

Abstract
Mauthner cells (M-cells) are large reticulospinal neurons located in the hindbrain of teleost fish. They are key neurons involved in a characteristic behavior known as the C-start or escape response that occurs when the organism perceives a threat. The M-cell has been extensively studied in adult goldfish where it has been shown to receive a wide range of excitatory, inhibitory and neuromodulatory signals1. We have been examining M-cell activity in embryonic zebrafish in order to study aspects of synaptic development in a vertebrate preparation. In the late 1990s Ali and colleagues developed a preparation for patch clamp recording from M-cells in zebrafish embryos, in which the CNS was largely intact2,3,4. The objective at that time was to record synaptic activity from hindbrain neurons, spinal cord neurons and trunk skeletal muscle while maintaining functional synaptic connections within an intact brain-spinal cord preparation. This preparation is still used in our laboratory today. To examine the mechanisms underlying developmental synaptic plasticity, we record excitatory (AMPA and NMDA-mediated)5,6 and inhibitory (GABA and glycine) synaptic currents from developing M-cells. Importantly, this unique preparation allows us to return to the same cell (M-cell) from preparation to preparation to carefully examine synaptic plasticity and neuro-development in an embryonic organism. The benefits provided by this preparation include 1) intact, functional synaptic connections onto the M-cell, 2) relatively inexpensive preparations, 3) a large supply of readily available embryos 4) the ability to return to the same cell type (i.e. M-cell) in every preparation, so that synaptic development at the level of an individual cell can be examined from fish to fish, and 5) imaging of whole preparations due to the transparent nature of the embryos.
Keywords: Neuroscience, Issue 79, Synapses, Zebrafish, Ligand-Gated Ion Channels, Neurosciences, Mauthner cells, reticulospinal neurons, Zebrafish, synapse, ion channels, AMPA receptors, NMDA receptors, action potentials, glycine receptors
Introduction
One of the outstanding questions in developmental neurobiology pertains to the role of synaptic plasticity phenomena during synaptic maturation. Our research focuses on understanding the plasticity mechanisms that underlie synaptic development with an overall goal of understanding animal behavior in the context of synaptic development. In order to do this, we developed a largely intact preparation in an organism (zebrafish, Danio rerio) that has gained considerable traction for developmental studies in the late 1990s.
The zebrafish offers several distinct advantages for neurodevelopmental studies. For instance, its genome is sequenced and one can perform morpholino knockdowns and zinc-finger nuclease knockouts to silence gene activity and protein expression. One can also perform overexpression studies and transgenic lines can be constructed7-9. Embryos are relatively easy and abundant to acquire, and the transparent nature of the embryo lends itself well to imaging and opto-genetic studies. Additionally, behavioral analysis can be performed, and electrophysiological experiments can be undertaken on muscle fibers, spinal cord neurons and hindbrain neurons in largely intact organisms, allowing one to examine cellular function in vivo. Importantly, we are able to examine synaptic activity in not just the same class of cell, but in the very same pair of neurons, from preparation to preparation. In other words, this is one of the rare vertebrate preparations in which one can return to the same neuron, in situ, to study its development.
In order to maintain neuronal circuitry as viable as possible, we developed a preparation in which the hindbrain and spinal cord were left intact, while patch clamping from the M-cells. In this preparation, the eyes, jaw and skin overlaying the brain are gently removed and the hindbrain is peeled away from the notochord, allowing access to the ventral surface of the hindbrain. The reticulospinal neurons are located a few micrometers deep and are relatively easily accessible. At 48 hr post fertilization (hpf; some abbreviations are listed in Table 1) the cells are electrically compact and one can faithfully record synaptic activity (both excitatory and inhibitory currents), voltage-gated currents and action potentials from them.
Our findings have suggested that many aspects of synaptic development and maturation occur in the 24 hr prior to hatching, and therefore much of our attention is focused on organisms ranging in age from 24 hpf to 72 hpf. However, by 72 hpf, M-cells are less electrically compact and are difficult to properly space clamp. The technique can be used to record not only from the M-cells, but also from its homologs, MiD2 cm and MiD3 cm. Theoretically, one may also perform double recordings, from a spinal cord neuron such as a primary motor neuron and from the M-cell, but this is difficult and it might be argued that the benefits gained from such a difficult technique may not be worth the extraordinary effort that is required.
Protocol
1. Preparation and Dissection
Prepare the dissecting dish lined with Sylgard. Recording chambers from WPI (Sarasota, Florida, USA; Suppliers are listed in Table 2) are used as dissecting dishes (Figure 1). The chambers are designed to accommodate glass slides and small plastic washers are used as molds for the Sylgard. The washers are first fixed to the slide with grease and liquid Sylgard is added to the center of the washer. The Sylgard will cure overnight and as it does so, it forms a small, circular bed on the glass slide, which becomes the base of the dissecting dish. The slide is placed into the recording chamber and serves as the base of the chamber to which the preparation is affixed (Figure 1).
Prepare the solutions listed in Table 3. Extracellular solutions should be left at room temperature while intracellular solutions should be kept sterile. One can also add Lucifer yellow (0.1%) to the intracellular solution so that visualization of M-cell morphology may be performed at the end of the experiment. This serves to confirm cell identity (Figure 2).
Raise wild type zebrafish embryos in egg water at 28.5 °C, and collect and stage according to Kimmel et al. 10. For dissections, transfer embryos to the dissecting dish that also serves as the recording chamber. Anesthetize for 7-10 min in an anesthetizing salt solution (0.02% tricaine; solution 1, Table 3) that closely approximates physiological saline. One can determine if they are adequately anesthetized by examining the response to a gentle tail pinch.
Pin the embryos through the notochord and onto the Sylgard-lined dish with 0.001" tungsten wire (Scientific Instrument Services. Inc., Ringoes, NJ, USA) using 25X magnification on a dissecting microscope (Figure 1). The tungsten wire is easily cut into small segments with fine dissecting scissors.
Adjust the magnification on the dissecting scope to 40-50X to perform the dissection. Once the embryo is pinned to the dish, the jaw, eyes and forebrain are removed using a fine pair of forceps (Dumont #5).
The hindbrain is gently peeled away from the underlying notochord by carefully working the forceps between the notochord and the brain. The hindbrain is then gently turned over so that the ventral surface faces up. This positioning provides good access to the M-cells, which are usually within 5-10 μm of the ventral surface in young embryos and ~20-50 μm in older larvae (Figure 2A).
Carefully move the preparation to the recording setup where the patch clamp experiment can be performed. The M-cells are visualized with Nomarski Differential Interference Contrast (DIC), although other modes of imaging (i.e. Hoffman modulation contrast or Dodt Gradient Contrast Imaging from Luigs and Neumann, Germany) may also be used.
2. Patch Clamp Recording (Whole Cell Mode)
All recordings are performed in the whole cell patch clamp mode11. Recording chambers are placed onto a microscope stage in an electrophysiology setup. Our stages are fixed and the upright patch clamp microscope is bolted to a movable microscope platform or a microscope translator.
Patch clamp pipettes are pulled on a horizontal puller (P-97; Sutter Instruments Co.) from thin-walled, borosilicate glass obtained from WPI. Pipette tip diameters are on the order of 0.2-0.4 μm after fire polishing to a smooth edge, and the shank taper is ~4 mm in length.
Attach the pipette to the amplifier head-stage in the electrophysiology setup. It is important to keep the head-stage at roughly 45° angle to the horizontal axis as this ensures an entry angle for the pipette that is suitable for the formation of high resistance seals onto the Mauthner cell.
Just before entering the bath solution, apply a small amount of positive pressure to the pipette to reduce the chance of dirtying the tip. When recording mEPSCs, bath solutions include 1 μM TTX to block action potentials, 5 μM strychnine to block glycine receptors and 10 μM bicuculline or 100 μM picrotoxin to block GABAA receptors. When recording mIPSCs, bath solutions include 1 μM TTX, kynurenic acid and either bicuculline or strychnine depending on the receptor activity of interest.
Approach the M-cell with a small amount of positive pressure in the pipette. The positive pressure gently pushes the cell from side to side and when positioned immediately over the cell, forms a small dimple on the cell membrane. Leave the pipette in place for a few seconds to gently clean the cell surface so that a strong seal between the pipette and the membrane can be formed. Releasing the positive pressure in the pipette allows the seal to be initiated and a small amount of negative pressure coupled with negative pipette potentials results in GigaΩ seals forming within a few seconds.
Change the holding potential on the amplifier to -60 mV. Rupture the cell membrane with a series of short pulses of suction. Immediately record membrane potentials and minimize capacitance artifacts. Compensate cell capacitance (Cm) and access (series) resistance (Ra) by 70-85%. Ra should be routinely monitored, every 30 sec to a minute, and if there is a change of 20% or more, abort the experiment.
Once the experiment has ended and enough data has been acquired, the preparation is sacrificed by removing the hindbrain with a pair of forceps. At this point, data analysis can begin.
Representative Results
In this manuscript we present a method for patch-clamp recording in the whole-cell mode from reticulospinal neurons in zebrafish, particularly focusing on the M-cell. Of course, it is also possible to record in other patch clamp modes such as cell-attached, inside-out and outside out. The type of data that one can obtain is not limited to what we have presented here, but we try to provide an example of both synaptic activity (excitatory and inhibitory) in voltage-clamp mode as well as action potentials in current clamp. As the organism ages and the M-cells develop more extensive and elaborate dendrites, the ability to adequately voltage clamp and space clamp the cell is compromised and the current clamp mode becomes the recording of choice.
Figure 3 shows representative recordings of excitatory AMPA and NMDA receptor currents obtained from 48 hpf embryos in the presence of TTX (1 μM) and pharmacological agents to block GABA and glycine receptors. When the M-cells are voltage clamped at -60 mV, the synaptic currents are due to the activation of AMPA receptors (Figure 3A). Acquisition and analysis of these events allows one to record parameters such as rise time, decay time, amplitude and frequency. AMPA events typically have a fast rise time (20-80% rise time of ~0.1 msec) and decay time (~0.5 msec), and exhibit an average amplitude of approximately 25 pA. When the M-cells are clamped at +40 mV, the resulting outward currents are due to AMPA and NMDA receptor activity (Figures 3C and 3D). NMDA receptor currents exhibit longer time courses than AMPA receptor currents, and need to be recorded at positive potentials or in Mg-free solution to remove the Mg block of the NMDA receptors2,6. The average mEPSC shown in Figure 3D represents mixed AMPA and NMDA currents recorded at +40 mV.
Figure 4 shows representative results when recording mIPSCs from Mauthner cells. These events occurred in the presence of TTX (1 μM) and kynurenic acid (1 mM) to block AMPA and NMDA receptors. These events exhibit reasonably fast rise and decay kinetics by 48 hpf and are frequent enough to acquire many of them over a 2-3 min recording3. Figure 5 shows action potentials recorded as a result of injecting depolarizing current into the cell. M-cells of 48 hpf embryos usually fire a single action potential (Figure 5B), although they sometimes fire multiple APs when injected with suprathreshold stimuli (Figure 5A).
| Abbreviation | Full Name | |
| 1. | mPSC | Miniature postsynaptic current |
| 2. | mEPSC | Miniature excitatory postsynaptic current |
| 2 | mIPSC | Miniature inhibitory postsynaptic current |
| 3. | MΩ | Mega Ohm (unit of resistance; 106 Ohm) |
| 4. | GΩ | Giga Ohm (unit of resistance; 109 Ohm) |
| 4. | MOsm | Milli osmolar (unit of osmolarity; 10-3 osmoles) |
| 4. | TTX | tetrodotoxin |
| 5. | ATP | adenosine triphosphate |
| 6. | GTP | guanosine triphosphate |
| 7. | GFP | Green fluorescent protein |
Table 1.
| Equipment Name | Company | Catalogue Number | |
| 1. | Dissecting Dish | Warner Instruments | 64-0291 |
| 2. | 0.001" Tungsten Wire | Scientific Instruments Services Inc. | W406 |
| 2 | Dissecting microscope | Leica Microsystems | MZ9.5 |
| 3. | Pipette Puller | Sutter Instruments Co. | P-97 |
| 4. | Microforge | Narishige Group | MF-900 |
| 5. | Upright Patch clamp microscope | Leica Microsystems | DMLFSA |
| 6. | Micromanipulator | Siskiyou Inc. | MX7500 |
| 7. | Digidata | Molecular Devices | 1322A |
| 8. | Amplifier | Molecular Devices | 200B |
| 9. | Acquisition software | Molecular Devices | pClamp9 |
| 10. | Axograph X | Axograph | Axograph X |
Table 2.
| Solution Name | Reagent and concentration (mM) | |
| 1. | Dissecting solution | 134 NaCl, 2.9 KCl, 2.1 CaCl2, 1.2 MgCl2, 10 HEPES, 10 glucose and 0.02% Tricaine |
| 2. | mPSC Extracellular solution | 134 NaCl, 2.9 KCl, 2.1 CaCl2, 1.2 MgCl2, 10 HEPES, 10 glucose and 0.001 TTX |
| 3. | mPSC Intracellular solution | 134 CsCl, 2 MgCl2, 10 HEPES, 10 EGTA, 4 Na2-ATP, 0.4 Li-GTP |
| 4. | Action potential Extracellular solution | 137 NaCl, 2.9 KCl, 2.1 CaCl2, 1.2 MgCl2, 10 HEPES and 10 Glucose (with 10 μM D-tubocurarine) |
| 5. | Action potential Intracellular solution | 130 KCl, 2 NaCl, 10 EGTA, 10 HEPES, 4Mg-ATP and 0.4 Li-GTP. |
Table 3. All extracellular solutions are adjusted to 290 mOsm and pH 7.8. All intracellular solutions are adjusted to 280 mOsm and pH 7.4.
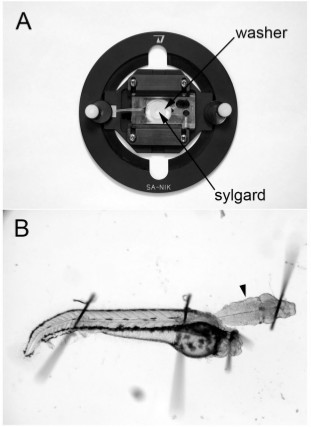 Figure 1. Dissecting dish and intact hindbrain-spinal cord preparation. (A) Dissecting and recording dish obtained from WPI. A thin washer is carefully sealed onto the glass slide at the bottom of the dish and Sylgard is applied to the well. (B) 48 hpf hindbrain-spinal cord preparation. The ventral surface of the hindbrain is facing up and the M-cell is located just beneath the surface of the tissue at the level of the arrowhead.
Figure 1. Dissecting dish and intact hindbrain-spinal cord preparation. (A) Dissecting and recording dish obtained from WPI. A thin washer is carefully sealed onto the glass slide at the bottom of the dish and Sylgard is applied to the well. (B) 48 hpf hindbrain-spinal cord preparation. The ventral surface of the hindbrain is facing up and the M-cell is located just beneath the surface of the tissue at the level of the arrowhead.
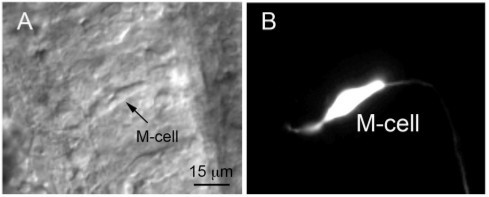 Figure 2. (A) Nomarski differential interference contrast image of a M-cell obtained from a 2 dpf embryo shortly after hatching. The image was taken after recording spontaneous excitatory synaptic activity from the M-cell. (B) The cell was filled with Lucifer yellow during the experiment. Adapted from12 with permission.
Figure 2. (A) Nomarski differential interference contrast image of a M-cell obtained from a 2 dpf embryo shortly after hatching. The image was taken after recording spontaneous excitatory synaptic activity from the M-cell. (B) The cell was filled with Lucifer yellow during the experiment. Adapted from12 with permission.
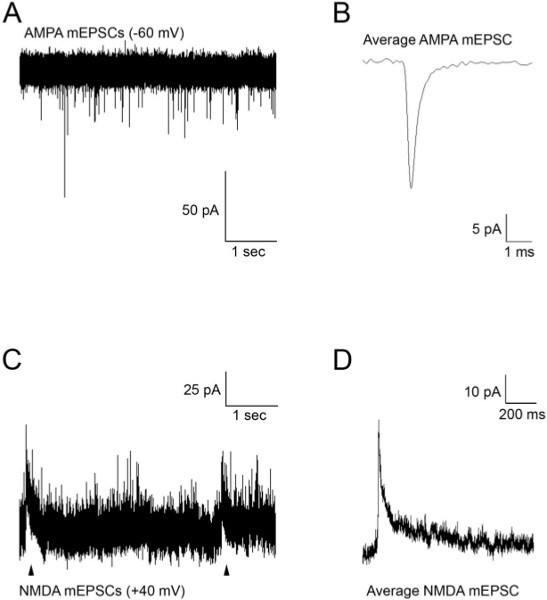 Figure 3. (A) Spontaneous AMPA mEPSCs recorded from an M-cell at a holding potential of -60 mV. (B) Average mEPSCs obtained from the recording in (A). (C) Spontaneous AMPA and NMDA (arrowheads) mEPSCs recorded from an M-cell at a holding potential of +40 mV. (D) Average mEPSCs obtained from the recording in (C). Glutamate mEPSCs were recorded in the presence of TTX (1 μM) to block action potentials, strychnine (5 μM) and picrotoxin (100 μM) to block glycine and GABA currents.
Figure 3. (A) Spontaneous AMPA mEPSCs recorded from an M-cell at a holding potential of -60 mV. (B) Average mEPSCs obtained from the recording in (A). (C) Spontaneous AMPA and NMDA (arrowheads) mEPSCs recorded from an M-cell at a holding potential of +40 mV. (D) Average mEPSCs obtained from the recording in (C). Glutamate mEPSCs were recorded in the presence of TTX (1 μM) to block action potentials, strychnine (5 μM) and picrotoxin (100 μM) to block glycine and GABA currents.
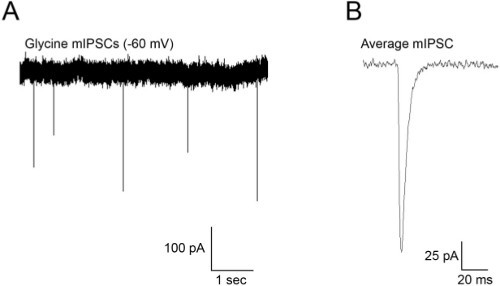 Figure 4. (A) Spontaneous mIPSCs recorded from an M-cell at a holding potential of -60 mV. (B) Average mEPSCs obtained from the recording in (A). mIPSCs were recorded in the presence of TTX (1 μM) to block action potentials, kynurenic acid (1 mM) to block glutamate receptor activity and bicuculline (10 μM) to block GABA currents.
Figure 4. (A) Spontaneous mIPSCs recorded from an M-cell at a holding potential of -60 mV. (B) Average mEPSCs obtained from the recording in (A). mIPSCs were recorded in the presence of TTX (1 μM) to block action potentials, kynurenic acid (1 mM) to block glutamate receptor activity and bicuculline (10 μM) to block GABA currents.
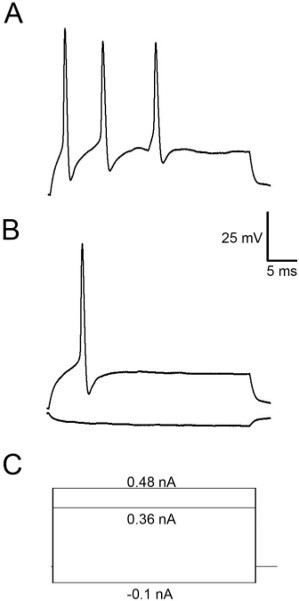 Figure 5.Action potentials recorded from an M-cell. In this particular cell a suprathreshold stimulus (0.48 nA) elicited several action potentials (A), but a stimulus to threshold only results in the production of a single action potential (B). (C) Current protocols for the traces shown in A and B.
Figure 5.Action potentials recorded from an M-cell. In this particular cell a suprathreshold stimulus (0.48 nA) elicited several action potentials (A), but a stimulus to threshold only results in the production of a single action potential (B). (C) Current protocols for the traces shown in A and B.
Discussion
The protocol described above allows one to patch clamp record from the M-cells and its homologs. The technique is challenging and care should be taken in several key areas throughout the procedure. We will now discuss some of these areas and will offer helpful hints to ensure success.
Construction of the dissecting/recording chamber is a critical aspect of the experiment. The M-cells are difficult to see unless differential interference contrast is used. Nomarski DIC works best with clean glass coverslips, but we have found that a very thin layer of Sylgard (~1-2 mm) will not distort the DIC to such an extent that it interferes with identification of the M-cells. Therefore, we use a recording chamber that has a thin glass coverslip on the bottom upon which is placed a small plastic washer (~5 mm diameter). We mix Sylgard 184 in a petri dish and carefully add the liquid Sylgard to the inside of the washer with a Pasteur pipette. The Sylgard can be cured at room temperature over a couple of days or can be heated for rapid curing. The washer may be removed after the Sylgard has cured, although this is not necessary. Once the dissection/recording chamber has been made, it can be used for several weeks before a fresh Sylgard-lined glass slide is needed.
The next step in the procedure is to make up all of the relevant solutions on the day of the experiment (Table 3). Intracellular solutions containing everything but ATP and GTP are made in advance and stored in 5 ml aliquots at -20 °C. On the day of recording an aliquot is thawed to room temperature and fresh ATP and GTP is added. The solution is adjusted to 280 mOsm and pH 7.4. In our hands, we find that addition of fresh ATP and GTP on a daily basis results in good recordings. All solutions need to be kept sterile for the best chance of success.
Intracellular solutions must be filtered through a 0.22 μm filter (Sigma) to remove small particles and debris that might affect the recording. To do this, we first draw the intracellular solution into a 1 ml syringe, affix the filter to the end of the syringe and add a plastic pipette tip that was heated and pulled to fine tip, to the end of the filter. This allows us to apply the filtered intracellular solution directly to the inside of the glass patch pipettes.
Once the solutions are prepared, the embryo can be anesthetized and dissected. Prior to the dissection, care should be taken to adequately assess the age of the organism. Each embryo within a clutch ages at a slightly different rate. Therefore when assessing the age of the organism it is important to do so according to established procedures 10,13, by using phenotypical cues such as the number of somites, the overall shape of the body and the time of egg fertilization. All dissections are performed with a Dumont #5 fine pair of forceps. These need to be sharp and in good working condition otherwise it will be very difficult to remove the skin overlaying the hindbrain and any connective tissue on the ventral surface of the hindbrain. We often have to sharpen the forceps after a few weeks of use, and do this ourselves with a sharpening stone.
To pin the fish to the thin layer of sylgard, we use pins fashioned from fine tungsten wire 0.001 inches in diameter. The pins are cut with an old, micro-dissection scissors under a dissecting microscope, and are small enough to fit right through the notochord. Pinning the embryos at any other point does not immobilize them and makes dissections almost impossible to perform. When first learning to perform the recordings, it can be difficult to identity the M-cell from its homologs (particularly MiD2 cm which is located in the neighboring rhombomere - rhombomere 5). In some embryos these two pairs of cells appear similar in size. The otoliths provide a convenient landmark to aid in the identification of M-cells. At 48 hpf the M-cells are located right next to the otoliths, and even though the otoliths are removed during the dissection, they leave behind a slightly damaged lateral region of the hindbrain, which serves as a marker for their original position. Transgenic fish that express GFP or some other fluorescent marker in the M-cell, provide excellent preparations for new experimenters. In our hands the M-cells have a membrane capacitance of approximately 18-24 pF at 48 hpf, while the smaller homologs are closer to 12-14 pF. Cells with greater surface area have larger membrane capacitance values (measured in pico Farads (pF)) compared with smaller cells. Thus, membrane capacitance can be used as a rough indicator of cell size, where larger cells have larger capacitance values. This electrophysiological characteristic serves as another property by which the M-cell can be identified from its homologs. Finally, we often use Lucifer yellow in our recording (intracellular) solutions, which allows us to conduct a firm identification of the cell type under investigation using fluorescence imaging once recordings are complete.
Pipette tip resistances in the range of 3.5-4.5 MΩ are the best compromise between tip size and low access resistance, which typically ranges from 8 to 15 MΩ. This is particularly important for events with fast kinetics; ~0.1 msec (20-80%) rise times and ~0.5 msec exponential decays 12,14,15 (Figures 3A and 3B). Data is acquired at a digitization rate of 50-100 kHz and filtered with a low pass frequency of 5-10 KHz. With regard to the patch pipettes, we have found that they do not need to be fire-polished to obtain a recording, but anecdotally, it appears to offer slightly better chance of obtaining good seals compared with unpolished pipettes. Since the pipette tips are relatively large, we do not add very much positive pressure to the pipette before entering the solution. It takes practice deciding upon the appropriate amount of positive pressure to apply as an excess of pressure will move the cell too much, and will prevent seal formation. It may also damage synaptic contacts as the cell is gently displaced from its original position. On the other hand, too little pressure results in dirty tips and does not clean the surface of the cell well enough to form good seals. A happy medium is only attained by significant practice and experience. Seal formation can be enhanced with applications of negative voltages to the pipette. We typically have seals of 1.2-2 GΩ, which are excellent for breakthroughs into the whole cell mode.
When pharmacological agents need to be applied to the cell cytoplasm, we include them in the intracellular solution prior to filling the pipette. Care should be taken when preparing the pipette solutions to avoid loss of the agents by having it bind to the 0.22 μm filter. Therefore, we filter the intracellular medium first, and then carefully add the pharmacological agent to the filtered solution. Application of compounds to the intracellular compartment has its own unique set of challenges. For instance, addition of drugs to the intracellular medium usually reduces the chances of forming a GΩ seal, but not to the point where it is a major hindrance to the experiment.
One should realize that application of pharmacological agents in this manner does not allow the experimenter to record the most appropriate control since the agent has immediate access to the cell from the start of the whole cell configuration. Therefore, while we record data throughout this type of experiment, one must be aware that the next best control is the intracellular application of an inactive form of the agent. In many instances this takes the form of a heat-inactivated compound, a scrambled peptide or an inactive isomer obtained from the supplier.
We acquire data in the form of synaptic currents (mPSCs), voltage-gated currents and action potentials. Miniature currents may be analyzed by a few excellent programs (pClamp, Axograph, etc.), although we prefer to use Axograph X (John Clements). Axograph X runs on both Windows and Macintosh platforms and can be used for both the acquisition and analysis of electrophysiological data. mEPCs are acquired with the aid of a template of the experimenter's choice, obtained from the recording itself. Individual events can be analyzed for properties such as rise time, amplitude, half width and decay time, etc. We export the spreadsheet of data to Kaleidagraph (Synergy Software)where we can produce figures, including copies of event traces from Axograph X.
One of the more difficult aspects of the experiment is determining whether the recording was performed well enough to provide clean, reliable data. For instance, space-clamping issues may arise when recording mPSCs from M-cells that have large axons and extensive dendritic processes. When voltage clamping, one can generally control the voltage at the tip of the pipette reasonably well (especially if the access resistance (Ra) is low), but may not have proper control of the membrane potential in dendritic processes or axons that are far away from the pipette tip. One method of determining if the cell was properly space clamped is to produce a scatter plot of the rise time vs the amplitude of the mPSCs that were recorded. A correlation between the data is suggestive of inadequate space clamping. In other words, if the space clamp is poor then mPSCs that occur close to the pipette will have quicker rise times and greater amplitudes than events occurring some distance away from the pipette. If a correlation does exist, then the data is problematic and cannot be used.
Another aspect of electrophysiological recordings that needs to be addressed is that of leak currents. This is especially true when voltage clamping a cell in order to record voltage-gated currents such as Na+ or K+ currents. When the membrane potential is deviated from rest, leak currents can be recorded along with voltage-gated activity. Leak currents (IL), a combination of potassium (IK), sodium (INa) and chloride (ICl) current through non-voltage-gated channels will obscure the activity of interest and must be subtracted from the recording. The amplifier is capable of performing this function online during the recording by running what is typically called a P/N protocol. During a P/N protocol the amplifier will run several small depolarizations (or hyperpolarizations) from rest and will record the resulting leak current that occurs. It is important to use small enough pulses that do not activate voltage-gated channels. The currents that occur during these pulses are recorded and averaged so that these leak currents can be subtracted from the voltage-gated channel activity.
Lastly, one needs to take into account the junction potential, which occurs at the tip of the electrode between the intracellular and extracellular solutions. Intracellular and extracellular solutions are typically composed of different ions, with differing activities and mobilities. Larger ions with lower mobilities will offer a greater resistivity to ion movement than smaller ones and this can result in a small but significant potential difference at the junction of the solutions. This junction potential must be taken into account when determining the appropriate voltages experienced by the cell.
The technique presented here is one used by our lab for many years. However, there are alternative methods of recording from the M-cell. Most notably, Joseph Fetcho and colleagues have developed an excellent preparation in which one can record from the M-cells of older larvae (4-5 days old) in a less invasive manner than presented here, where the M-cell is approached from the dorsal surface16. We had tried a similar technique but found it easier to approach the M-cell from the ventral side of the hindbrain, where the cell is closer to the surface of the brain, particularly in young fish (i.e. 24 hpf to 72 hpf). Furthermore, an approach from the dorsal surface usually requires the use of a transgenic line that expresses a fluorescent tag in the M-cell, which has the potential to introduce additional variables into the recording. This issue is not present when using wild type fish. However, with the technique presented here, we are limited to studying younger animals (24 hpf to 72 hpf). Thus, each approach has its benefits and limitations.
Student's t-test can be used to compare two groups of data while an analysis of variance (ANOVA) can be used to compare multiple groups. Care must be taken to choose the appropriate post-hoc test for parametric vs non-parametric data.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
This work was supported by grants from the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Canadian Foundation for Innovation (CFI) to DWA.
References
- Korn H, Faber DS. The Mauthner cell half a century later: a neurobiological model for decision-making. Neuron. 2005;47:13–28. doi: 10.1016/j.neuron.2005.05.019. [DOI] [PubMed] [Google Scholar]
- Ali DW, Buss RR, Drapeau P. Properties of miniature glutamatergic EPSCs in neurons of the locomotor regions of the developing zebrafish. J. Neurophysiol. 2000;83:181–191. doi: 10.1152/jn.2000.83.1.181. [DOI] [PubMed] [Google Scholar]
- Ali DW, Drapeau P, Legendre P. Development of spontaneous glycinergic currents in the Mauthner neuron of the zebrafish embryo. J. Neurophysiol. 2000;84:1726–1736. doi: 10.1152/jn.2000.84.4.1726. [DOI] [PubMed] [Google Scholar]
- Drapeau P, Ali DW, Buss RR, Saint-Amant L. In vivo recording from identifiable neurons of the locomotor network in the developing zebrafish. J. Neurosci. Methods. 1999;88:1–13. doi: 10.1016/s0165-0270(99)00008-4. [DOI] [PubMed] [Google Scholar]
- Patten SA, Ali DW. AMPA receptors associated with zebrafish Mauthner cells switch subunits during development. J. Physiol. 2007;581:1043–1056. doi: 10.1113/jphysiol.2007.129999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patten SA, Ali DW. PKCgamma-induced trafficking of AMPA receptors in embryonic zebrafish depends on NSF and PICK1. Proc. Natl. Acad. Sci. U.S .A. 2009;106:6796–6801. doi: 10.1073/pnas.0811171106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekker SC. Morphants: a new systematic vertebrate functional genomics approach. Yeast. 2000;17:302–306. doi: 10.1002/1097-0061(200012)17:4<302::AID-YEA53>3.0.CO;2-#. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasevicius A, Ekker SC. Effective targeted gene 'knockdown' in zebrafish. Nat. Genet. 2000;26:216–220. doi: 10.1038/79951. [DOI] [PubMed] [Google Scholar]
- Tallafuss A, et al. Turning gene function ON and OFF using sense and antisense photo-morpholinos in zebrafish. Development. 2012;139:1691–1699. doi: 10.1242/dev.072702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Archiv : European Journal of Physiology. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Patten SA, Ali DW. AMPA receptors associated with zebrafish Mauthner cells switch subunits during development. J. Physiol. 2007;581:1043–1056. doi: 10.1113/jphysiol.2007.129999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M. The Zebrafish Book: A Guide for the Laboratory use of Zebrafish (Danio rerio) 5. Vol. 1. University of oregon Press; 2007. [Google Scholar]
- Patten SA, Ali DW. PKCgamma-induced trafficking of AMPA receptors in embryonic zebrafish depends on NSF and PICK1. Proc. Natl. Acad. Sci. U.S.A. 2009;106:6796–6801. doi: 10.1073/pnas.0811171106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patten SA, Roy B, Cunningham ME, Stafford JL, Ali DW. Protein kinase Cgamma is a signaling molecule required for the developmental speeding of alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptor kinetics. The European Journal of Neuroscience. 2010;31:1561–1573. doi: 10.1111/j.1460-9568.2010.07216.x. [DOI] [PubMed] [Google Scholar]
- Kinkhabwala A, et al. A structural and functional ground plan for neurons in the hindbrain of zebrafish. Proc. Natl. Acad. Sci. U.S.A. 2011;108:1164–1169. doi: 10.1073/pnas.1012185108. [DOI] [PMC free article] [PubMed] [Google Scholar]