

Abstract
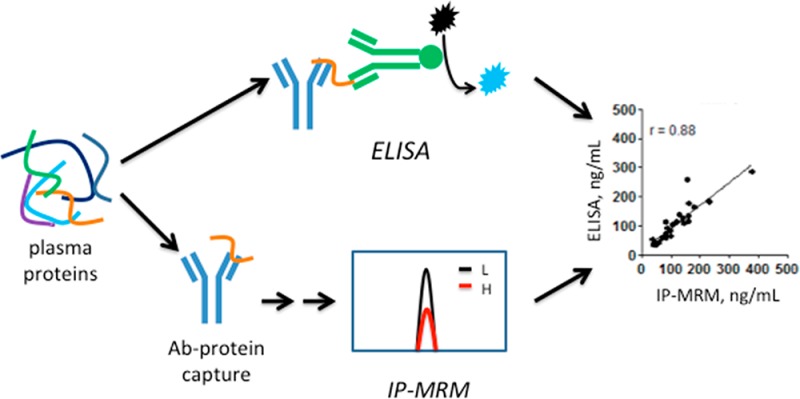
Quantitative analysis of protein biomarkers in plasma is typically done by ELISA, but this method is limited by the availability of high-quality antibodies. An alternative approach is protein immunoprecipitation combined with multiple reaction monitoring mass spectrometry (IP-MRM). We compared IP-MRM to ELISA for the analysis of six colon cancer biomarker candidates (metalloproteinase inhibitor 1 (TIMP1), cartilage oligomeric matrix protein (COMP), thrombospondin-2 (THBS2), endoglin (ENG), mesothelin (MSLN) and matrix metalloproteinase-9 (MMP9)) in plasma from colon cancer patients and noncancer controls. Proteins were analyzed by multiplex immunoprecipitation from plasma with the ELISA capture antibodies, further purified by SDS-PAGE, digested and analyzed by stable isotope dilution MRM. IP-MRM provided linear responses (r = 0.978–0.995) between 10 and 640 ng/mL for the target proteins spiked into a “mock plasma” matrix consisting of 60 mg/mL bovine serum albumin. Measurement variation (coefficient of variation at the limit of detection) for IP-MRM assays ranged from 2.3 to 19%, which was similar to variation for ELISAs of the same samples. IP-MRM and ELISA measurements for all target proteins except ENG were highly correlated (r = 0.67–0.97). IP-MRM with high-quality capture antibodies thus provides an effective alternative method to ELISA for protein quantitation in biological fluids.
Keywords: immunoprecipitation, MRM, plasma biomarkers, biomarker verification, colon cancer
Quantitative analysis of protein biomarkers is one of the most challenging tasks in biomedical research.1 Enzyme-linked immunosorbent assay (ELISA) is widely used for protein quantitation in human serum or plasma owing to its high sensitivity and throughput. However, the availability of high-quality ELISAs for biomarker candidates is limited, and the performance characteristics of many commercially marketed ELISAs are poorly documented or unknown.2 Development of ELISAs is also expensive and time-consuming. The limitations of ELISA, combined with the large numbers of biomarker candidates emerging from genomic and proteomic discovery studies, have created a need for alternative means of targeted protein quantitation.1
Multiple reaction monitoring (MRM) mass spectrometry has emerged as a versatile platform for systematic development of targeted protein assays and which can serve as an alternative to ELISA in biomarker research.3,4 MRM assays target sequence-specific tandem MS fragmentations of proteotypic peptides, thereby providing highly selective measurements for distinct proteins. Without fractionation or enrichment strategies, MRM assays allow for the quantitation of protein in the low μg/mL or high ng/mL concentration range,5−7 whereas immunoaffinity depletion of abundant blood proteins and minimal protein fractionation can enable quantitation in the low ng/mL range.8−10 A recently described method called PRISM combined targeted peptide-level preselection with MRM to achieve high sensitivity measurements without antibody capture.2
Immunoaffinity capture of intact proteins or their peptides after digestion can dramatically enhance the sensitivity of MRM assays. Anderson and colleagues introduced an immuno-MRM assay approach (Stable Isotope Standards with Capture by Antipeptide Antibodies; SISCAPA), in which proteotypic tryptic peptides and their corresponding spiked stable isotope-labeled internal standards are captured by antibodies raised against the peptides.11 This approach has been extensively developed by several laboratories10,12−14 and used to systematically develop and implement targeted assays for candidate biomarkers15,16 and implement, as a prototype, clinical assay for thyroglobulin.17
An alternate approach was described by Berna et al., who used immunoaffinity enrichment of intact proteins followed by digestion and MRM to quantify protein biomarkers of cardiovascular disease.18,19 Nicol et al.20 demonstrated that multiple antibodies immobilized on hydrazide beads could simultaneously enrich several candidate lung cancer biomarkers in serum for MRM measurements in the low ng/mL range. Targeted quantitation can be extended to sequence variant proteins using the same approach.21 Protein-capture-based immuno-MRM (IP-MRM) assays have been less thoroughly explored than peptide-capture-based assays.
We asked how the analytical performance of IP-MRM would compare to that for ELISA. To address this question, we employed six commercially available ELISAs to measure candidate biomarker proteins for colon cancer in plasma of cancer patients and noncancer controls. We obtained the capture antibodies used in the ELISAs from the manufacturer and then configured immuno-MRM assays for the six proteins. Our analyses provide the first reported comparison of an IP-MRM assay to ELISA with the same samples and reagents.
Experimental Procedures
Chemicals and Reagents
Trypsin (sequencing grade) was purchased from Promega (Madison, WI). Isotope-labeled peptides were obtained from New England Peptide (Gardner, MA) with either U–13C6, U–15N2-lysine (+8 Da) or U–13C6, U–15N4-arginine (+10 Da) at the peptide C terminus. Chemical purity was ranged from 95 to 99% and isotopic purity was greater than 99%. Peptide concentrations were benchmarked by amino acid analysis performed by the supplier. TIMP1, THBS2, COMP, MSLN, ENG, MMP9 antibodies and ELISA kits were purchased from R&D Systems (Minneapolis, MN). ELISAs were performed according to the manufacturer’s recommendations. TIMP1, ENG and MMP9 recombinant proteins were purchase from Sino Biological (Beijing, China). THBS2, COMP and MSLN recombinant proteins were from R&D Systems. Identities of all recombinant proteins were confirmed by SDS-PAGE (Figure S1A [Supporting Information (SI)]) and tandem mass spectrometry (Figure S2–S7 [SI]) analysis of tryptic digests. All other chemical reagents were purchased from commercial sources and were used without further purification.
Collection and Storage of Plasma
Human plasma samples were collected during surgery for either colon carcinoma or for inguinal hernia repair in accordance with the Ayers Institute protocol at Vanderbilt University Medical Center (IRB#110877). Samples from colon cancer patients undergoing surgery at Vanderbilt University between August 2011 and June 2012 with a successful collection were used in the study. Control samples were chosen from a larger group of inguinal hernia repair patients who underwent surgery during the same time period. Peripheral whole blood samples were collected preoperatively in EDTA lavender top vacutainer tubes (BD Vacutainer, catalog number 366643) and gently mixed by inverting the tube 8–10 times. Plasma was separated by centrifugation at 1500g for 10 min at 4 °C. Aliquots (0.2 mL) were taken and stored at −80 °C until needed.
Antibody Immobilization
Antibodies were immobilized on aldehyde beads (Thermo Scientific, catalog number 26148) according to the manufacturer’s protocol with minor modifications. Briefly, antibodies were dissolved in PBS buffer (0.01 M sodium phosphate, 0.15 sodium chloride, pH 7.2) and incubated with coupling resin and 75 μM sodium cyanoborohydride at room temperature on a rotator. An aliquot was collected before and after binding for determination of binding efficiency by protein bicinchoninic acid assay. After immobilization, the active aldehyde sites on the resin were blocked with 1 M Tris buffer and 75 μM sodium cyanoborohydride followed by several washes with PBS to remove any nonbound antibody. After determining the binding efficiency, the immobilized resins for all antibodies were either combined or directly aliquoted such that ∼1 μg of each immobilized antibody was used for each immunoprecipitation.
Protein Capture and Sample Preparation for MRM
Plasma (50 μL) was diluted 5-fold with RIPA buffer containing a protease inhibitor cocktail (Roche, catalog number 11873580001). Diluted plasma was incubated with the immobilized antibody resin overnight at 4 °C with gentle shaking. The resin was washed three times with 0.5 mL RIPA buffer, and the bound proteins were eluted into 15 μL of 2X NuPAGE lithium dodecyl sulfate loading buffer (Invitrogen, Carsbad, CA) containing 50 mM DTT by incubation at 95 °C for 5 min. The eluted proteins then were loaded and separated by SDS-PAGE on a NuPAGE Novex 10% Bis Tris mini gel (Invitrogen NP0301BOX). A protein molecular weight standard (Precision Plus Protein Kaleidoscope Standard, Bio-Rad, Hercules, CA) was loaded in one lane on each gel and used for estimation of relative mass determination of captured proteins.
After electrophoresis at a constant 180 V for 20 min, gels were washed three times with deionized water, stained with SimplyBlue SafeStain (Invitrogen) for 1 h, and destained with deionized water at 4 °C overnight. From each gel lane, fractions were taken to enable targeted analysis of the target proteins. For TIMP1, a molecular weight fraction of 25–37 kDa was collected. For analysis of the remaining five proteins, a molecular weight range of 75–200 kDa THBS2, COMP and MMP9 and another of 37–75 kDa for ENG and MSLN were excised from the gel, cut into 1 mm cubes, and placed in 100 μL of 100 mM ammonium bicarbonate. Samples were reduced with 5 μL of 100 mM DTT for 15 min at 50 °C and alkylated with 15 μL of 100 mM iodoacetamide for 30 min at room temperature in the dark. Excess dye was removed from gel slices with two exchanges of 100 μL 50% acetonitrile/50 mM ammonium bicarbonate and subsequently dehydrated with 100% acetonitrile. The solvent was removed from the gel pieces under vacuum. The residue was resuspended in 0.01 μg/μL MS grade trypsin (Promega, Madison, WI) in 25 mM ammonium bicarbonate containing a standard mixture of heavy isotope-labeled peptides for the analytes (20 fmol/peptide) and incubated at 37 °C overnight. Peptides were extracted with 60% acetonitrile containing 1% formic acid, and then each fraction was evaporated under vacuum. Samples were redissolved for MRM analysis in 30 μL of 5% acetonitrile containing 0.1% formic acid.
MRM Analysis
MRM analyses were performed on a TSQ Vantage triple quadrupole mass spectrometer (ThermoFisher Scientific, San Jose, CA) equipped with an Eksigent Ultra nanoLC solvent delivery system, microautosampler and a nanospray source. Sample peptides (3 μL injection volume) were loaded onto a 75 μm × 11 cm PicoFrit column (PF360-75-10-N-5, New Objective, Inc., Woburn, MA) packed with 3 μm, ReproSil-Pur C18-AQ (Dr. Maisch, Ammerbuch-Entringen, Germany). Liquid chromatography was carried out at a flow rate of 300 nL/min with a mobile phase consisting of 0.1% formic acid in either HPLC grade water (solvent A) or 90% acetonitrile (solvent B). An elution gradient was programmed from 97% A for 1 min, then increased to 7% B over 4 min, 25% B over 15 min, 40% B over 7 min, 90% B by 40 min, and then held at that composition for 10 min before returning to 97% solvent A over 1 min. Mobile phase composition then was held at this initial condition for 29 min prior to the next analysis. Instrument parameters included Q2 gas 1.5 mTorr, scan width 0.005 Th, scan time 10 ms, and both Q1 and Q3 resolution fwhm 0.7. Proteotypic peptides and MRM transitions were selected with the Skyline software utility22 and were further optimized by analyses of tryptic digests of recombinant proteins on the TSQ Vantage triple quadrupole mass spectrometer.
Selection of Peptides and MRM Transitions
Target peptides were chosen according to previously published criteria.20,23,24 We digested the recombinant proteins and determined the resultant peptides by monitoring all possible tryptic peptides between 8 and 22 amino acids in LC–MS experiments using the product scan mode. Where recombinant protein was not available, selection of peptides and transitions were selected on the basis of observations from previous discovery experiments, observed peptides in open proteomics databases, or computational predicted peptides through algorithms such as enhanced signature peptide (ESP) predictor.25 Peptides containing methionine residues as well as those containing post-translational modification sites such as glycosylation listed in the UniProt database (to minimize interference on digestion) were excluded. All peptides were run through BLASTP (Uniprot) to ensure their uniqueness to the protein of interest. Peptides and transitions selected for each protein are shown in Table S1 (SI).
Data Analysis
MRM data acquired on the TSQ Vantage triple quadrupole mass spectrometer were analyzed with Skyline software.22 Peak integrations were manually reviewed and transitions from peptides measured were confirmed by the same retention times and transition patterns of the light peptides and synthetic heavy, stable isotope-labeled peptides. Peak areas for the four most intense MRM transitions were integrated and summed to generate a peptide peak area, which was divided by the peptide peak area for the internal standard heavy peptide. For each protein, the unique peptide that generated the highest summed peak area signal was used to quantify the protein of interest.
Results
Biomarker Candidate Proteins and Overview of Approach
Six biomarker candidate proteins (TIMP1, ENG, MSLN, THBS2, MMP9 and COMP) were selected on the basis of literature reports, which suggest that they are overexpressed in colon cancers or because differential expression of these proteins in blood was associated with cancer.26−32 We also chose these candidates because well-characterized, commercially available ELISAs were available for each. Moreover, the same capture antibodies used in the ELISAs were available to us for evaluation in IP-MRM assays. To analyze the biomarker candidates in plasma, we performed either single-protein or multiplexed immunoprecipitations and then resolved the target proteins into molecular weight fractions by SDS-PAGE (Figure S1B [SI]). The gel bands were digested and the digests were spiked with isotope-labeled standards and analyzed by MRM. The MRM signals from each peptide were shown to be consistent with their corresponding isotope labeled peptides in both retention time and transient patterns (Figure S8 [SI]).
Initial Characterization of IP-MRM and ELISA for Analysis of TIMP1
Recombinant TIMP1 protein was spiked into a “mock plasma” matrix consisting 60 mg/mL BSA in PBS to mimic the human plasma environment and provide a defined analyte concentration for estimation of recovery. A 2-fold dilution series ranging from 640 to 10 ng/mL was constructed and a plot showing the theoretical TIMP1 concentration versus the calculated protein concentration is shown in Figure 1A (the theoretical TIMP1 concentration versus the ratio L/H is also shown in Figure S9A (SI). The calculated protein concentration was calculated from the measured peak area ratio of the light to heavy peptides. The slope (0.329) indicated TIMP1 recovery after all process steps including immunoaffinity capture, trypsin digestion and peptide extraction. The limit of detection (LOD) was 2.5 ng/mL (Table 1).
Figure 1.

Response curves for IP-MRM analyses of recombinant TIMP1, COMP, MMP9, THBS2, MSLN and ENG proteins. Proteins were spiked at 10–640 ng/mL in a background matrix of 60 mg/mL BSA in DPBS and analyzed by IP-MRM as described in Experimental Procedures. Values plotted are mean ± standard deviation (n = 3).
Table 1. Measurement summaries for biomarker candidates by IP-MRM assay and ELISA.
| protein | TIMP1 | COMP | MMP9 | THBS2 | MSLN | ENG | |
|---|---|---|---|---|---|---|---|
| IP-MRM | peptide | GFQALGDAADIR | ELQETNAALQDVR | AVIDDAFAR | ACVGDVQER | TDAVLPLTVAEVQK | VLPGHSAGPR |
| linearity r | 0.979 | 0.995 | 0.979 | 0.984 | 0.991 | 0.978 | |
| recovery (%) | 33 | 50 | 41 | 13.5 | 55 | 16 | |
| LLODa (ng/mL) | 2.5 | 5.1 | 3.0 | 2.0 | 8.9 | 5.6 | |
| CV at 10 ng/mL (%) | 2.3 | 17 | 10 | 7 | 12 | 19 | |
| normalb (n = 12) | 141 ± 41 | 153 ± 62 | 111 ± 46 | 10.5 ± 7.9 | 8.3 ± 3.5 | 10.1 ± 3.9 | |
| cancer (n = 12) | 212 ± 59c | 221 ± 117 | 141 ± 96c | 21 ± 10 | 14.1 ± 8.5 | 8.6 ± 1.5 | |
| P valued | 0.0028 | 0.091 | 0.34 | 0.010 | 0.041 | 0.26 | |
| ELISA | LLOQe (ng/mL) | 0.313 | 0.156 | 0.313 | 0.313 | 0.156 | 0.156 |
| CV at LLOQ (%) | 13.4 | 0.0 | 2.8 | 2.9 | 8.8 | 2.3 | |
| normal (n = 12) | 97 ± 16 | 178 ± 59 | 117 ± 61 | 26 ± 6 | 22 ± 8 | 3.9 ± 0.8 | |
| cancer (n = 12) | 120 ± 35c | 208 ± 124 | 107 ± 73c | 33 ± 9 | 22 ± 10 | 4.5 ± 0.9 | |
| P valued | 0.060 | 0.44 | 0.73 | 0.035 | 1.0 | 0.10 | |
LLOD, lower limit of detection.
Values are mean ± SD.
n = 11.
Unpaired t test.
The ELISA lower limit of quantitation (LLOQ) was based on the lowest concentration for the manufacturer’s specified calibration curve. Plasma samples were analyzed at the dilution recommended by the manufacturer.
Next, IP-MRM was used to measure TIMP1 in 12 50 μL plasma aliquots from patients with colon cancer and in 12 samples from noncancer controls. The same samples were also analyzed with a commercial ELISA kit with the same capture antibody (Figure S10A [SI]). The mean values in IP-MRM analyses in plasma samples from cancer patients and controls were 212 ng/mL and 141 ng/mL, respectively, which was a significant difference (unpaired t test, p = 0.0028) (Table 1). In ELISA analyses, the mean TIMP1 levels in plasma samples from cancer patients and controls were 120 ng/mL and 97 ng/mL, which were not significantly different (unpaired t test, p = 0.06).
Comparison of Multiplexed IP-MRM Assay with ELISA
A key advantage of MRM-based methods is the capacity for multiplexed analyses, which can increase analysis throughput and minimize sample consumption. We performed a multiplexed IP-MRM analysis of the remaining proteins (ENG, MSLN, THBS2, MMP9 and COMP), which were spiked into a “mock plasma” matrix consisting of 60 mg/mL BSA in PBS to produce concentrations ranging from 640 to 10 ng/mL. Antibodies for the five proteins (one antibody for each protein) were immobilized on the resin, mixed and aliquoted. The mixed antibody resin (1 μg/antibody/analyte) was incubated with solutions of BSA matrix containing the spiked target proteins. Each concentration point and blank was prepared in triplicate. Plots of calculated protein concentration determined by the peak area ratio of the light peptide to heavy peptide (L/H) showed a linear increase in measured protein across the concentration range (Figure 1B–F) (the theoretical protein concentration versus the ratio L/H is also shown in Figure S9B–F [SI]).
Recoveries were determined for each of the five proteins based on the slope of the response curve and ranged from 13.5% (THBS2) to 55% (MSLN). We note that both THBS2 and ENG yielded lower slopes than the other proteins. This could reflect less efficient capture or digestion for these proteins, or some combination of both effects. LOD values were determined by comparing the variance of the blank samples (with no analyte spiked in) to the variance of the lowest level spiked sample (analyte at 10 ng/mL).9 The LOD values were between 2 and 10 ng/mL (Table 1). On the basis of triplicate measurements at the lowest spiked concentration (10 ng/mL) for each protein, CV values were all below 20% (Table 1).
All five proteins were analyzed individually by ELISA in plasma samples from 12 cancer patients and from 12 controls (11 cancer plasma samples were analyzed for TIMP1 and MMP9). The mean concentrations of these five proteins determined by ELISA in plasma samples from normal and cancer patients ranged from 3.9 ng/mL (ENG) to 208 ng/mL (COMP) (Figure S10B–F [SI]), Table 1). For IP-MRM analyses, the mixed antibody resin (1 μg antibody/analyte) was used to simultaneously capture all five protein targets. Three aliquots of each colon cancer and normal plasma sample were analyzed by IP-MRM. The mean concentrations of these five proteins determined by IP-MRM ranged from 8.3 ng/mL (ENG) to 221 ng/mL (COMP) (Figure S10B–F [SI]), Table 1). Comparison of measured values between normal and cancer plasma samples with either ELISA or IP-MRM yielded only two significant differences (p < 0.05, unpaired student t test) for TIMP1 and THBS2, but only when measured by IP-MRM (Table 1).
Comparison of IP-MRM and ELISA Measurements across Individual Samples
We asked how the IP-MRM and ELISA methods compared for measurement of the six biomarker candidate proteins across individual samples. Figure 2 shows paired comparisons of measurements with the two analysis methods for each sample. The data indicate that the concordance of measurements depends on both the analyte and the method. Analysis of the average measured plasma concentrations for all analytes and all samples indicated that the IP-MRM measurements were higher by 8.6 ng/mL at (p = 0.005, paired t test; 95% CI: 2.7–14.5), across all markers and that the two measurements were highly correlated (r = 0.93). Comparison of the average difference in plasma concentrations measured by IP-MRM versus ELISA for individual candidates indicated that differences were significant (p < 0.05, paired t test) for TIMP1 (67.1 ng/mL), THBS2 (−13.4 ng/mL), MSLN (−11.7 ng/mL) and ENG (5.2 ng/mL), but not for COMP (−6.4 ng/mL), MMP9 (13.5 ng/mL). We also examined correlation of IP-MRM and ELISA for the individual analytes (Figure 3). Correlation coefficients for the two methods were high for MMP9, COMP and TIMP1 and moderate for THBS2 and MSLN. However there was no apparent correlation between the methods for ENG, as ELISA measurements all were approximately 5 ng/mL, whereas IP-MRM measurements varied from 5 to 20 ng/mL.
Figure 2.

Quantitation of six biomarker candidate proteins in plasma samples from colon cancer patients and noncancer controls by IP-MRM and ELISA. Plasma levels of TIMP1, COMP, MMP9, THBS2, MSLN and ENG were measured in controls (circles) and colon cancer patients (squares). Results are from the average of triplicate IP-MRM measurements and duplicate ELISA measurements.
Figure 3.

Correlation of protein expression levels measured by ELISA and IP-MRM in plasma samples. Mean concentrations from triplicate analyses of TIMP1, COMP, MMP9, THBS2, MSLN and ENG in plasma samples from 24 subjects (23 for TIMP1 and MMP9) by ELISA and IP-MRM are plotted. Pearson correlation coefficients are indicated in the figure.
Discussion
The targeted analysis of protein biomarker candidates in plasma or serum requires highly sensitive and specific assays. Although ELISA provides the gold standard platform for such analyses, the selection of high-quality ELISAs is limited. Configuration of a new ELISA is often costly and time-consuming and may fail for lack of high-quality reagents. The rationale for hybrid immuno–MS assays was first proposed by Anderson and colleagues11 and has been developed through the peptide capture (SISCAPA) approach.10,12−14 The key advantages of an immuno–MS approach are the ability to systematically configure MRM assays based on proteotypic peptides and the requirement for only one antibody for target capture. Protein-capture-based immuno–MS analysis has been reported,18,19 but has not been as thoroughly explored. A potential advantage of protein capture is that the antibodies might be employed to transition the IP-MRM assay to an ELISA platform. With that consideration in mind, we asked how IP-MRM and ELISA would compare when the same capture antibodies are used.
Our results indicate that the two methods offer equivalent performance with a few exceptions. A global comparison of the IP-MRM and ELISA measurements in our study indicated a slight systematic bias in favor of higher measurement values for IP-MRM. However, this difference was not predictive for individual analytes, as absolute measurement differences between the methods varied in either direction by approximately 5–10 ng/mL. Measurement variation (CV at LOD) for IP-MRM ranged from 2.3 to 19%, which was similar to variation for ELISAs of the same samples (Table 1). IP-MRM and ELISA measurements were well-correlated, with coefficients for all of the analytes except ENG = −.017. Poor correlation of IP-MRM and ELISA for ENG appears to be related to limited range of the ELISA measurements, which were near the LOD of the assay at the dilution used (Figure S10F [SI]). Because the same capture antibody was used for both assays, it appears unlikely that differential capture would explain the difference in ENG measurement by the two methods. The difference may reflect selective recognition of a subset of ENG proteins in plasma samples by the detection antibody, perhaps due to unanticipated modifications. Such modifications may not have affected peptide-based quantitation in IP-MRM assays.
We used plasma samples from individuals with colon cancers and from noncancer controls in our study, and the biomarker candidate proteins were selected on the basis of existing literature. However, these experiments were designed only to compare the performance of the assay platforms, not to validate biomarker candidates for colon cancer detection. Although we cannot draw any conclusions about the utility of the biomarker candidates, the data illustrate the importance of reliable, precise assays for biomarker validation studies. We observed a high degree of overlap in measurement distributions for all of the candidates in control and cancer plasma samples. This is expected for most biomarker candidates that, despite being overexpressed in tumors, also may be derived from other tissues. Assays capable of validating cancer biomarkers will be expected to precisely measure small concentration differences for proteins present at ng/mL levels or lower.
Our study affirmed a key advantage of IP-MRM, which is the ability of the assay platform to perform multiplexed protein capture, as reported by Nico et al.20 This advantage is particularly important when amounts of plasma or serum samples are limited. In our study, all proteins could be captured and quantified from a single aliquot of 50 μL plasma, while input quantities for single ELISA measurements were between 2 and 66 μL. The capture antibody for IP-MRM approach should have a relatively high affinity for the targeted protein. Our experiments represent a “best case” example, because the capture antibodies were of sufficiently high quality to support robust and sensitive ELISA kits. We have done other IP-MRM experiments with antibodies found to be unsatisfactory for ELISA, and the same antibodies also failed to provide significant protein enrichment for IP-MRM measurements (unpublished observations). IP-MRM might be used to analyze different isoforms or post-translational modifications of proteins if they are bound with similar affinity by the capture antibody.
Another potentially advantageous feature of IP-MRM is the detection of proteins via multiple peptides. While this offers flexibility in assay development and confirmation of measurements made on single peptides, previous work demonstrates that one peptide usually provides the greatest measurement sensitivity.10,13 For this reason, we chose not to explore comparison MRM measurements with multiple peptides and instead focused on analysis of peptides that generated the strongest signals. In addition, higher throughput IP-MRM assays can utilize magnetic beads for antibody capture in a 96-well-plate format.33
IP-MRM directed at intact proteins provides an effective approach to systematically configure targeted protein measurements. The protein-targeted capture compares favorably to both peptide-targeted IP-MRM and ELISA. For novel targets, the choice of methods depends primarily on the availability of antibodies and their performance. Protein-based IP-MRM assays offer a faster, less costly approach to targeted protein measurement than ELISAs and could dramatically expand the scope of targeted protein quantitation in biology and medicine.
Acknowledgments
This work was supported by a cooperative agreement award 5U24CA126479 from the National Cancer Institute through the Clinical Proteomic Technology Assessment for Cancer (CPTAC) program.
Glossary
Abbreviations
- CPTAC
Clinical Proteomic Technology Asssessment for Cancer
- MS
mass spectrometry
- MRM
multiple reaction monitoring
- IP-MRM
immunoprecipitation GeLC MS
- ELISA
enzyme-linked immunosorbent assay
- SISCAPA
stable isotope standards and capture by antipeptide antibodies
- CV
coefficient of variation
- SID
stable isotope dilution
- CRC
colorectal cancer
- fwhm
full width at half-maximum
- LOD
limit of detection
- SCX
strong cation exchange
- IEF
isoelectric focusing
- HPLC
high performance liquid chromatography
- SDS
sodium dodecyl sulfate
Supporting Information Available
This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Rifai N.; Gillette M. A.; Carr S. A. Protein biomarker discovery and validation: The long and uncertain path to clinical utility. Nat. Biotechnol. 2006, 248971–983. [DOI] [PubMed] [Google Scholar]
- Shi T.; Fillmore T. L.; Sun X.; Zhao R.; Schepmoes A. A.; Hossain M.; Xie F.; Wu S.; Kim J. S.; Jones N.; Moore R. J.; Pasa-Tolic L.; Kagan J.; Rodland K. D.; Liu T.; Tang K.; Camp D. G. 2nd; Smith R. D.; Qian W. J. Antibody-free, targeted mass-spectrometric approach for quantification of proteins at low picogram per milliliter levels in human plasma/serum. Proc. Natl. Acad. Sci. U.S.A. 2012, 1093815395–15400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T.; Su D.; Liu T.; Tang K.; Camp D. G. 2nd; Qian W. J.; Smith R. D. Advancing the sensitivity of selected reaction monitoring-based targeted quantitative proteomics. Proteomics 2012, 1281074–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J. Y.; Dann G. P.; Shi T.; Wang L.; Gao X.; Su D.; Nicora C. D.; Shukla A. K.; Moore R. J.; Liu T.; Camp D. G. 2nd; Smith R. D.; Qian W. J. Simple sodium dodecyl sulfate-assisted sample preparation method for LC–MS-based proteomics applications. Anal. Chem. 2012, 8462862–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnidge D. R.; Goodmanson M. K.; Klee G. G.; Muddiman D. C. Absolute quantification of the model biomarker prostate-specific antigen in serum by LC–MS/MS using protein cleavage and isotope dilution mass spectrometry. J. Proteome Res. 2004, 33644–652. [DOI] [PubMed] [Google Scholar]
- Anderson L.; Hunter C. L. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol. Cell. Proteomics 2006, 54573–588. [DOI] [PubMed] [Google Scholar]
- Kuzyk M. A.; Smith D.; Yang J.; Cross T. J.; Jackson A. M.; Hardie D. B.; Anderson N. L.; Borchers C. H. Multiple reaction monitoring-based, multiplexed, absolute quantitation of 45 proteins in human plasma. Mol. Cell. Proteomics 2009, 881860–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T.; Zhou J. Y.; Gritsenko M. A.; Hossain M.; Camp D. G. 2nd; Smith R. D.; Qian W. J. IgY14 and SuperMix immunoaffinity separations coupled with liquid chromatography-mass spectrometry for human plasma proteomics biomarker discovery. Methods 2012, 562246–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addona T. A.; Abbatiello S. E.; Schilling B.; Skates S. J.; Mani D. R.; Bunk D. M.; Spiegelman C. H.; Zimmerman L. J.; Ham A. J.; Keshishian H.; Hall S. C.; Allen S.; Blackman R. K.; Borchers C. H.; Buck C.; Cardasis H. L.; Cusack M. P.; Dodder N. G.; Gibson B. W.; Held J. M.; Hiltke T.; Jackson A.; Johansen E. B.; Kinsinger C. R.; Li J.; Mesri M.; Neubert T. A.; Niles R. K.; Pulsipher T. C.; Ransohoff D.; Rodriguez H.; Rudnick P. A.; Smith D.; Tabb D. L.; Tegeler T. J.; Variyath A. M.; Vega-Montoto L. J.; Wahlander A.; Waldemarson S.; Wang M.; Whiteaker J. R.; Zhao L.; Anderson N. L.; Fisher S. J.; Liebler D. C.; Paulovich A. G.; Regnier F. E.; Tempst P.; Carr S. A. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 2009, 277633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshishian H.; Addona T.; Burgess M.; Mani D. R.; Shi X.; Kuhn E.; Sabatine M. S.; Gerszten R. E.; Carr S. A. Quantification of cardiovascular biomarkers in patient plasma by targeted mass spectrometry and stable isotope dilution. Mol. Cell. Proteomics 2009, 8102339–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson N. L.; Anderson N. G.; Haines L. R.; Hardie D. B.; Olafson R. W.; Pearson T. W. Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA). J. Proteome Res. 2004, 32235–244. [DOI] [PubMed] [Google Scholar]
- Kuhn E.; Addona T.; Keshishian H.; Burgess M.; Mani D. R.; Lee R. T.; Sabatine M. S.; Gerszten R. E.; Carr S. A. Developing multiplexed assays for troponin I and interleukin-33 in plasma by peptide immunoaffinity enrichment and targeted mass spectrometry. Clin. Chem. 2009, 5561108–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshishian H.; Addona T.; Burgess M.; Kuhn E.; Carr S. A. Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol. Cell. Proteomics 2007, 6122212–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razavi M.; Frick L. E.; LaMarr W. A.; Pope M. E.; Miller C. A.; Anderson N. L.; Pearson T. W. High-throughput SISCAPA quantitation of peptides from human plasma digests by ultrafast, liquid chromatography-free mass spectrometry. J. Proteome Res. 2012, 11125642–5649. [DOI] [PubMed] [Google Scholar]
- Whiteaker J. R.; Lin C.; Kennedy J.; Hou L.; Trute M.; Sokal I.; Yan P.; Schoenherr R. M.; Zhao L.; Voytovich U. J.; Kelly-Spratt K. S.; Krasnoselsky A.; Gafken P. R.; Hogan J. M.; Jones L. A.; Wang P.; Amon L.; Chodosh L. A.; Nelson P. S.; McIntosh M. W.; Kemp C. J.; Paulovich A. G. A targeted proteomics-based pipeline for verification of biomarkers in plasma. Nat. Biotechnol. 2011, 297625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addona T. A.; Shi X.; Keshishian H.; Mani D. R.; Burgess M.; Gillette M. A.; Clauser K. R.; Shen D.; Lewis G. D.; Farrell L. A.; Fifer M. A.; Sabatine M. S.; Gerszten R. E.; Carr S. A. A pipeline that integrates the discovery and verification of plasma protein biomarkers reveals candidate markers for cardiovascular disease. Nat. Biotechnol. 2011, 297635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoofnagle A. N.; Becker J. O.; Wener M. H.; Heinecke J. W. Quantification of thyroglobulin, a low-abundance serum protein, by immunoaffinity peptide enrichment and tandem mass spectrometry. Clin. Chem. 2008, 54111796–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berna M.; Ott L.; Engle S.; Watson D.; Solter P.; Ackermann B. Quantification of NTproBNP in rat serum using immunoprecipitation and LC/MS/MS: A biomarker of drug-induced cardiac hypertrophy. Anal. Chem. 2008, 803561–566. [DOI] [PubMed] [Google Scholar]
- Berna M. J.; Zhen Y.; Watson D. E.; Hale J. E.; Ackermann B. L. Strategic use of immunoprecipitation and LC/MS/MS for trace-level protein quantification: myosin light chain 1, a biomarker of cardiac necrosis. Anal. Chem. 2007, 79114199–4205. [DOI] [PubMed] [Google Scholar]
- Nicol G. R.; Han M.; Kim J.; Birse C. E.; Brand E.; Nguyen A.; Mesri M.; FitzHugh W.; Kaminker P.; Moore P. A.; Ruben S. M.; He T. Use of an immunoaffinity-mass spectrometry-based approach for the quantification of protein biomarkers from serum samples of lung cancer patients. Mol. Cell. Proteomics 2008, 7101974–1982. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Chaerkady R.; Wu J.; Hwang H. J.; Papadopoulos N.; Kopelovich L.; Maitra A.; Matthaei H.; Eshleman J. R.; Hruban R. H.; Kinzler K. W.; Pandey A.; Vogelstein B. Mutant proteins as cancer-specific biomarkers. Proc. Natl. Acad. Sci. U.S.A. 2011, 10862444–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B.; Tomazela D. M.; Shulman N.; Chambers M.; Finney G. L.; Frewen B.; Kern R.; Tabb D. L.; Liebler D. C.; MacCoss M. J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 267966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange V.; Picotti P.; Domon B.; Aebersold R. Selected reaction monitoring for quantitative proteomics: a tutorial. Mol. Syst. Biol. 2008, 4, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteaker J. R.; Zhao L.; Abbatiello S. E.; Burgess M.; Kuhn E.; Lin C.; Pope M. E.; Razavi M.; Anderson N. L.; Pearson T. W.; Carr S. A.; Paulovich A. G. Evaluation of large scale quantitative proteomic assay development using peptide affinity-based mass spectrometry. Mol. Cell. Proteomics 2011, 104M110 005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusaro V. A.; Mani D. R.; Mesirov J. P.; Carr S. A. Prediction of high-responding peptides for targeted protein assays by mass spectrometry. Nat. Biotechnol. 2009, 272190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Su Y.; Fingleton B.; Acuff H.; Matrisian L. M.; Zent R.; Pozzi A. Increased plasma MMP9 in integrin α1-null mice enhances lung metastasis of colon carcinoma cells. Int. J. Cancer 2005, 116152–61. [DOI] [PubMed] [Google Scholar]
- Creaney J.; Francis R. J.; Dick I. M.; Musk A. W.; Robinson B. W.; Byrne M. J.; Nowak A. K. Serum soluble mesothelin concentrations in malignant pleural mesothelioma: Relationship to tumor volume, clinical stage and changes in tumor burden. Clin. Cancer Res. 2011, 1751181–1189. [DOI] [PubMed] [Google Scholar]
- Hawinkels L. J.; Kuiper P.; Wiercinska E.; Verspaget H. W.; Liu Z.; Pardali E.; Sier C. F.; ten Dijke P. Matrix metalloproteinase-14 (MT1-MMP)-mediated endoglin shedding inhibits tumor angiogenesis. Cancer Res. 2010, 70104141–4150. [DOI] [PubMed] [Google Scholar]
- Holten-Andersen M. N.; Christensen I. J.; Nielsen H. J.; Stephens R. W.; Jensen V.; Nielsen O. H.; Sorensen S.; Overgaard J.; Lilja H.; Harris A.; Murphy G.; Brunner N. Total levels of tissue inhibitor of metalloproteinases 1 in plasma yield high diagnostic sensitivity and specificity in patients with colon cancer. Clin. Cancer Res. 2002, 81156–164. [PubMed] [Google Scholar]
- Jubb A. M.; Hurwitz H. I.; Bai W.; Holmgren E. B.; Tobin P.; Guerrero A. S.; Kabbinavar F.; Holden S. N.; Novotny W. F.; Frantz G. D.; Hillan K. J.; Koeppen H. Impact of vascular endothelial growth factor-A expression, thrombospondin-2 expression, and microvessel density on the treatment effect of bevacizumab in metastatic colorectal cancer. J. Clin. Oncol. 2006, 242217–227. [DOI] [PubMed] [Google Scholar]
- Klee E. W.; Bondar O. P.; Goodmanson M. K.; Dyer R. B.; Erdogan S.; Bergstralh E. J.; Bergen H. R. 3rd; Sebo T. J.; Klee G. G. Candidate serum biomarkers for prostate adenocarcinoma identified by mRNA differences in prostate tissue and verified with protein measurements in tissue and blood. Clin. Chem. 2012, 583599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen N. M.; Bystrom P.; Christensen I. J.; Berglund A.; Nielsen H. J.; Brunner N.; Glimelius B. TIMP-1 is significantly associated with objective response and survival in metastatic colorectal cancer patients receiving combination of irinotecan, 5-fluorouracil, and folinic acid. Clin. Cancer Res. 2007, 13144117–4122. [DOI] [PubMed] [Google Scholar]
- Whiteaker J. R.; Zhao L.; Anderson L.; Paulovich A. G. An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry-based quantification of protein biomarkers. Mol. Cell. Proteomics 2010, 91184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.