MO). Supernatants were then incubated while rocking (1 hour, 4°C) prior to centrifugation (2 min, 5K, 4°C) to collect the Sepharose beads.
After removing the supernatant by vacuum, the Sepharose was washed twice by the addition of 1 mL of mRIPA buffer followed by centrifugation (2 min, 5K, 4°C), with the supernatant removed after each step. The supernatant from the final wash was removed and the Sepharose resuspended in 50 μl protein sample dye. After boiling the samples (10 min, 95°C) and collecting the supernatant from the Sepharose, 40 μl of Sepharose supernatant (IP) and 2.7 μg cell lysate (WCL) was analyzed by Western blot using mouse α-FLAG peroxidase-conjugated antibody (1:3000 M2 α-FLAG HRP; Sigma-Aldrich). Blots were developed with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific, Waltham, MA).
2.3. Assessment of antibodies for the immunoprecipitation of endogenous laforin
For each antibody tested, HepG2 cells grown to 90% confluence in three 100 mm plates were collected in 500 μl mRIPA buffer supplemented with protease inhibitors. 10 μl of each laforin antibody was then incubated with the lysates (2 hours, 4°C) followed by rocking with 35 μl Protein A Sepharose (2 hours, 4°C). 40 μl of IPs and 50 μg of WCL samples were analyzed by Western blot using 1:1000 of either α-laforin antibody N84/37.1 or #113 to avoid same species cross-reactivity of the secondary antibody. Either 1:3000 goat α-mouse HRP (Invitrogen) or 1:3000 goat α-rabbit HRP (Calbiochem, Billerica, MA) secondary was used.
2.4. Phosphatase assays
para-nitrophenylphosphate (pNPP) assays were performed in 50 μl replicates containing 1X phosphatase buffer (0.1 M sodium acetate, 0.05 M Bis-Tris, 0.05 M Tris-HCl, pH 5), 2 mM dithiothreitol (DTT), and 50 mM pNPP. Following the addition of laforin, assays were incubated at 37 C for the specified time followed by the addition of 200 μL of 0.25 M NaOH to terminate the reactions. The absorbance of the reactions was then measured at 410 nm. Malachite green assays were performed in 20 μl replicates containing 1X phosphatase buffer (pH 7), 45 μg amylopectin, and 2 mM DTT. Following addition of laforin, reactions were incubated at 37 C for the specified time, and terminated by the addition of 20 μL of 0.1 M N-ethylmaleimide. 80 μL of malachite green reagent was then added to the reactions and the absorbance measured at 620 nm [27].
2.5. Assessment of antibody impact on recombinant laforin activity
Recombinant wild-type laforin-HIS6 was purified using Ni2+-affinity chromatography [11, 28] in the presence of 15% glycerol and used for subsequent assays. For pNPP assays, 100 ng of laforin was first incubated with 2 μl α-laforin antibody #113 (30 min, 4°C) followed by the addition of 10 μl Protein A Sepharose (30 min, 4°C). For malachite green assays, 100 ng of laforin was incubated with 1 μl α-laforin antibody #113 (30 min, 4°C) followed by 3 μl of Protein A Sepharose (30 min, 4°C). Reaction components were then added to the immunoprecipitated laforin and reactions incubated at 37°C for 10 minutes prior to termination. 100 ng of inactive C266S laforin was used as a control. As commercial preparations of Protein A Sepharose can contain contaminating phosphate, malachite green assays were performed with 3 μl of Protein A Sepharose in the absence of laforin.
2.6. Immunoprecipitation of overexpressed laforin for activity assay
Two 100 mm plates of HEK293 cells grown to 80% confluence were transfected with pcDNA3.1 FLAG wild-type laforin, inactive C266S laforin, or empty vector [13]. After 24 hours, lysates were collected in 500 μl mRIPA supplemented with protease inhibitors for each construct. Lysates were then incubated with 10 μl of α-laforin antibody #113 (1 hour, 4°C) prior to the addition of 35 μl of Protein A Sepharose (1 hour, 4°C). 50 μl of mRIPA buffer was then added to resuspend the Protein A Sepharose, giving a final volume of 80 μL. 10 μl and 5 μl of immunoprecipitated laforin was used for pNPP and malachite green assays (2 hours, 37°C), respectively. 20 μl of the Protein A Sepharose was also boiled (15 min, 95°C) and analyzed by Western blot with 1:1000 α-laforin antibody N84/37.1. 15 μg of WCL was probed with 1:5000 mouse α-β actin (Sigma-Aldrich).
For FLAG peptide elution assays, HEK293 cells were transfected with pcDNA3.1 FLAG human wild-type laforin, inactive C266S laforin, or wild-type VHR. After 24 hours, cells were lysed in 500 μl mRIPA buffer supplemented with protease inhibitors and the lysates incubated with 35 μl M2 α-FLAG agarose (Sigma-Aldrich) (2 hours, 4°C). After washing the agarose beads twice with mRIPA buffer, half of the collected agarose beads was subjected to incubation with 100 μg/mL FLAG peptide (Sigma-Aldrich; 12 hours, 4°C). Laforin eluate was then obtained by centrifugation of the α-FLAG agarose (3 min, 4°C) and collection of the supernatant. Immunoprecipitated laforin bound to α-FLAG agarose or FLAG-peptide eluate was then subjected to pNPP and glucan phosphatase assays.
2.7. Immunoprecipitation and activity assay of endogenous laforin from HepG2 cells
1.4 × 106 cells/mL of HepG2 liver cells were plated in four 150 mm plates and grown to 90% confluence. The lysate of two plates was immunoprecipitated with either α-laforin antibody #113 in conjunction with Protein A Sepharose or only Protein A Sepharose as a control. HepG2 lysates were first collected in 2 mL HIS buffer (300 mM NaCl, 50 mM Tris, 15% glycerol, 0.5% Triton X-100, 3 mM tris(2-carboxyethyl)phosphine (TCEP), pH 8) supplemented with protease inhibitors. Lysates were cleared by centrifugation (10 min, 4K, 4°C), filtered through a 0.22 μm filter, and 60 μl of α-laforin antibody #113 was then added to the supernatant (1 hour, 4°C). 150 μl of Protein A Sepharose was then added (1 hour, 4°C) and the beads washed three times with 1 mL of 1X phosphatase buffer (pH 7) to remove contaminating cellular phosphate.
From the final volume of 80 μL of immunoprecipitated laforin, pNPP and malachite green assays were then performed in quadruplicate with 10 μl and 5 μl immunoprecipitated laforin as described previously, with reactions incubated for 1.5 and 18 hours at 37°C, respectively. 20 μl of the Protein A Sepharose was boiled (15 min, 95°C) and analyzed by Western blot with 1:1000 α-laforin antibody N84/37.1. 80 μg of WCL was probed with 1:5000 mouse α-β actin.
2.8. Immunoprecipitation and activity assay of endogenous laforin from mouse skeletal muscle and human skin tissue
For the mouse tissue experiments, 0.1 g of skeletal muscle collected from a wild-type C57BL/6 mouse was homogenized in 4 mL of HIS buffer supplemented with protease inhibitors. The mouse muscle lysate was then cleared by centrifugation (10 min, 4K, 4°C), filtered through a 0.22 μm filter, and the supernatant collected. This process resulted in 4 mL of 3.8 mg/mL of soluble mouse skeletal muscle lysate. Similarly, soluble human skin tissue lysate from a normal adult (Novus Biologicals, Littleton, CO) was diluted in HIS buffer supplemented with protease inhibitors. Then, 0.02 – 1.0 mg of mouse skeletal muscle lysate and 0.1 – 0.75 mg of human skin tissue lysate was immunoprecipitated with 20 μl of α-laforin antibody #113 (1 hour, 4°C) in conjunction with 60 μl of Protein A Sepharose (1 hour, 4°C) in a final volume of 500 μL. As a negative control, mouse lysate supernatant and human lysate was immunoprecipitated with only Protein A Sepharose. The Protein A Sepharose was then washed three times with 1 mL of 1X phosphatase buffer (pH 7).
The final volume of 60 μL of immunoprecipitated mouse or human laforin was divided into three 20 μL aliquots, with one aliquot utilized in malachite green assays, a second utilized in immunoblotting experiments, and a third aliquot reserved for any necessary subsequent experiments (i.e. repeat experiments). The malachite green assays were performed in quadruplicate with 5 μl of the immunoprecipitated laforin for each replicate, with reactions incubated for 12 hours at 37°C. As a control, muscle tissue from a laforin deficient mouse was prepared as above and malachite green assays were performed following the same protocol. The laforin-deficient samples were utilized to define the amount of contaminating phosphate from the immunoprecipitation protocol. The immunoblotting aliquot was generated by boiling 20 μl of α-laforin-Protein A Sepharose lysate from the immunoprecipitation. The mouse tissue aliquot was then analyzed by Western blot with 1:1000 α-laforin antibody M01 (Abnova, Walnut, CA), which is specific for an epitope in both mouse and human laforin (amino acids 101-199, which share 99% identity). The human skin cell lysate was processed in the same manner and was analyzed by Western blot with 1:1000 α-laforin antibody N84/37.1.
3. Results
In order to generate a bioassay for endogenous laforin activity, we first optimized assay conditions using overexpressed human laforin. The basis of this bioassay is to immunoprecipitate laforin and then assess its glucan phosphatase activity. Therefore, it was crucial to determine if the activity of laforin bound to an α-laforin antibody-agarose complex is inhibited. Our experimental design was to first express FLAG-tagged wild-type laforin, immunoprecipitate laforin using αFLAG agarose, and then perform activity assays of laforin both bound to and eluted from the immunoprecipitation complex (Fig. 1A). We chose to employ two assays to assess laforin function. First, we used the pNPP assay, which utilizes the exogenous phosphatase substrate para-nitrophenylphosphate (pNPP), to assess general phosphatase activity. Dephosphorylation of pNPP generates a product that reacts with base to form an ion that can be detected at 410 nm. In addition to the pNPP assay, we used a malachite green assay utilizing the phosphorylated glucan polymer amylopectin to determine biologically relevant glucan phosphatase activity [9, 27]. This assay detects free phosphate liberated from amylopectin, which forms a complex with ammonium molybdate and malachite green that can be measured at 620 nm. This assay is specific for glucan phosphatase activity because it employs amylopectin as the substrate. As a control for these assays, we used a DSP that dephosphorylates proteinaceous substrates, vaccinia virus VH1-related DSP (VHR) [5, 27, 29]. VHR possesses pNPP activity, but cannot liberate phosphate from amylopectin [9, 27]. We also utilized an enzymatically inactive laforin mutant in which the catalytic cysteine residue is mutated to serine (C266S), abolishing enzymatic activity against pNPP and amylopectin [5]. We found that following immunoprecipitation with FLAG-agarose, FLAG-laforin both bound to and eluted from the FLAG-agarose exhibited pNPP and malachite green activity (Fig. 1B and C). Therefore, the activity of laforin is not impacted by immunoprecipitation. Conversely, C266S laforin did not demonstrate pNPP or malachite green activity and VHR demonstrated only pNPP activity, each as expected.
Figure 1. Laforin exhibits phosphatase activity both bound to and free of the antibody-agarose complex.

A. Schematic of experimental design. Before attempting to test endogenous laforin, we first optimized our experimental conditions using overexpressed laforin. We immunoprecipitated FLAG-laforin and assessed its pNPP and glucan phosphatase activity both bound to and free of the FLAG antibody-agarose complex (“on beads” and “off beads”) in order to determine if antibody binding to laforin inhibits activity. B. pNPP assay of immunoprecipitated FLAG-laforin. FLAG-tagged human wild-type laforin, catalytically inactive C266S laforin, and wild-type VHR were expressed in HEK293 cells and immunoprecipitated with α-FLAG agarose. VHR, a DSP that acts upon protein substrates and not glucan substrates, was included as a control. Immunoprecipitated proteins were either left bound to the antibody-agarose complex (“on beads”) or eluted from the complex using FLAG peptide (“off beads”). The absorbance of reactions at 410 nm was measured. Reactions were performed in quadruplicate. Error bars indicate ± standard error. C. Glucan phosphatase assay of immunoprecipitated FLAG-laforin. The absorbance of reactions at 620 nm was measured. Reactions were performed in quadruplicate. Error bars indicate ± standard error. Proteins are as indicated as in Figure 1B. The experiments above were performed a minimum of three times.
We then analyzed seven monoclonal and polyclonal α-laforin antibodies for the ability to immunoprecipitate overexpressed human laforin. Following immunoprecipitation of FLAG-laforin with each α-laforin antibody, we separated the immunoprecipitated proteins by SDS-PAGE, and immunoblotted using α-FLAG. We found that the best antibodies for the immunoprecipitation of overexpressed laforin were the Protein A purified α-laforin antibodies N84/37.1, #113, and #139 (Fig. 2A).
Figure 2. Laforin antibody selection.
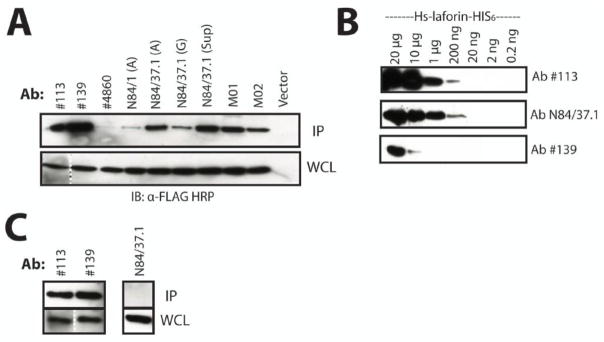
A. Immunoprecipitation of overexpressed laforin using available antibodies. FLAG-laforin was immunoprecipitated using a variety of α-laforin antibodies in conjunction with Protein A Sepharose. We tested protein A-purified rabbit polyclonal α-laforin antibodies #113, #139, #4860, protein A- and G-purified mouse monoclonal α-laforin antibodies N84/1 and N84/37.1 from NeuroMabs, and several commercially available mouse monoclonal α-laforin antibodies (M01 and M02, Abnova). “(A)” or “(G)” indicates affinity purification of an antibody with either protein A or protein G, respectively. “Sup” indicates unpurified tissue culture supernatant. Cells containing empty vector were immunoprecipitated with α-FLAG agarose. The depicted image is a representation. The dotted line indicates where an image portion is a composite of the same image due to the presence of molecular weight marker in between the lanes. B. Detection limit of laforin antibodies. Serial dilutions of recombinant laforin (20.0 μg to 0.2 ng) were probed with the α-laforin antibodies. Only α-laforin antibodies #113, N84/37.1, and #139 are shown, as they displayed the most sensitive detection of recombinant laforin. Primary antibody was used at 1:1000 and secondary antibody at 1:3000. The depicted image is a representation. C. Immunoprecipitation of endogenous laforin from HepG2 cultures. Only the polyclonal α-laforin antibodies #113 and #139 were able to immunoprecipitate endogenous laforin. While the other antibodies did not immunoprecipitate laforin, laforin was detected in their WCL samples. Only the result from α-laforin antibody N84/37.1 is shown to depict this negative result. The depicted image is a representation. The dotted line indicates where an image portion is a composite of the same image due to the presence of molecular weight marker in between lanes. The above experiments were performed a minimum of three times.
Next, we sought to assess the detection limit of each α-laforin antibody using serial dilutions of recombinant human laforin. A range of recombinant laforin protein from 20 μg to 0.02 ng per lane was loaded onto identical gels and then subjected to SDS-PAGE. Following Western transfer, each blot was then incubated with one of the seven α-laforin antibodies. The α-laforin antibodies #113 and N84/37.1 were among the best antibodies for blotting, easily detecting as little as 200 ng of recombinant laforin (Fig. 2B). While equally efficient at immunoprecipitating overexpressed laforin as α-laforin antibody #113 (Fig. 2A), α-laforin antibody #139 was less efficient at detecting recombinant laforin (Fig. 2B).
Laforin antibodies were then tested for the ability to immunoprecipitate endogenous laforin from HepG2 liver cells, as laforin is present in many tissues of the human body, including brain, heart, skeletal muscle, liver, and kidney [7, 10]. HepG2 cell lysates were incubated with each α-laforin antibody followed by incubation with Protein A Sepharose. The amount of laforin immunoprecipitated by each α-laforin antibody was then assessed following SDS-PAGE and immunoblot using either α-laforin antibody #113 or N84/37.1. We found that only the polyclonal α-laforin antibodies (#113 and #139) were able to efficiently immunoprecipitate endogenous laforin (Fig. 2C). Based on our findings, α-laforin antibody N84/37.1 was selected for blotting and α-laforin antibody #113 for immunoprecipitation purposes.
As α-laforin antibody #113 is polyclonal, one concern of our design was that polyclonal antibody binding to laforin could inhibit enzymatic activity. Therefore, we tested if laforin bound by polyclonal α-laforin antibodies displays reduced phosphatase activity. We incubated recombinant laforin with α-laforin antibody #113 followed by Protein A Sepharose and then performed both pNPP and malachite green assays. We compared the activity of Sepharose-bound laforin with laforin lacking antibody and Sepharose, and found that the immunoprecipitation of recombinant laforin with polyclonal α-laforin antibody #113 did not inhibit pNPP or glucan phosphatase activity (Fig. 3A, left and middle panels). It should be noted that the malachite green assay detects phosphate released from amylopectin as well as any free phosphate present in assay reagents. Therefore, we performed malachite green assays with only Protein A Sepharose in the absence of laforin to confirm that the Protein A Sepharose was not a source of phosphate contamination (Fig. 3A, right panel). These results demonstrate that laforin is active both bound and free from the polyclonal antibody-Sepharose complex.
Figure 3. Immunoprecipitation and activity assay of recombinant and overexpressed laforin.
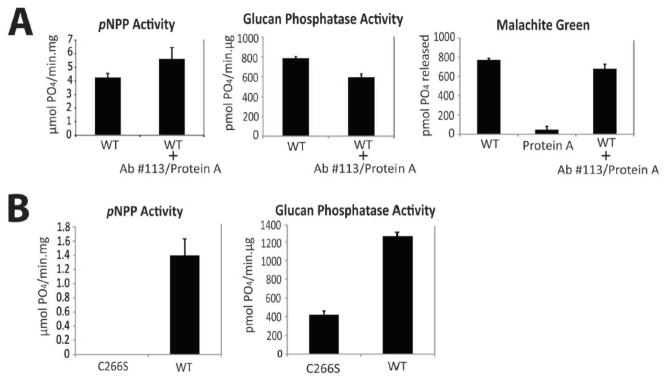
A. Polyclonal α-laforin antibody binding does not inhibit recombinant laforin activity. Recombinant laforin was incubated with α-laforin antibody #113 and Protein A Sepharose. Then, the phosphatase activity of non-incubated and incubated recombinant laforin was assayed using pNPP and malachite green assays. Identical malachite green reactions were carried out containing only Protein A Sepharose to test for the presence of phosphate contamination. Protein A Sepharose (Sigma-Aldrich) was utilized, as free phosphate was present in other commercial preparations (data not shown). Reactions containing inactive C266S recombinant laforin were utilized as a negative control. Reactions were performed in quadruplicate. Error bars indicate ± standard error. B. Immunoprecipitated overexpressed laforin exhibits pNPP and glucan phosphatase activity. Human wild-type and inactive C266S FLAG-laforin was expressed in HEK293 cells and then immunoprecipitated using α-laforin antibody #113. In order to determine protein amounts, immunoprecipitated laforin was analyzed by SDS-PAGE and Western blotted alongside known amounts of recombinant human laforin. pNPP and glucan phosphatase activity of immunoprecipitated laforin was then determined using the amount of immunoprecipitated laforin calculated by comparison to the known recombinant protein amounts. Reactions were performed in quadruplicate. Error bars indicate ± standard error. The experiments above were repeated a minimum of three times.
We then attempted to immunoprecipitate and assess the activity of laforin overexpressed in cell culture and immunoprecipitated with α-laforin antibody #113. We first expressed wild-type and inactive C266S FLAG-laforin in HEK293 cells, immunoprecipitated laforin with α-laforin antibody #113 in conjunction with Protein A Sepharose, and then assessed the activity of the immunoprecipitated laforin. pNPP and malachite green assays were performed with 10 μl and 5 μl of immunoprecipitated laforin, respectively. We found that only wild-type FLAG-laforin exhibited pNPP activity (Fig. 3B, left panel) and glucan phosphatase activity (Fig. 3B, right panel), with very low background from C266S FLAG-laforin (Fig. 3B, right panel). These data indicate that our experimental design can function when using cell lysate and can differentiate between active and inactive laforin.
Next, we determined if we could immunoprecipitate endogenous laforin from HepG2 liver cells and detect its phosphatase activity. We grew HepG2 cells to 90% confluence and lysed them in the same buffer used to purify recombinant laforin, as this buffer provided us with higher yields of immunoprecipitated endogenous laforin than other buffers (data not shown). We found that we could immunoprecipitate at least ~20 ng of laforin from HepG2 cells using α-laforin antibody #113 in conjunction with Protein A Sepharose (Fig. 4A). After immunoprecipitating endogenous laforin, we performed pNPP and malachite green assays with the Sepharose-bound laforin. We found that the Protein A Sepharose control exhibited a similar pNPP absorbance as Protein A Sepharose with bound laforin (Fig. 4B). We then pre-cleared the HepG2 cell lysates with Protein A Sepharose in addition to immunoprecipitating with Protein A Sepharose incubated first with BSA, but control pNPP activity was not diminished (data not shown). These results indicate that the pNPP assay is not suitable for use as a bioassay, likely due to the interaction of Protein A Sepharose with other cellular phosphatases. In contrast to these results, the Protein A Sepharose control exhibited very little background when the malachite green assay was utilized, with only immunoprecipitated laforin exhibiting robust glucan phosphatase activity (Fig. 4C). Thus, the malachite green assay with amylopectin substrate is specific for the quantification of endogenous laforin activity, and when used in conjunction with the α-laforin antibody #113 is suitable to assess the functionality of endogenous laforin.
Figure 4. Immunoprecipitated endogenous laforin exhibits specific glucan phosphatase activity.

Laforin was immunoprecipitated from HepG2 cells using α-laforin antibody #113 and Protein A Sepharose or Protein A Sepharose alone as a control. A. Western analysis of immunoprecipitated endogenous laforin from HepG2 cells. The laforin immunoprecipitate sample was divided into three equal aliquots and one aliquot was separated via SDS-PAGE, Western transferred, and immunoblotted with α-laforin antibody N84/37.1. Known amounts of recombinant laforin were ran alongside the immunoprecipitated laforin to allow for quantification of the immunoprecipitated laforin. The depicted image is a representation. B. pNPP assay of immunoprecipitated endogenous laforin from HepG2 cells. Reactions were performed in quadruplicate. Error bars indicate ± standard error. C. Glucan phosphatase assay of immunoprecipitated endogenous laforin from HepG2 cells. Reactions were performed in quadruplicate. Error bars indicate ± standard error. The experiments above were repeated a minimum of three times.
Finally, we tested our bioassay for the detection of endogenous laforin activity in both mouse and human tissue lysate. We generated and obtained lysates from C57BL/6 mouse skeletal muscle and human skin tissue, respectively. Next, we immunoprecipitated laforin from different amounts of lysate based on total protein, ranging from 0.02 mg 1.0 mg for the mouse skeletal muscle lysate and from 0.1 mg – 0.75 mg of human skin tissue lysate. We divided the immunoprecipitated laforin into three 20 μl aliquots to use the material for: Western analysis, malachite green assays, and a third aliquot to repeat both assays. We detected ~4 ng of laforin in the 1.0 mg lane of mouse skeletal muscle lysate (Fig 5A). Since we divided the immunoprecipitation from the 1.0 mg sample into three aliquots, we immunoprecipitated ~12 ng total from 1.0 mg of the lysate. Similarly, we detected ~10 ng of laforin from the 0.75 mg sample of human skin lysate (Fig. 5B). Thus, we immunoprecipitated ~30 ng in total from the 0.75 mg sample. We then tested the immunoprecipitated laforin for glucan phosphatase activity using the assay established above. Laforin immunoprecipitated from at least 0.5 mg of total protein from mouse skeletal muscle lysate and from at least 0.1 mg of total protein from human skin lysate demonstrated robust glucan phosphatase activity above the Protein A Sepharose control (Fig. 5C and 5D). Therefore, we successfully immunoprecipitated endogenous laforin from both mouse and human tissue and measured its glucan phosphatase activity.
Figure 5. Endogenous laforin activity from mouse and human tissue lysate.

Laforin was immunoprecipitated from increasing amounts of mouse skeletal muscle lysate and human skin lysate using α-laforin antibody #113 in conjunction with Protein A Sepharose and tested for glucan phosphatase activity. A. Western analysis of immunoprecipitated laforin from mouse skeletal muscle. Increasing amounts of mouse skeletal muscle lysate from 0.02 mg – 1.0 mg of total protein was used to immunoprecipitate laforin. The immunoprecipitate sample was divided into three equal aliquots and one aliquot was separated via SDS-PAGE, Western transferred, and immunoblotted with α-laforin antibody M01. Known amounts of recombinant laforin were ran alongside the immunoprecipitated laforin to allow for quantification of the immunoprecipitated laforin. As a negative control, antibody was excluded from a 1.0 mg sample [lane: (−) Ab#113]. The depicted image is a representation. B. Western analysis of immunoprecipitated laforin from human skin lysate. Increasing amounts of human skin lysate from 0.1 mg – 0.75 mg of total protein was used to immunoprecipitate laforin. The immunoprecipitate sample was divided into three equal aliquots and one aliquot was separated via SDS-PAGE, Western transferred, and immunoblotted with α-laforin antibody N84/37.1. Known amounts of recombinant laforin were ran alongside the immunoprecipitated laforin to allow for quantification of the immunoprecipitated laforin. As a negative control, antibody was excluded from a 0.75 mg sample [lane: (−) Ab#113]. The depicted image is a representation. C. Glucan phosphatase assay of laforin immunoprecipitated from mouse skeletal muscle. Increasing amounts of mouse skeletal muscle lysate from 0.02 mg – 1.0 mg of total protein was used to immunoprecipitate laforin. The immunoprecipitated sample was divided into three equal aliquots and one aliquot was used to perform glucan phosphatase assays in quadruplicate. Error bars indicate ± standard error. D. Glucan phosphatase assay of laforin immunoprecipitated from human skin. Increasing amounts of human skin lysate from 0.1 mg – 0.75 mg of total protein was used to immunoprecipitate laforin. The immunoprecipitated sample was divided into three equal aliquots and one aliquot was used to perform glucan phosphatase assays in quadruplicate. Error bars indicate ± standard error. The experiments above were repeated a minimum of three times.
4. Discussion
We sought to characterize α-laforin antibodies capable of detecting and immunoprecipitating endogenous laforin in order to generate a bioassay for laforin activity. First, we explored the conditions for the immunoprecipitation and assay of overexpressed laforin while also determining if antibody binding to laforin could inhibit laforin activity. We began by testing the activity of overexpressed FLAG-laforin immunoprecipitated using α-FLAG agarose, and found that overexpressed laforin did demonstrate pNPP and glucan phosphatase activity. This activity was not impacted by whether laforin was bound to or eluted from the FLAG-agarose.
We then identified two α-laforin antibodies, N84/37.1 and #113, that were capable of detecting and immunoprecipitating endogenous laforin, respectively. Following immunoprecipitation of overexpressed laforin with α-laforin antibody #113, we found that binding of this polyclonal antibody to laforin did not negatively impact activity. These results reflected those obtained with recombinant laforin, where α-laforin antibody #113 and Protein A Sepharose binding did not impact either pNPP or malachite green activity. Following immunoprecipitation of endogenous laforin using α-laforin antibody #113, we found that the pNPP assay gave significant background, likely due to the interaction of another cellular phosphatase with the Protein A Sepharose. However, using the malachite green assay with amylopectin substrate, we showed that glucan phosphatase activity was specific to successful immunoprecipitation of endogenous laforin. Finally, we demonstrated that this methodology can be successfully employed using mouse or human tissue lysate.
It is important to note that Western analysis alone is likely not sufficient to accurately assess if a given treatment is yielding functional laforin. Western analysis is unable to discriminate between wild type and mutant laforin, unless the antibody epitope is itself mutated. Therefore, one needs an enzymatic assay to quantify the activity of functional laforin rather than just total protein. The assay that we have developed faithfully reports the activity of functional endogenous laforin.
While we utilized finite supplies of a polyclonal α-laforin antibody in our study, we found that rabbit polyclonal antibodies created in different animals were as successful in the immunoprecipitation of endogenous laforin as α-laforin antibody #113. Thus, after a brief assessment for quality, stocks of polyclonal antibodies against laforin could be continuously maintained. The bioassay we have developed in this work will no doubt prove useful in the assessment of future therapeutic interventions for LD.
Laforin is widely expressed in tissues throughout the human body [7, 10]. Prior to PCR analysis, LD was diagnosed using light microscopy of a skin biopsy in conjunction with hematoxylin eosin or periodic acid Schiff (PAS) staining [1, 6]. A similar skin biopsy of LD patients following therapeutic intervention in conjunction with the immunoprecipitation method we have identified would allow for detection of endogenous laforin activity. Alternatively, this bioassay could be utilized on cerebrospinal fluid (CSF) collected from patients.
Highlights.
This study develops a bioassay for laforin glucan phosphatase activity
Immunoprecipitated laforin exhibits unique glucan phosphatase activity
The bioassay faithfully reports laforin activity from mouse and human tissue
This bioassay should prove useful with future efforts to treat Lafora disease patients
Acknowledgments
This work was supported by National Institutes of Health Grants P20RR020171 and R01NS070899 and University of Kentucky College of Medicine startup funds to M.S.G. This publication was also supported by grant number TL1 RR033172 from the National Center for Research Resources (NCRR), funded by the Office of the Director, National Institutes of Health (NIH) and supported by the NIH Roadmap for Medical Research. The content is solely the responsibility of the authors and does not necessarily represent the official views of NCRR and NIH. We wish to thank Drs. Carol Beach and Martin Chow in the Molecular Basis of Human Disease COBRE Proteomics core and Protein Analytical Core, respectively, for technical assistance as well as members of the Gentry lab for fruitful discussions.
The monoclonal antibody N84/37.1 was developed by and/or obtained from the UC Davis/NIH NeuroMab Facility, supported by NIH grant U24NS050606 and maintained by the Department of Neurobiology, Physiology and Behavior, College of Biological Sciences, University of California, Davis, CA 95616. This paper is subject to the NIH Public Access Policy. This work was carried out in accordance with the Uniform Requirements for Manuscripts Submitted to Biomedical Journals and the Code of Ethics of the World Medical Association (Declaration of Helsinki) for Experiments Involving Humans. All work involving human tissue has been cleared by the University of Kentucky College of Medicine Institution Review Board.
Abbreviations
- BSA
bovine serum albumin
- CBM
carbohydrate binding module
- CSF
cerebrospinal fluid
- DSP
dual specificity phosphatase
- DTT
dithiothreitol
- HRP
horseradish peroxidase
- HEK
human embryonic kidney
- LB
Lafora body
- LD
Lafora disease
- mRIPA
modified RIPA buffer
- pcDNA3.1 FLAG
pcDNA3.1 with N-terminal FLAG tag
- PAS
periodic acid Schiff
- pNPP
para-nitrophenylphosphate
- TBS
Tris-buffered saline
Footnotes
Competing Interests: The authors declare no competing interests.
Author contribution: The primary author Amanda Sherwood wrote the manuscript, generated the figures, performed the experiments, and analyzed the experimental data. The secondary author Mary Beth Johnson performed initial antibody characterizations that facilitated in the development of this work. The third author Dr. Antonio Delgado-Escueta aided in the conception of the project. The senior author Dr. Matthew Gentry also conceived the project, aiding in experimental planning, analysis of the experimental data as well as performing experiments, and preparation/revision of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Minassian BA. Lafora’s disease: Towards a clinical, pathologic, and molecular synthesis. Pediatr Neurol. 2001;25:21–9. doi: 10.1016/s0887-8994(00)00276-9. [DOI] [PubMed] [Google Scholar]
- 2.Gentry MS, Dixon JE, Worby CA. Lafora disease: Insights into neurodegeneration from plant metabolism. Trends Biochem Sci. 2009;34:628–39. doi: 10.1016/j.tibs.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yokoi S, Austin J, Witmer F, Sakai M. Studies in myoclonus epilepsy (lafora body form). I. Isolation and preliminary characterization of lafora bodies in two cases. Arch Neurol. 1968;19:15–33. doi: 10.1001/archneur.1968.00480010033002. [DOI] [PubMed] [Google Scholar]
- 4.Tagliabracci VS, Girard JM, Segvich D, Meyer C, Turnbull J, Zhao X, et al. Abnormal metabolism of glycogen phosphate as a cause for lafora disease. J Biol Chem. 2008 doi: 10.1074/jbc.M807428200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gentry MS, Dowen RH, 3rd, Worby CA, Mattoo S, Ecker JR, Dixon JE. The phosphatase laforin crosses evolutionary boundaries and links carbohydrate metabolism to neuronal disease. J Cell Biol. 2007;178:477–88. doi: 10.1083/jcb.200704094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delgado-Escueta AV, Ganesh S, Yamakawa K. Advances in the genetics of progressive myoclonus epilepsy. Am J Med Genet. 2001;106:129–38. doi: 10.1002/ajmg.1575. [DOI] [PubMed] [Google Scholar]
- 7.Minassian BA, Lee JR, Herbrick JA, Huizenga J, Soder S, Mungall AJ, et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet. 1998;20:171–4. doi: 10.1038/2470. [DOI] [PubMed] [Google Scholar]
- 8.Tagliabracci VS, Turnbull J, Wang W, Girard JM, Zhao X, Skurat AV, et al. Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proc Natl Acad Sci U S A. 2007;104:19262–6. doi: 10.1073/pnas.0707952104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Worby CA, Gentry MS, Dixon JE. Laforin: A dual specificity phosphatase that dephosphorylates complex carbohydrates. J Biol Chem. 2006;281:30412–8. doi: 10.1074/jbc.M606117200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serratosa JM, Gomez-Garre P, Gallardo ME, Anta B, de Bernabe DB, Lindhout D, et al. A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the lafora type (epm2) Hum Mol Genet. 1999;8:345–52. doi: 10.1093/hmg/8.2.345. [DOI] [PubMed] [Google Scholar]
- 11.Gentry MS, Worby CA, Dixon JE. Insights into lafora disease: Malin is an e3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc Natl Acad Sci U S A. 2005;102:8501–6. doi: 10.1073/pnas.0503285102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan EM, Young EJ, Ianzano L, Munteanu I, Zhao X, Christopoulos CC, et al. Mutations in nhlrc1 cause progressive myoclonus epilepsy. Nat Genet. 2003;35:125–7. doi: 10.1038/ng1238. [DOI] [PubMed] [Google Scholar]
- 13.Wang J, Stuckey JA, Wishart MJ, Dixon JE. A unique carbohydrate binding domain targets the lafora disease phosphatase to glycogen. J Biol Chem. 2002;277:2377–80. doi: 10.1074/jbc.C100686200. [DOI] [PubMed] [Google Scholar]
- 14.Sherwood AR, Dukhande VV, Gentry MS. Laforin: Function and action of a glucan phosphatase. In: Choi S, editor. Encyclopedia of Signaling Molecules. XLVIII. Springer; New York: 2012. pp. 1003–10. [Google Scholar]
- 15.Gentry MS, Roma-Mateo C, Sanz P. Laforin, a protein with many faces: Glucan phosphatase, adapter protein, et alii. FEBS J. 2013;280:525–37. doi: 10.1111/j.1742-4658.2012.08549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gentry MS, Pace RM. Conservation of the glucan phosphatase laforin is linked to rates of molecular evolution and the glycogen metabolism of the organism. BMC Evol Biol. 2009;9:138. doi: 10.1186/1471-2148-9-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh S, Ganesh S. Lafora progressive myoclonus epilepsy: A meta-analysis of reported mutations in the first decade following the discovery of the epm2a and nhlrc1 genes. Hum Mutat. 2009;30:715–23. doi: 10.1002/humu.20954. [DOI] [PubMed] [Google Scholar]
- 18.Ganesh S, Delgado-Escueta AV, Sakamoto T, Avila MR, Machado-Salas J, Hoshii Y, et al. Targeted disruption of the epm2a gene causes formation of lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum Mol Genet. 2002;11:1251–62. doi: 10.1093/hmg/11.11.1251. [DOI] [PubMed] [Google Scholar]
- 19.Tagliabracci VS, Heiss C, Karthik C, Contreras CJ, Glushka J, Ishihara M, et al. Phosphate incorporation during glycogen synthesis and lafora disease. Cell Metab. 2011;13:274–82. doi: 10.1016/j.cmet.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nitschke F, Wang P, Schmieder P, Girard JM, Awrey DE, Wang T, et al. Hyperphosphorylation of glucosyl c6 carbons and altered structure of glycogen in the neurodegenerative epilepsy lafora disease. Cell Metab. 2013;17:756–67. doi: 10.1016/j.cmet.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 21.Monaghan TS, Delanty N. Lafora disease: Epidemiology, pathophysiology and management. CNS Drugs. 2010;24:549–61. doi: 10.2165/11319250-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 22.Shi N, Pardridge WM. Noninvasive gene targeting to the brain. Proc Natl Acad Sci U S A. 2000;97:7567–72. doi: 10.1073/pnas.130187497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Linde L, Kerem B. Introducing sense into nonsense in treatments of human genetic diseases. Trends Genet. 2008;24:552–63. doi: 10.1016/j.tig.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 24.Turnbull J, DePaoli-Roach AA, Zhao X, Cortez MA, Pencea N, Tiberia E, et al. Ptg depletion removes lafora bodies and rescues the fatal epilepsy of lafora disease. PLoS Genet. 2011;7:e1002037. doi: 10.1371/journal.pgen.1002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valles-Ortega J, Duran J, Garcia-Rocha M, Bosch C, Saez I, Pujadas L, et al. Neurodegeneration and functional impairments associated with glycogen synthase accumulation in a mouse model of lafora disease. EMBO Mol Med. 2011;3:667–81. doi: 10.1002/emmm.201100174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berkovic SF, So NK, Andermann F. Progressive myoclonus epilepsies: Clinical and neurophysiological diagnosis. J Clin Neurophysiol. 1991;8:261–74. [PubMed] [Google Scholar]
- 27.Sherwood AR, Paasch BC, Worby CA, Gentry MS. A malachite green-based assay to assess glucan phosphatase activity. Anal Biochem. 2013;435:54–6. doi: 10.1016/j.ab.2012.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vander Kooi CW, Taylor AO, Pace RM, Meekins DA, Guo HF, Kim Y, et al. Structural basis for the glucan phosphatase activity of starch excess4. Proc Natl Acad Sci U S A. 2010;107:15379–84. doi: 10.1073/pnas.1009386107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Denu JM, Zhou G, Wu L, Zhao R, Yuvaniyama J, Saper MA, et al. The purification and characterization of a human dual-specific protein tyrosine phosphatase. J Biol Chem. 1995;270:3796–803. doi: 10.1074/jbc.270.8.3796. [DOI] [PubMed] [Google Scholar]