

Abstract
Core histone octamers that are repetitively spaced along a DNA molecule are called nucleosomal arrays. Nucleosomal arrays are obtained in one of two ways: purification from in vivo sources, or reconstitution in vitro from recombinant core histones and tandemly repeated nucleosome positioning DNA. The latter method has the benefit of allowing for the assembly of a more compositionally uniform and precisely positioned nucleosomal array. Sedimentation velocity experiments in the analytical ultracentrifuge yield information about the size and shape of macromolecules by analyzing the rate at which they migrate through solution under centrifugal force. This technique, along with atomic force microscopy, can be used for quality control, ensuring that the majority of DNA templates are saturated with nucleosomes after reconstitution. Here we describe the protocols necessary to reconstitute milligram quantities of length and compositionally defined nucleosomal arrays suitable for biochemical and biophysical studies of chromatin structure and function.
Keywords: Cellular Biology, Issue 79, Chromosome Structures, Chromatin, Nucleosomes, Histones, Microscopy, Atomic Force (AFM), Biochemistry, Chromatin, Nucleosome, Nucleosomal Array, Histone, Analytical Ultracentrifugation, Sedimentation Velocity
Introduction
Eukaryotic genomes do not exist as naked DNA, but rather are compacted and organized by bound proteins. These complexes of DNA and protein are known as chromatin. The basic repeating unit of chromatin is the nucleosome. A nucleosome consists of histone octamer and 146 base pairs of DNA wrapped around the histone octamer about 1.6 times1. The histone octamer is composed of two copies each of the core histones H2A, H2B, H3, and H4. Core histone octamers that are repetitively spaced along a DNA molecule are called nucleosomal arrays. The extended structure of nucleosomal arrays has been referred to as the 10 nm fiber or the "beads on a string" structure and is present in vitro under low salt conditions2. The 10 nm fiber is capable of condensing into higher order structures through intra-array compaction and/or inter-array oligomerization2. These higher order structures can be induced in the presence of salts, or can be influenced through the binding of chromatin architectural proteins to the nucleosomal array3,4. Levels of chromatin compaction are inversely correlated with rate of transcription in vivo5,6. Recent research highlights the importance of the structural organization of genomes in processes such as differentiation, cancer development, and others7,8. The use of nucleosomal arrays to study chromatin structure and function has become widespread. Here we describe a method for the assembly of nucleosomal arrays from recombinant core histones and nucleosome positioning DNA.
Using recombinant DNA with tandem repeats of nucleosome positioning sequences allows for the reconstitution of arrays that contain regularly spaced nucleosomes. Two of the more popular positioning sequences are the 5S rRNA gene sequence and the "601" sequence9,10. The 601 sequence was derived from SELEX experiments and more strongly positions nucleosomes than the 5S sequence11. Consequently, the linker DNA length of the 601 arrays is more homogeneous. Tandemly repeated nucleosome positioning DNA is obtained by gel filtration4,12. Recombinant histones are purified from E. coli under denaturing conditions13. The use of recombinant histones allows one to carefully control the histone composition of the nucleosomal arrays. For example, core histones bearing specific mutations14 or post-translational modifications15,16 can be substituted for wild type core histones.
Sedimentation velocity experiments monitor the rate of sedimentation of macromolecules in solution under an applied centrifugal force17. This yields information about the size and shape of macromolecules in a sample. Sedimentation velocity experiments are thus an appropriate tool for studying solution state changes in chromatin fiber structure due to chromatin condensation18. Importantly, it is first necessary to use sedimentation velocity experiments as a quality control step in nucleosomal array reconstitution. If DNA and nucleosomes are not combined at the proper molar ratio, the arrays may be under- or over-saturated with core histones. Thus, the information gained from sedimentation velocity experiments is used to ensure that the DNA is properly saturated with nucleosomes. It is important to use alternative methods to estimate the saturation of DNA with nucleosomes, especially if working with a previously uncharacterized DNA template. Therefore, we also describe a method for analysis of nucleosomal arrays using atomic force microscopy (AFM). AFM is a powerful technique that allows visualization of the effects of a number of parameters, such as the level of saturation, effect of the presence of histone variants or the effects of MgCl2 19,20. AFM has also been applied to study nucleosome dynamics using time lapse imaging 21. In vitro assembled nucleosomal 12-mer arrays are particularly amenable to AFM studies because they belong in the right size range for AFM imaging 22. In the present study we have used AFM of nucleosomal arrays as a quality control as well as a means of affirming the data from AUC ("seeing is believing"). In addition to simple visualization, AFM allows measurement of height profiles of samples as an additional metric.
Protocol
1. Assembly of Recombinant Core Histones into Octamers
Rationale: The first step in nucleosomal array reconstitution is to prepare native core histone octamers from lyophilized recombinant core histones. Histone proteins are combined in equal molar amounts and assembled into histone octamers by dialyzing the samples out of a denaturing buffer into refolding buffer.
Purify and lyophilize recombinant core histones (H2A, H2B, H3, H4) as described13.
Dissolve approximately 5 mg of each lyophilized core histone in 3 ml of unfolding buffer (6 M guanidinium HCl, 20 mM Tris pH 7.5, 5 mM DTT). Allow each aliquot to dissolve for at least one hour. Ensure that no protein remains on the sides of the container.
Measure the absorbance of each histone at 276 nm using unfolding buffer as a reference.
Calculate the molarity of each histone using their extinction coefficients (See Table 1 for Xenopus histone extinction coefficients). Compare the absorbance determined concentration of histone to the lyophilized dry weight, and estimate if the majority of the protein is dissolved.
Determine which histone you have the fewest moles of, as this will be the limiting number of moles for all histones.
Combine the histones in equal molar amounts and dilute with unfolding buffer to a final concentration of 1 mg/ml.
Place the sample into 6-8 kDa MWCO dialysis tubing. Seal the tubing and place into 2 L of cold refolding buffer (2 M NaCl, 10 mM Tris pH 7.5, 1 mM EDTA, 5 mM β-mercaptoethanol).
Dialyze the sample for 18 hr at 4 °C with stirring. Ensure the dialysis tubing can rotate freely and vigorously, or else the histones may precipitate.
Change the dialysis buffer twice in the 18 hr period (i.e. change the refolding buffer about every 6 hr until done). Even if precipitate is formed do not discard the sample, some octamer may be recovered although the yield will be decreased.
Note: See step 2.1-2.2 to prepare column equilibration for the next day.
2. Purification of Histone Octamers by Size Exclusion Chromatography
Rationale: After concluding with section 1, samples will contain histone octamers as well as other histone complexes such as aggregates, H3/H4 tetramers, and H2A/H2B dimers. Histone octamers will be purified away from these other complexes using size exclusion chromatography (SEC).
Connect a HiLoad 16/60 Superdex200 16/60 (S200) column to an FPLC system. Ensure that air bubbles do not enter the system.
Clean the S200 column with 0.2 μm filtered water. Use a flow rate of 0.3 ml/min and set a back pressure limit of 0.5 MPa. Ensure you have enough water and allow the column to clean overnight.
Equilibrate the S200 column and sample loop of the FPLC using refolding buffer. Use 1 L of 0.2 μm filtered refolding buffer. Flow refolding buffer through the column at a rate of 1 ml/min and a maximum pressure of 0.5 MPa for about 2 hr.
Equilibrate a Vivaspin centrifugal concentrator (50 kDa MWCO) with 1 ml of refolding buffer. Remove the sample from dialysis tubing and centrifuge the sample at 4 °C to remove any precipitates. Pipette the sample supernatant into the concentrator. Concentrate the sample to a volume of approximately 500 μl.
Remove the concentrated octamer sample to a new container and rinse the concentrator with 1 ml refolding buffer. Concentrate the rinse down to 500 μl, and add it to the octamer sample. This helps to salvage any octamer remaining in the concentrator.
Spin the sample in a 1.5 ml microfuge tube for 5 min at 10,000 rpm at 4 °C. Collect the supernatant and transfer to a new tube. This helps to remove any precipitate before loading the sample on the FPLC.
Load the sample onto the S200 column. Load a maximum of 1.5 ml total volume or 15 mg octamer in a single purification.
Elute the sample using freshly made refolding buffer. Use a flow rate of 1 ml/min and a back pressure limit of 0.5 MPa. Use two column volumes (400 ml) refolding buffer and collect 2 ml fractions. Monitor the absorbance at 280 nm during elution. The different species will elute in order of decreasing size. Histone aggregates usually elute at approximately 45 ml, octamer at about 65 ml, and dimer at around 84 ml (Figure 1).
Analyze the elution fractions from peaks of interest by running samples on an 18-20% SDS-PAGE. Fractions containing purified octamers should have equal molar amounts of the four core histone proteins (Figure 1).
Pool the fractions that contain purified octamers, and concentrate to ≤15 mg/ml with a Vivaspin centrifugal filter. If left dilute, octamers may disassociate into H2A/H2B dimers and H3/H4 tetramers.
Determine the octamer concentration by measuring the absorbance at 280 nm and calculate using the extinction coefficient from Table 1.
Histone octamers can be kept short term in refolding buffer at 4 °C. If being stored long term, the octamers will be more stable at -20 °C and in a 50% glycerol solution. Octamers stored in glycerol should be dialyzed into fresh refolding buffer before absorbance measurements and use in nucleosomal array reconstitutions.
3. Reconstitution of Nucleosomal Arrays from DNA and Purified Octamers
Rationale: Reconstitution of nucleosomal arrays requires that histone octamers and template DNA be combined at specific molar ratios. Mixtures of DNA and histone octamers in 2 M NaCl are step dialyzed into buffers of decreasing ionic strength. A gradual change to lower salt ensures proper nucleosome formation. Obtaining nucleosomal arrays with the desired level of histone octamer saturation of the template DNA requires small scale test reconstitutions followed by a large scale preparative reconstitution. The appropriate conditions defined by the small scale reconstitutions are used to guide the preparative reconstitution.
Purify tandemly repeated nucleosome positioning DNA as described 4,12. Briefly, isolate the plasmid DNA using a Qiagen GigaPrep kit or a similar product. Set-up a restriction enzyme digest in order to release the template DNA and digest the plasmid DNA to smaller sizes (≤700bp). The template DNA can then be purified away from the small plasmid DNA remnants using size exclusion chromatography (SEC). A gravity fed column, with a height of about 115 cm packed with Sephacryl S-1000 beads is sufficient for purification of 601207bp x 12mer DNA.
Notes: For plasmid DNA purification at decreased cost but increased effort, one can use alkaline lysis with phenol/chloroform DNA purification in order to isolate plasmid DNA from E. coli23,24. Strategies for separating template DNA from plasmid DNA may vary with changes in template DNA size. For SEC it is important to set up the restriction enzyme digest in a fashion which maximizes the difference in size between the template and plasmid DNA.
Determine which molar ratios (r) to use for reconstitutions. r is equal to the ratio of moles of octamer to moles of DNA repeat. For initial small scale trials, r values of 0.9, 1, and 1.1 are appropriate if the intent is to obtain saturated nucleosomal arrays.
Prepare the small scale samples by calculating the amount of octamer to add to approximately 18 μg of DNA for each molar ratio to be tested. Mix the DNA and histone octamers. The final concentration of NaCl in the sample should be ≥2 M, and the final concentration of DNA should be around 0.3 μg/μl. The final sample buffer conditions should also include 10 mM Tris pH 7.8, and 1 mM EDTA.
The samples are now ready for dialysis. Load the samples into 12k-14k MWCO dialysis tubing. Dialyze the samples against 2L of buffer of decreasing ionic strength as follows.
Components in all buffers: 10 mM Tris pH 7.8, 1 mM EDTA. Buffer 1 (add 1 M NaCl, 1 mM DTT) for 5-6 hr. Buffer 2 (add 0.75 M NaCl, 1 mM DTT) overnight. Buffer 3 (add 2.5 mM NaCl, 1 mM DTT) for 5-6 hr. Buffer 4 (add 2.5 mM NaCl, 0.1 mM PMSF) overnight.
Remove the samples from the dialysis tubing and proceed to sections 4-6. If the small-scale samples yield the desired results, repeat the steps in sections 3-6 on the large scale.
Note: In order to conserve resources, small scale samples should have the minimum volume necessary to suffice for downstream screening assays. In order to have enough sample for the sedimentation velocity and AFM experiments listed here, 50 μl samples at 0.3 mg/ml DNA are appropriate. The size of large scale, preparative samples are dependent on their intended use. A large scale sample should be prepared in the same manner as the small scale samples.
4. Sedimentation Velocity Analysis of Reconstituted Nucleosomal Arrays
Rationale: Sedimentation velocity experiments in the Beckman Xl-A/I analytical ultracentrifuge yield information on the size and shape of molecules in solution. Sedimentation velocity experiments performed under low salt conditions are used to determine the extent to which the reconstituted arrays are saturated with nucleosomes.
Dilute the sample to an absorbance around 0.5 at 260 nm using TEN buffer (10 mM Tris pH 7.8, 1 mM EDTA, 2.5 mM NaCl). Load 400 μl of sample into a cell with a two sector centerpiece, with the sample on one side and the reference (TEN buffer) on the other25. Keep the reference meniscus higher than the sample meniscus (i.e. add 420 μl reference solution).
Load the cells into the rotor, and align the cells properly using the hash marks on the bottom of the cells and rotor25. Gently dust the lenses of the cells using compressed air.
Turn on the XL-A/XL-I centrifuge, insert the rotor, and attach and secure the optics as described in the manual. Open the Proteome Lab software and select file: new.
Set up a single scan run at 3,000 rpm and a temperature of 25 °C. Under options, select stop after last scan, and run radial calibration (radial calibration is not necessary if the rotor was the last rotor used in the AUC).
For each cell, name the samples, select a wavelength (260 nm), select absorbance, and choose a save file location on your computer. Begin the single scan run.
Use the single scan to determine the appropriate radial scan length (Figure 2A). Decreasing the length of the cell that will be scanned decreases the run time. However, be sure to include the sample meniscus and extend the end very close to, or at the bottom of the cell.
Note: Single scans allow the measurement area to be shortened by starting measurements just before the sample meniscus (Figure 2A). Most of the scans generated during the AUC run should have boundary fractions past the meniscus as well as stable plateaus (Figure 2B). Dirty lenses on the AUC cells can generate scans with large spikes, these may affect analysis of the data. The UltraScan software includes a manual which is a great resource for information regarding the analysis of analytical ultracentrifugation data26.
Using the XL-A/XL-I control panel, enter a speed of 0 and press start. This will prompt the centrifuge chamber to pull a vacuum and allow the temperature of the chamber to equilibrate. Wait 1 hr to allow for temperature equilibration before beginning a run.
Use the software to set the run up for the desired speed and number of scans. Higher speeds increase resolution, but one should collect at least 20 scans (preferably more) before the sample has sedimented to the bottom of the cell. It is convenient to overlay the last several scans (accomplished in the options menu) in order to monitor the progress of the run and check for potential problems such as leaking cells. Monitor the first couple of scans to ensure proper operation.
5. Editing and Analyzing Sedimentation Velocity Data
Rationale: Raw sedimentation velocity data should be analyzed by a program that yields a diffusion-corrected sedimentation coefficient distribution. This information in turn indicates the fraction of a reconstituted sample that contains a given saturation level, e.g. if using a 12-mer DNA template it will be possible to determine the fraction of reconstituted arrays that has 10, 11 and 12 nucleosomes/DNA.
Use AUC data analysis software such as UltraScan to edit the data26. Editing of scan data must be done for each cell and creates a data set for subsequent analysis. Proper editing of scans involves defining the sample meniscus, reducing the data to a region of interest, and defining baseline as well as plateau regions (Figure 2). Unwanted scans can also be removed from the data set at this point. However, since it is possible to remove unwanted scans during analysis, it is recommended that all scans be kept at this point.
We strongly recommend that the enhanced van Holde-Weischet method be used to analyze the data27. This analysis method corrects for the effects of diffusion over the course of the run and yields the integral distribution of sedimentation coefficients. In particular, the enhanced van Holde-Weischet method (as implemented in UltraScan) will allow for the inclusion of scans which lack stable plateaus, or which contain boundary fractions which have not cleared the meniscus in the analysis28.
Determine the distribution of sedimentation coefficients for the assembled arrays. Representative results for 601-12 (207bp, 12-mer) nucleosomal arrays are shown in Figure 3.
If one of the small scale samples contains arrays with the desired range of sedimentation coefficients, then repeat the process at an increased scale starting with section 3. If none of the small scale samples has a proper distribution of sedimentation coefficients then repeat the small scale tests with a new r range starting at section 3.
6. Visualizing Nucleosomal Arrays Using Atomic Force Microscopy (AFM)
Rationale: AFM allows visualization of the level of saturation of nucleosomal arrays. This technique complements sedimentation velocity by AUC and restriction enzyme digestion as a quality control assay.
Using methods described above, obtain a well saturated nucleosome array (Figure 5A).
Prepare APTES treated mica slides by placing ~30 μl 1:1,000 dilution of commercial (Sigma-Aldrich (3-Aminopropyl)triethoxysilane A3648-100ML) in 0.22 μm filtered nanopure water for 30 min.
After 30 min rinse the APTES off with filtered water and gently dry them in a Nitrogen flow.
Place appropriately diluted nucleosome array sample (~1.5 ng/μl) on the slide, and incubate covered at room temperature for 15 min.
Rinse sample off with filtered sample buffer, dry as before, and place slide on the stage of AFM (in this case an Asylum Research MFP-3D Atomic Force Microscope).
Start by imaging 2 x 2 μm scans with 512 scan lines. Count the number of nucleosomes to ascertain level of saturation (Figure 5B).
Ideally you should image areas on the slide with well separated nucleosome arrays.
For higher resolution images, a 500 x 500 nm scan can be obtained (Figure 5C).
Images can be flattened and analyzed using the MFP-3D software provided by Asylum.
Each image can be divided into four quadrants and zoomed in digitally to obtain a clearer view of well-separated arrays and multiple free hand lines can be drawn through the arrays and height profiles are obtained for the nucleosomes (Figure 5C, right panel-height trace and 5D).
Several such images should be analyzed for each type of array encompassing several hundreds of nucleosomes. Height profiles are recorded, categorized and plotted in MS Excel.
Representative Results
To illustrate the protocol we reconstituted nucleosomal arrays from recombinant Xenopus core histones and DNA consisting of 12 tandem 207 bp repeats of the 601 positioning sequence (601207 x 12). We first assembled native octamers from lyophilized core histones and then purified the octamers by FPLC using an S200 column (Figure 1A). Larger complexes elute earlier from the S200 column. Histones generally elute in this order: non-specific histone aggregates, histone octamer, H3/H4 tetramer, and H2A/H2B dimer (Figure 1A). The peaks from the S200 column were analyzed by SDS-PAGE. The gels of purified octamer fractions should indicate equimolar amounts of the four histone proteins (Figure 1B). Note that Xenopus H2A and H2B have molecular weights that differ by only about 200 Da and hence they appear as a single band on SDS gels. H3/H4 tetramers and H2A/H2B dimers will elute after, but very close to the histone octamer peak. When selecting which fractions to keep it is prudent to discard fractions appearing toward the end of the octamer peak in the chromatogram. These fractions may have an increased amount of non-octameric histone complexes. In this case fractions 64-67 were pooled and saved for nucleosomal array reconstitution (Figure 1).
Within UltraScan there are two different ways of viewing the diffusion-corrected distribution of sedimentation coefficients that is obtained from the enhanced van Holde-Weischet analysis. The red line represents the integral distribution of sedimentation coefficients. For any given point on the graph, the y-axis indicates the fraction of the sample that has a sedimentation coefficient equal to or less than the value indicated on the x-axis. Thus, a vertical line is indicative of a homogenous sample, while a heterogeneous sample will have a curve with positive slope. The green line is the derivative of the integral distribution. The area under a peak is proportional to the fraction of the sample that has that sedimentation coefficient. Fully saturated 601207 x 12 nucleosomal arrays have a sedimentation coefficient of 29S 29. For the sample shown in Figure 3, the data indicate that approximately 70% of the arrays are saturated and 30% are over saturated.
Atomic force microscopy has gone from an emerging technique to a popular complementary approach to study chromatin organization in vitro. Here we have used AFM alongside of analytical ultracentrifugation to establish the extent of template saturation after reconstitution. In Figure 6 the majority of the ~27S arrays (Figure 4A) used for imaging contained 10-11 nucleosomes (Figure 4B), demonstrating excellent agreement between the AFM and AUC results. The AFM results obtained here thus validate this approach and make it a reliable technique for characterizing nucleosomal arrays.
| Histone Protein | Σ276, Unfolded Proteins cm-1M-1 | Molecular Weight Da |
| H2A | 4,350 | 13,960 |
| H2B | 7,250 | 13,774 |
| H3 | 4,640 | 15,273 |
| H4 | 5,800 | 11,236 |
| Octamer | 44,080 | 108,486 |
Table 1. Extinction coefficients and molecular weights of Xenopus laevis core histones and renatured histone octamer.
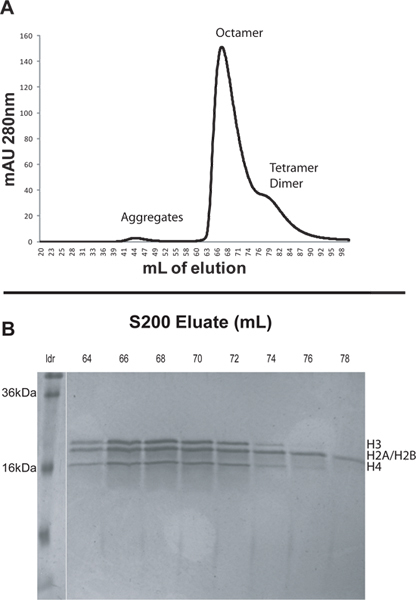 Figure 1. A. Elution profile from the S200 column as described in section 2. The small peak at about 44 ml is due to large histone aggregates. The prominent peak at 67 ml is the histone octamers. The broad shoulder from 76-90 ml contains the H3/H4 tetramers and H2A/H2B dimers. B. 20% SDS-PAGE of selected fractions from the S200 column. Early fractions from the octamer peak should be collected as they are least likely to be contaminated by the non-octameric histone complexes. In this case fractions 64-67 were pooled and saved for nucleosomal array reconstitution. Click here to view larger figure.
Figure 1. A. Elution profile from the S200 column as described in section 2. The small peak at about 44 ml is due to large histone aggregates. The prominent peak at 67 ml is the histone octamers. The broad shoulder from 76-90 ml contains the H3/H4 tetramers and H2A/H2B dimers. B. 20% SDS-PAGE of selected fractions from the S200 column. Early fractions from the octamer peak should be collected as they are least likely to be contaminated by the non-octameric histone complexes. In this case fractions 64-67 were pooled and saved for nucleosomal array reconstitution. Click here to view larger figure.
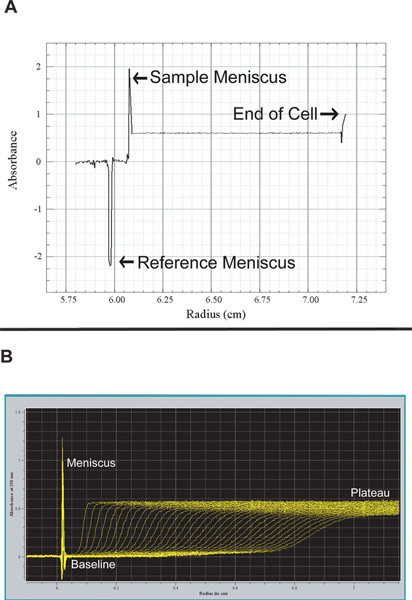 Figure 2. A. ProteomeLab software screen capture of a single scan collected at 3,000 rpm (with labels added). Note that the sample does not sediment at this low speed. The single scan is used to set the range of measurements for the AUC cell scan (step 4.6). B. Screen capture of a series of sedimentation velocity scans obtained using the UltraScan program (with labels added)26. This series of scans is edited to generate a data set for analysis (see step 5.1).
Figure 2. A. ProteomeLab software screen capture of a single scan collected at 3,000 rpm (with labels added). Note that the sample does not sediment at this low speed. The single scan is used to set the range of measurements for the AUC cell scan (step 4.6). B. Screen capture of a series of sedimentation velocity scans obtained using the UltraScan program (with labels added)26. This series of scans is edited to generate a data set for analysis (see step 5.1).
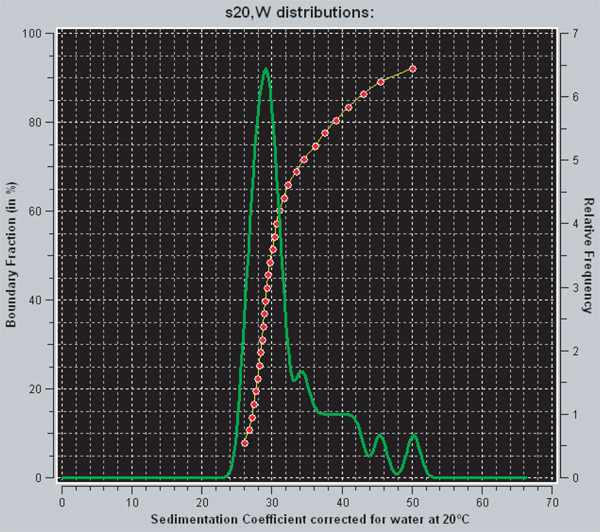 Figure 3. A screen capture from UltraScanII. The red and green lines are two different methods for viewing the distribution of sedimentation coefficients in UltraScan. The red line is the integral distribution of sedimentation coefficients, while the green is the derivative. Extra information on interpretation can be found in the results section.
Figure 3. A screen capture from UltraScanII. The red and green lines are two different methods for viewing the distribution of sedimentation coefficients in UltraScan. The red line is the integral distribution of sedimentation coefficients, while the green is the derivative. Extra information on interpretation can be found in the results section.
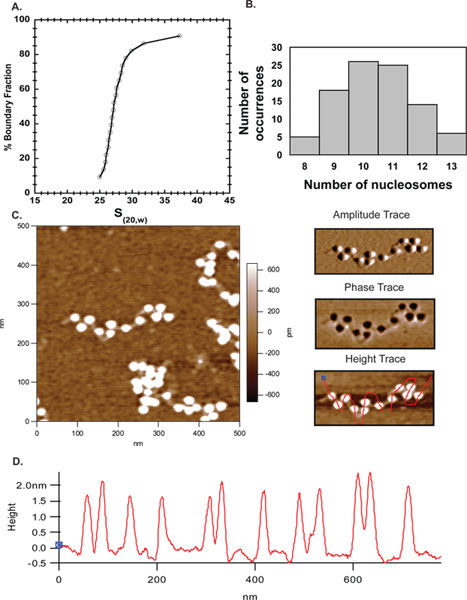 Figure 4. AFM. A. Sedimentation velocity profile for 601207 x 12 arrays assembled with mouse octamer indicating the average sedimentation coefficient is 27S. B. The same arrays in A were imaged by AFM and the number of nucleosomes counted in several images were plotted in MS Excel. The plot indicated that a majority of arrays had 10-11 nucleosomes corroborating the AUC data. C. A 500 x 500 nm scan of 601207 x 12 nucleosome arrays with the linker DNA clearly visible. The top right panel is the amplitude trace and the middle right panel is the phase trace for the array shown in C. The bottom right panel is the same height trace shown on the left but with a free hand line drawn through the nucleosomes to determine the height profile of the nucleosomes. D. Height profiles of the nucleosomes in the above image demonstrate that all nucleosomes range within 1.5-2.5 nm height as previously reported 40.
Figure 4. AFM. A. Sedimentation velocity profile for 601207 x 12 arrays assembled with mouse octamer indicating the average sedimentation coefficient is 27S. B. The same arrays in A were imaged by AFM and the number of nucleosomes counted in several images were plotted in MS Excel. The plot indicated that a majority of arrays had 10-11 nucleosomes corroborating the AUC data. C. A 500 x 500 nm scan of 601207 x 12 nucleosome arrays with the linker DNA clearly visible. The top right panel is the amplitude trace and the middle right panel is the phase trace for the array shown in C. The bottom right panel is the same height trace shown on the left but with a free hand line drawn through the nucleosomes to determine the height profile of the nucleosomes. D. Height profiles of the nucleosomes in the above image demonstrate that all nucleosomes range within 1.5-2.5 nm height as previously reported 40.
Discussion
Model nucleosomal arrays are a very useful tool for the in vitro study of chromatin structure and function. For example, they have been widely used to study the mechanism of chromatin fiber condensation in solution 30-34, and made it possible to obtain an x-ray structure of a tetranucleosome 35. More recently they have proven useful in deciphering the structural effects of specific core histone variants, mutants and posttranslational modifications14-16,36. Here we describe a general method for the assembly of model nucleosomal arrays from nucleosome positioning DNA and recombinant core histones.
The reconstitution of nucleosomal arrays from purified octamers and 601207 x 12 DNA is straightforward, involving several dialysis steps that sequentially lower the NaCl concentration from 2 M to 2.5 mM. The most difficult part of the protocol is to use an r-value that yields the desired template saturation level. The appropriate r-value is first determined empirically on a small scale and then repeated on a larger scale to generate the nucleosomal arrays used for experiments. In our case, we were attempting to obtain 601207 x 12 DNA templates mostly saturated with 12 nucleosomes per DNA. The sedimentation coefficient of a fully saturated 601207 x 12 nucleosomal array is 29 S, while the same DNA template with only 11 nucleosomes per DNA sediments at ~27S 29. Thus, sedimentation velocity in the analytical ultracentrifuge provides a very sensitive method for determining the nucleosome saturation level after reconstitution. We analyzed our data using the enhanced van Holde-Weischet method, which yields a diffusion-corrected sedimentation coefficient distribution. This information is essential because it tells one how homogeneous or heterogeneous the sample is after reconstitution. In other words, for a 601207 x 12 DNA template, it indicates the fraction of the sample that has 12 nucleosomes per DNA, 11 nucleosomes per DNA, etc. Figure 3 shows the results of the enhanced van Holde-Weischet analysis of 601207 x 12 nucleosomal arrays reconstituted at an r of 1.1, in which roughly 30% of the arrays are over saturated. The utility of the sample is usually tied to the percentage of saturated arrays, but is dependent on the experiments the arrays will be used in. Some applications may require very homogenous and/or saturated arrays. A number of methods exist for improving the homogeneity and saturation of nucleosomal arrays.
In this case the oversaturated arrays could be removed by selective precipitation upon the addition of MgCl237. More homogenous arrays can also be obtained by purifying the sample using sucrose gradient centrifugation, preparative gel electrophoresis, and ion exchange chromatography10,13. A modified nucleosomal array reconstitution method calls for adding short competitor DNA to the samples before reconstitution through salt dialysis38,39. This allows one to assemble nucleosomal arrays with an excess of histone octamer without over saturating the DNA template. Nucleosomal arrays reconstituted using competitor DNA will likely require a purification step for the removal of the competitor DNA and extra histones.
Even after small scale pilot array assemblies, it is possible that nucleosomal arrays reconstituted at a large scale will not be properly saturated. In order to avoid wasting the template DNA and histone octamer in the sample, it is possible to fix over or under saturated arrays. If the arrays are over saturated, extra DNA can be added to the sample. If the arrays are under saturated, extra octamer can be added to correct it. However, adding octamer to arrays already dialyzed into low salt can result octamer dissociation. If adding extra octamer or DNA to arrays, dialyze the arrays back into 2 M NaCl before adding extra octamer or DNA to the bulk sample. Repeat step dialysis for fixed samples from high to low salt as per step 3.4. The amount of octamer to add in order to correct the arrays can be estimated from sedimentation coefficients for under saturated arrays29. After adjusting the sample of nucleosomal arrays, measurements of saturation level should be made once again.
While AFM is a powerful method for characterizing nucleosomal arrays, it is complicated and a labor intensive process. This makes it a poor technique for screening small scale reconstitutions, but an excellent technique for the characterization of large-scale samples and for the study of chromatin organization. Previously we have used AFM to visualize "macro" particles generated by a macroH2A deletion construct that was "hyper-responsive" to even low MgCl2 concentrations. Likewise, Montel et al. (2009) have shown that H2A Bbd variant causes the nucleosome arrays to be more "open" compared to wild type arrays. Therefore, AFM is a reliable technique for quality control, as well as for the study of chromatin fiber structure in general.
Disclosures
The authors have no conflicts of interest.
Acknowledgments
This work was supported by NIH grants GM45916 and GM66834 to J.C.H. and a fellowship from the International Rett Syndrome Foundation to A.K. This work was also supported by NIH grant GM088409 and Howard Hughes Medical Institute contributions to K.L.
References
- Luger K, Mader A, Richmond R, Crystal Sargent D. structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;7 doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Hansen JC. Conformational dynamics of the chromatin fiber in solution: determinants, mechanisms, and functions. Annual Review of Biophysics and Biomolecular Structure. 2002;31:361–392. doi: 10.1146/annurev.biophys.31.101101.140858. [DOI] [PubMed] [Google Scholar]
- McBryant S, Adams V, Hansen J. Chromatin architectural proteins. Chromosome Research. 2006;14(1):39–51. doi: 10.1007/s10577-006-1025-x. [DOI] [PubMed] [Google Scholar]
- Hansen JC, Ausio J, Stanik VH, van Holde KE. Homogeneous reconstituted oligonucleosomes, evidence for salt-dependent folding in the absence of histone H1. Biochemistry. 1989;28(23):9129–9136. doi: 10.1021/bi00449a026. [DOI] [PubMed] [Google Scholar]
- Szerlong HJ, Prenni JE, Nyborg JK, Hansen JC. Activator-dependent p300 acetylation of chromatin in vitro: enhancement of transcription by disruption of repressive nucleosome-nucleosome interactions. The Journal of Biological Chemistry. 2010;285(42):31954–31964. doi: 10.1074/jbc.M110.148718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Molecular Cell. 2002;9(2):279–289. doi: 10.1016/s1097-2765(02)00459-8. [DOI] [PubMed] [Google Scholar]
- Nguyen C, Gonzales F. chromatin structure associated with methylation-induced gene silencing in cancer cells: correlation of accessibility, methylation, MeCP2 binding and acetylation. Nucleic Acids Research. 2001;29(22):4598–4606. doi: 10.1093/nar/29.22.4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuesta I, Zaret KS, Santisteban P. The forkhead factor FoxE1 binds to the thyroperoxidase promoter during thyroid cell differentiation and modifies compacted chromatin structure. Molecular and Cellular Biology. 2007;27(20):7302–7314. doi: 10.1128/MCB.00758-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson RT, Stafford DW. Structural features of a phased nucleosome core particle. Proceedings of the National Academy of Sciences of the U S A. 1983;80(1):51–55. doi: 10.1073/pnas.80.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. Journal of Molecular Biology. 1998;276(1):19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- Lowary P, Widlund H, Cao H. Sequence motifs and free energies of selected natural and non-natural nucleosome positioning DNA sequences. Journal of Molecular Biology. 1999;288:213–229. doi: 10.1006/jmbi.1999.2686. [DOI] [PubMed] [Google Scholar]
- Gordon F, Luger K, Hansen JC. The Core Histone N-terminal Tail Domains Function Independently and Additively during Salt-dependent Oligomerization of Nucleosomal Arrays *. The Journal of Biological Chemistry. 2005;280(40):33701–33706. doi: 10.1074/jbc.M507048200. [DOI] [PubMed] [Google Scholar]
- Luger K, Rechsteiner TJ, Richmond TJ. Expression and purification of recombinant histones and nucleosome reconstitution. Methods in Molecular Biology (Clifton, N.J.) 1999;119(4):1–16. doi: 10.1385/1-59259-681-9:1. [DOI] [PubMed] [Google Scholar]
- McBryant SJ, Klonoski J, et al. Determinants of histone H4 N-terminal domain function during nucleosomal array oligomerization: roles of amino acid sequence, domain length, and charge density. The Journal of Biological Chemistry. 2009;284(25):16716–16722. doi: 10.1074/jbc.M109.011288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Shogren-Knaak , Fry CJ, Peterson CL. A native peptide ligation strategy for deciphering nucleosomal histone modifications. The Journal of Biological Chemistry. 2003;278(18):15744–158. doi: 10.1074/jbc.M301445200. [DOI] [PubMed] [Google Scholar]
- Lu X, Simon M. The effect of H3K79 dimethylation and H4K20 trimethylation on nucleosome and chromatin structure. Nat Struct Mol Biol. 2008;15(10):1122–1124. doi: 10.1038/nsmb.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausio J. Analytical Ultracentrifugation for the Analysis of Chromatin Structure. Biophysical Chemistry. 2000;86(2-3):141–153. doi: 10.1016/s0301-4622(00)00144-7. [DOI] [PubMed] [Google Scholar]
- Hansen J, Kreider J, Demeler B, Fletcher T. Analytical ultracentrifugation and agarose gel electrophoresis as tools for studying chromatin folding in solution. Methods. 1997;12(1):62–72. doi: 10.1006/meth.1997.0448. [DOI] [PubMed] [Google Scholar]
- Montel F, Menoni H, et al. The dynamics of individual nucleosomes controls the chromatin condensation pathway: direct atomic force microscopy visualization of variant chromatin. Biophysical Journal. 2009;97(2):544–5453. doi: 10.1016/j.bpj.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthurajan UM, McBryant SJ, Lu X, Hansen JC, Luger K. The linker region of macroH2A promotes self-association of nucleosomal arrays. The Journal of Biological Chemistry. 2011;286(27):23852–23864. doi: 10.1074/jbc.M111.244871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyakhtenko LS, Lushnikov AY, Lyubchenko YL. Dynamics of nucleosomes revealed by time-lapse atomic force microscopy. Biochemistry. 2009;48(33):7842–7848. doi: 10.1021/bi900977t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr D, Bash R, Wang H, Yodh J, Lindsay S. Using atomic force microscopy to study chromatin structure and nucleosome remodeling. Methods (San Diego, Calif) 2007;41(3):333–341. doi: 10.1016/j.ymeth.2006.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer PN, Edayathumangalam RS, et al. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods in Enzymology. 2004;375:23–44. doi: 10.1016/s0076-6879(03)75002-2. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell D. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Balbo A, Zhao H, Brown PH, Schuck P. Assembly, loading, and alignment of an analytical ultracentrifuge sample cell. J. Vis. Exp. 2009. p. e1530. [DOI] [PMC free article] [PubMed]
- Demeler B. UltraScan: a comprehensive data analysis software package for analytical ultracentrifugation experiments. Modern Analytical Ultracentrifugation: Techniques. 2005. pp. 210–230.
- Holde KV, Weischet W. Boundary analysis of sedimentation velocity experiments with monodisperse and paucidisperse solutes. Biopolymers. 1978;17(6):1387–1403. [Google Scholar]
- Demeler B, van Holde KE. Sedimentation velocity analysis of highly heterogeneous systems. Analytical Biochemistry. 2004;335:279–288. doi: 10.1016/j.ab.2004.08.039. [DOI] [PubMed] [Google Scholar]
- Hansen J, Lohr D. Assembly and structural properties of subsaturated chromatin arrays. Journal of Biological Chemistry. 1993;8:5840–5848. [PubMed] [Google Scholar]
- Routh A, Sandin S, Rhodes D. Nucleosome repeat length and linker histone stoichiometry determine chromatin fiber structure. Proceedings of the National Academy of Sciences of the U S A. 2008;105(26):8872–8877. doi: 10.1073/pnas.0802336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Fan JY, Rangasamy D, Tremethick DJ. The nucleosome surface regulates chromatin compaction and couples it with transcriptional repression. Nature Structural & Molecular Biology. 2007;14(11):1070–1076. doi: 10.1038/nsmb1323. [DOI] [PubMed] [Google Scholar]
- Dorigo B, Schalch T, Kulangara A, Duda S, Schroeder RR, Richmond TJ. Nucleosome arrays reveal the two-start organization of the chromatin fiber. Science (New York, N.Y.) 2004;306(5701):1571–1573. doi: 10.1126/science.1103124. [DOI] [PubMed] [Google Scholar]
- Correll SJ, Schubert MH, Grigoryev S. a Short nucleosome repeats impose rotational modulations on chromatin fibre folding. The EMBO Journal. 2012;31(10):2416–2426. doi: 10.1038/emboj.2012.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcbryant SJ, Krause C, Woodcock CL, Hansen JC. The Silent Information Regulator 3 Protein , SIR3p , Binds to Chromatin Fibers and Assembles a Hypercondensed Chromatin Architecture in the Presence of Salt. Molecular and Cellular Biology. 2008;28(11):3563–3572. doi: 10.1128/MCB.01389-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalch T, Duda S, Sargent DF, Richmond TJ. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature. 2005;436(7047):138–1341. doi: 10.1038/nature03686. [DOI] [PubMed] [Google Scholar]
- Fan JY, Gordon F, Luger K, Hansen JC, Tremethick DJ. The essential histone variant H2A.Z regulates the equilibrium between different chromatin conformational states. Nature Structural Biology. 2002;9(3):172–176. doi: 10.1038/nsb767. [DOI] [PubMed] [Google Scholar]
- Carruthers LM, Bednar J, Woodcock CL, Hansen JC. Linker histones stabilize the intrinsic salt-dependent folding of nucleosomal arrays: mechanistic ramifications for higher-order chromatin folding. Biochemistry. 1998;37(42):14776–14787. doi: 10.1021/bi981684e. [DOI] [PubMed] [Google Scholar]
- Huynh VAT, Robinson PJJ, Rhodes D. A Method for the In Vitro Reconstitution of a Defined "30 nm" Chromatin Fibre Containing Stoichiometric Amounts of the Linker Histone. Journal of Molecular Biology. 2005;345(5):957–968. doi: 10.1016/j.jmb.2004.10.075. [DOI] [PubMed] [Google Scholar]
- Dorigo B, Schalch T. Chromatin fiber folding: requirement for the histone H4 N-terminal tail. J. Mol. Biol. 2003;2836(03):85–96. doi: 10.1016/s0022-2836(03)00025-1. [DOI] [PubMed] [Google Scholar]
- Qian RL, Liu ZX, et al. Visualization of chromatin folding patterns in chicken erythrocytes by atomic force microscopy (AFM. Cell Research. 1997;7(2):143–150. doi: 10.1038/cr.1997.15. [DOI] [PubMed] [Google Scholar]