

Significance
Retroviruses, such as HIV, are important pathogens of vertebrates, including humans. They are capable of crossing species barriers to infect new hosts, but knowledge about the evolutionary history of retroviruses is limited. However, genomic traces of past retrovirus activities known as “endogenous retroviruses” can be screened from sequenced genomes and analyzed to improve understanding of retrovirus evolution. Here we use a unique approach to address the evolution of one group of retroviruses in a screen of 60 diverse vertebrate host genomes. We find evidence of rampant host-switching across mammalian orders by members of this group throughout their evolutionary history. We also find evidence that the spread of infective retroviruses from this group may be facilitated by rats.
Abstract
Genomic data provide an excellent resource to improve understanding of retrovirus evolution and the complex relationships among viruses and their hosts. In conjunction with broad-scale in silico screening of vertebrate genomes, this resource offers an opportunity to complement data on the evolution and frequency of past retroviral spread and so evaluate future risks and limitations for horizontal transmission between different host species. Here, we develop a methodology for extracting phylogenetic signal from large endogenous retrovirus (ERV) datasets by collapsing information to facilitate broad-scale phylogenomics across a wide sample of hosts. Starting with nearly 90,000 ERVs from 60 vertebrate host genomes, we construct phylogenetic hypotheses and draw inferences regarding the designation, host distribution, origin, and transmission of the Gammaretrovirus genus and associated class I ERVs. Our results uncover remarkable depths in retroviral sequence diversity, supported within a phylogenetic context. This finding suggests that current infectious exogenous retrovirus diversity may be underestimated, adding credence to the possibility that many additional exogenous retroviruses may remain to be discovered in vertebrate taxa. We demonstrate a history of frequent horizontal interorder transmissions from a rodent reservoir and suggest that rats may have acted as important overlooked facilitators of gammaretrovirus spread across diverse mammalian hosts. Together, these results demonstrate the promise of the methodology used here to analyze large ERV datasets and improve understanding of retroviral evolution and diversity for utilization in wider applications.
Retroviruses typically infect somatic cells and must integrate into the host genome to produce new viruses. When a germ-line cell is infected, an integrated provirus may be passed on to the host organism’s offspring. Thus, for millions of years retroviruses have colonized vertebrates, leaving traces in their genetic makeup as endogenous retroviruses (ERVs) (1, 2). These genetic traces of past retroviral activity provide unique opportunities to study the biology and evolution of viruses and hosts. Although known exogenous retrovirus (XRV) diversity is low, with just 53 species described among vertebrates (3), ERVs are highly diverse, adding value to their use as a resource with which to study retroviral evolution (4, 5). For this purpose, the growing catalog of reference genome assemblies permits detailed ERV phylogenomic studies across the genomes of diverse host species.
The Gammaretrovirus genus is one of seven genera that collectively constitute the Retroviridae (3). ERVs have been divided into classes, and gammaretroviral XRVs, such as murine leukemia virus (MLV), cluster with class I ERVs in phylogenetic analyses (ref. 4 and references therein) (Fig. 1). Historically, gammaretroviruses have attracted significant attention due to their occurrence in several vertebrates being linked with disease symptoms such as malignancies, immunosuppression, and neurological disorders (6). Interest has been stimulated further by concerns regarding the possibility of cross-species transmission (7, 8), particularly to humans via xenotransplantation (9) and by subsequently disputed hypotheses that a novel gammaretrovirus related to MLV was the causative agent of human disease (10). The potential of gammaretroviruses to switch host species is indicated both by close phylogenetic relationship between viruses in distantly related host taxa (7) and by experimental cross-species infections performed in vitro and in vivo (11–13). However, despite the interest surrounding gammaretroviruses, little is known about their evolutionary relationships across host species.
Fig. 1.
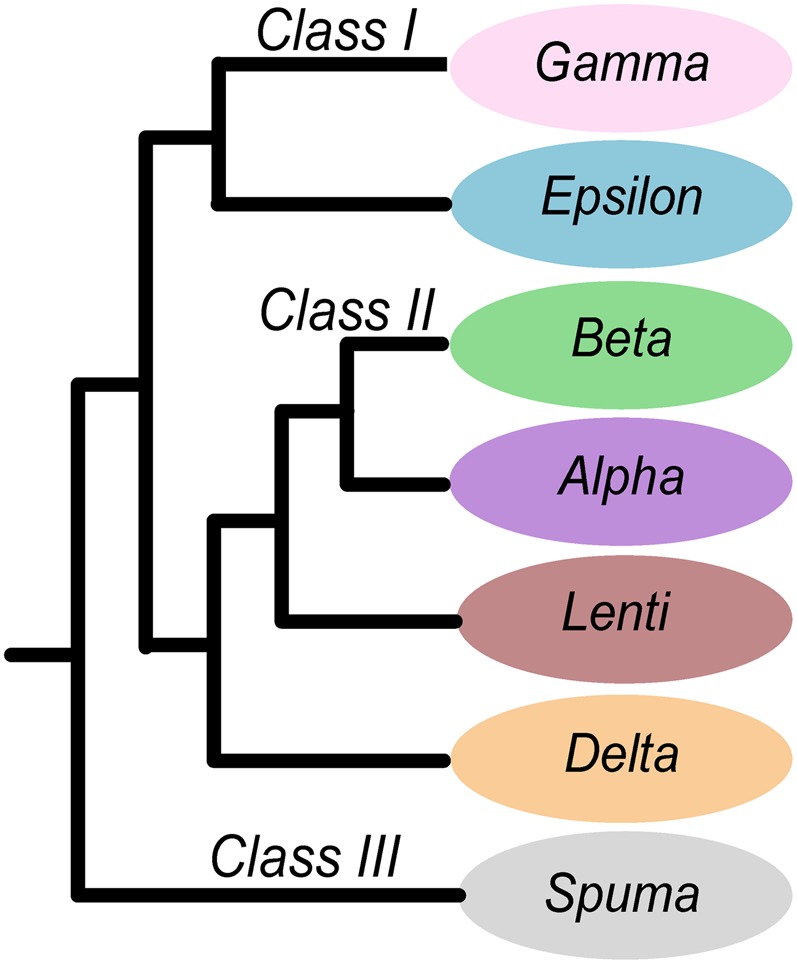
Retrovirus genera and ERV classes. Schematic illustrates the relationship among retroviral sequences, as recovered in Fig. S1. ERV classes associated with retroviral genera are indicated on branches.
The advent of improved sequencing technologies has facilitated in silico screening of ERVs from complete and near-complete host genomes using software such as RepeatMasker (http://repeatmasker.org) and the more specialized RetroTector (14), which can detect single or low-copy-number ERVs and attempts retroviral protein recontruction with collected data accessible for downstream analyses. Consequently, it is now feasible to examine broad-scale patterns in retrovirus evolution by using XRV and ERV sequences from a wide range of host taxa in phylogenomic analyses. Such analyses permit inferences of varied questions—from basic retroviral taxonomy to patterns of cross-species transfer—and thus open new avenues in the study of retrovirus biology to improve understanding of retrovirus evolution and spread. However, significant barriers to the use of retroviral data in phylogenetic analyses remain. Specifically, retroviral sequences are short (∼10 kb) and can transmit as either infectious units or genomic ERVs. Consequently, the selective pressures to which XRVs and ERVs are exposed differ considerably. XRVs are subject to rapid evolution arising from selection on infectious ability, whereas ERVs evolve under much slower postintegration host genomic mutation rates (4, 15). These differences can lead to considerable complications when establishing hypotheses of homology from sequence comparisons. Furthermore, the sheer volume of ERV sequences in vertebrate genomes presents a challenge to data assessment.
Here we use a phylogenetic methodology designed by us to reduce the impact of the above problems, and we perform a broad-scale phylogenomic analysis of gammaretroviruses and associated class I ERVs. For this analysis, we screen 60 currently available vertebrate genomes, sampled from across vertebrate diversity (Table S1). We use the resultant ERVs to address several issues of current importance in gammaretrovirus biology and draw unique insights in gammaretrovirus–host evolution. As an example, we focus on koala retrovirus (KoRV), which is widespread among wild and captive koala populations and is linked with disease symptoms (16, 17). The source of the infection of koalas with KoRV is unknown, but KoRV is hypothesized to have arisen as a consequence of a recent cross-species transmission event during the last 200 y, possibly from Asian mice (7, 18). We include sequences from these taxa and examine this hypothesis.
Results and Discussion
ERV Detection and Designation.
In silico screening of 60 vertebrate genomes recovered 87,750 ERVs meeting the baseline quality threshold of 300 in RetroTector (14) (Table S1). ERVs are frequently truncated from postintegrational rearrangements in the host genome, and the resulting missing data can present a major difficulty in multiple sequence comparisons. After exclusion of sequences with a high proportion of missing data, the number of ERVs was reduced to 36,765 for downstream analyses, including 11,922 class I ERVs. Here, we define class I ERVs phylogenetically as those forming a well-supported [1.00 Shimodaira–Hasegawa (SH)-like local support values using the SH test (19)] sister clade to the genus Epsilonretrovirus (Fig. S1, I, bottom). Within the class I clade, we define gammaretroviruses as those forming a well-supported (0.89 SH-like) monophyletic clade containing the known infectious gammaretrovirus reference sequences (Fig. S1, V). The gamma clade contains 3,653 ERVs and shares a sister-group relationship (1.00 SH-like) with a well-supported (1.00 SH-like) major class I monophyletic clade composed of ERVs closely related to porcine ERV-E (PERV-E), human ERV-E (HERV-E), HERV-R, and retinoic acid responsive human ERV-I (RRHERV-I) (Fig. S1, IV), strengthening the class I designation.
Great apes and rodents dominate in terms of number of ERVs detected per host taxon above the RetroTector quality threshold (Table S1), together making up 37.2% (32,682 ERVs) of the total despite representing just 16.7% of the host taxa (10 genomes). However, several outlier taxa are notable for hosting high ERV numbers, including the hedgehog (4,440 ERVs or 5.1% of total), armadillo (6,367 ERVs or 7.3% of total), and opossum (6,686 ERVs or 7.6% of total). This result equates to 20% of all ERVs from just 5% of host taxa, with the mean number of detected ERVs per genome being 799, excluding these three host taxa, the hominids and rodents. Why the opossum, armadillo, and hedgehog possess such elevated relative numbers of ERVs in their genomes presents a question for further investigation. A caveat for comparison of ERV quotients among genomes is that quality and completeness of genomic assemblies affect total number of detected ERVs, although it does not follow that relative ERV ratios within host taxa, as indicated in Fig. 2, are affected (e.g., compare results for high-quality human vs. low-quality gorilla genomes).
Fig. 2.

ERV distribution among vertebrate host taxa. Vertebrate host phylogeny with corresponding pie charts illustrate the proportion of analyzed ERVs (outer circle), class I ERVs (inner circle), and the fraction of gamma-ERVs (red sector) for each genome. Circle size correlates with ERV counts according to the scale.
Gamma and Class I ERV Origin.
The distribution of ERVs across host taxa and the proportion of analyzed gammaretroviruses and class I ERVs in each host genome are illustrated in Fig. 2 with details in Table S1. With reference to Fig. 2, it is apparent that class I ERVs are major components of the total ERV quotient of the majority of genomes examined. This finding is particularly so for the primates. The majority of gamma-ERVs occur in genomes from the Euarchontoglires, with 75% of all gamma-ERVs analyzed found in members of this group. Furthermore, 53% of all gamma-ERVs occur in rodent taxa, indicating that these taxa are a major reservoir for these viruses. An interesting trend is the marked decline in the proportion of gamma-ERVs to other class I ERVs in the simian primates (Saimiri to Homo). With the exception of the cat, hyrax, and opossum, the remaining genomes contain relatively few gamma-ERVs, with none detected in host genomes branching-off earlier than the Theria (placental mammals and marsupials), whereas other class I ERVs generally remain well represented (Fig. 2 and Table S1). These findings suggest a Gammaretrovirus origin early in mammalian evolution, after the appearance of the monotremes (Fig. 2). Thus, the occurrence of avian reticuloendotheliosis virus (REV), a class I gammaretrovirus, within a clade of hedgehog and bat ERVs (Fig. S1, V, Chicken) supports the occurrence of a relatively recent horizontal transmission event from mammal to bird, as suggested by another recent study (20). However, limitations in available data prevent more conclusive tests of these hypotheses. An earlier study detected that an echidna ERV was most closely related to REV using phylogenetic analyses of MLV-like retroviral sequences (7). Thus, analysis of echidna genomes, as well as additional bird and reptile genomes, will be particularly useful to confirm the taxonomic host range of gammaretroviruses.
It was recently suggested that gammaretroviruses may have originated in bats because a bat XRV, RfRV from Rhinolophus ferrumequinum (the greater horseshoe bat), was found to originate earlier than other mammalian gammaretroviral sequences in phylogenetic analyses (21). In our pan-phylogenomic analysis, which includes a wider host taxonomic sampling of ERVs, the RfRV is recovered well within the gamma-ERV clade (Fig. S1, V, Bat). Furthermore, our analyses suggest at least six independent origins of bat gammaretroviruses (Fig. S1, V) compared with the previous estimate of two (21). This finding highlights the importance of including a wide sampling of ERVs in evolutionary contexts of XRVs, because they arise from the same evolutionary continuum and provide a rich data source to complement evolutionary gaps in XRV data. We also note that ERV sequences originating from the little brown bat (Myotis lucifugus) and megabat (Pteropus vampyrus) branch off earlier than REV in our phylogeny (Fig. S1, V), potentially supporting an alternative origin in bats. However, pig and tenrec sequences branch off earlier than these, so the caveat of greater host taxon sampling applies, and additional genomes may further improve confidence in the origin of the gammaretrovirus clade. Here we extend the group to include a clade containing HERVS71, because this grouping forms a clear monophyletic evolutionary unit that has a sister-group relationship with another highly supported class I clade (see above). In relation to this finding, it is interesting to note that amplified fragments of a newly identified gamma-XRV from the bat Pteropus alecto were found to branch off earlier than REV in other phylogenetic analyses (21). The reasons for this result were unclear, because it was assumed that REV represented the most primitive Gammaretrovirus (20). Our results explain this observation by demonstrating that the limits of the gammaretrovirus group extend beyond REV (Fig. S1, V). With additional experimental data, it may be possible to apply our methodology to test whether the P. alecto XRV is the most primitive infectious Gammaretrovirus so far identified and whether it clusters within the same clade as REV, in the HERVS71 clade, or in a hitherto-unsampled clade.
Class I ERVs other than the gammaretroviruses were also detected in platypus, avian, turtle, and lizard genomes. However, none were detected in fish genomes, which instead harbor numerous related ERVs and retrovirus sequences from the Epsilonretrovirus genus. With reference to Fig. S1 (I, Aves), it is apparent that ERVs detected from diverse avian genomes branch off early in the sister clades that make up the class I ERVs, supporting an avian origin for the spread of class I retroviral sequences.
ERV Distributions Among Host Species.
Strikingly, we find that six large vegetarian mammals (cattle, llama, horse, panda, sloth, and elephant) possess multiple class I ERVs, but no representative gamma-ERVs (Fig. 2 and Table S1). Additionally, in our screening, the vegetarian wallaby has only a single gamma-ERV, and the manatee—which feeds on marine plants, fish, and invertebrates—also harbors no gamma-ERVs. Conversely, the only true carnivores included in our analysis (the cat, tarsier, and, to some extent, the dog) contain an overrepresentation of gamma-ERVs relative to other class I ERVs. Notably, the panda (Ailuropoda melanoleuca), a strictly vegetarian member of the Carnivora, does not show this pattern (Fig. 2). Together, these findings indicate that diet may play an important role in the exposure of different vertebrate taxa to retroviral infection. This suggestion is similar to arguments regarding murine-to-porcine transmission based on shared environments between pig and mouse, invoked to explain the close relationship observed between MLVs and PERVs (9). Although other vegetarian host taxa possess gamma-ERVs (gorilla, orangutan, guinea pig, mole rat, pika, rabbit, and megabat), all these host taxa, with the exception of megabat, belong to the Euarchontoglires (Fig. 2), which is the taxonomic group containing the highest overall representation of gamma-ERVs. It may be possible that these euarchontoglire hosts are not strictly vegetarian because recent studies suggest that several of these taxa do occasionally eat meat (22, 23), given that even rare carnivorous acts may be sufficient to expose hosts to retroviral infection. Other life-history and ecological traits may also influence susceptibility to infection by gammaretroviruses. One such characteristic may relate to living close to the ground, because small, ground-dwelling mammals such as members of rodentia and the afrotherian rock hyrax (Procavia capensis) tend to have high numbers of gamma-ERVs in their genomes (Fig. 2). However, additional genomes are required to improve support for this hypothesis. Other ecological traits, such as lifespan, body size, distribution, and sociality, do not appear to correlate with gamma-ERV content. From these observations, it is evident that additional scope for understanding retrovirus biology exists in examining further correlations with ecological and life-history traits.
Cross-Species Transmission.
Notably, our results suggest a history of frequent horizontal transmission of gammaretroviruses and associated class I retroviral sequences during evolution. In accordance with an earlier study based on PCR screens of vertebrate taxa (7), the results presented here suggest that interclass transmission, such as from birds to mammals is infrequent, with no cases identified among clades containing infectious retroviruses and only a few such cases implied across our class I ERV phylogeny (and mainly toward the base of the tree) (Fig. S1, I, Aves). However, the phylogenetic pattern for ERVs from different host genomes suggests a striking mode of evolution in which interorder transmission—for example, between primates and rodents—is common (Fig. 3 and Fig. S1). Here we show that this pattern may represent the default mode of evolution for the gammaretroviruses, because retroviral sequences from diverse mammals repeatedly occur adjacently in our phylogeny (Fig. S1). These results imply an inherent capacity for gammaretroviruses to switch across diverse mammalian hosts.
Fig. 3.

Gammaretrovirus phylogeny apex. Sequences from color-coded host genomes follow the short nomenclature outlined in Table S1. Hash marks in nomenclature precede the number of ERV sequences represented by each taxon. The expanded clades 1–3 are illustrated in Fig. S2, with loci details in Table S2. Colored circles indicate node support. Rat ERVs indicated by filled black circles frequently lie basally compared with the more numerous mouse sequences which are indicated by open circles.
A further result of our study is the finding that ERVs from rat as well as mouse genomes are distributed across the apex of the gamma-ERV clade (Fig. 3). When a tanglegram of host–virus relationships is constructed, it is evident that host and virus evolution is not congruent and that there have been multiple host-switching events by mouse and rat taxa (Fig. 4). Additionally, a smaller number of rat ERVs are often found to branch off earlier compared with the more numerous mouse sequences in the phylogenetic trees, suggesting that rats may have transmitted gammaretroviruses to mice (Fig. 3; clades 1–3 are expanded in Fig. S2, with ERV loci details in Table S2). Importantly, analyses of orthologous mouse and rat ERV loci support the assertion that phylogenetically related sequences represent cross-species transmission events, rather than integrations inherited from a shared ancestor. For all 431 mouse gamma-ERVs in clade 3 (Fig. 3), we determined the orthologous loci in the rat genome through genome-wide comparisons, none of which were found in the rat genome. Likewise, none of the eight rat ERVs had orthologous counterparts in the mouse genome. These results imply that rats have acted as important and overlooked spreaders of gammaretroviruses across diverse mammalian hosts. The “true rats” form the large genus Rattus, which contains 66 species in 7 species groups (24). Molecular-clock estimates suggest that the genus underwent initial diversification some 3.5 million years ago (25), probably in Asia where most species are found. Although all Rattus species have a native distribution in the Old World or Australasia, several species are highly invasive, particularly the black rat (Rattus rattus) and the brown rat (Rattus norvegicus), which are now found worldwide. Consequently, both these rat species overlap in range with other mammals known to carry gammaretroviruses and/or gamma-ERVs.
Fig. 4.

Host–gammaretrovirus relationships. Tanglegram of host species and retroviruses illustrating host-switching events. ERVs present in rodent genomes suggest a pattern of extensive host switching. Rat ERVs are highlighted in boxes. Loci details for rat and mouse ERVs are presented in Table S2.
An intriguing finding is the occurrence of cetacean sequences in a highly supported clade (100% posterior probability, 0.99 SH) otherwise composed solely of mouse and rat ERVs. These sequences cluster with the recently indentified killer whale ERV(26), and here we also recover an adjacent dolphin sequence (Fig. S1, V, Killer whale). If genome sequencing artifacts are ruled out, given that these are separate studies, our results thus suggest that cetaceans may have been exposed to this lineage as a consequence of rodent retrovirus transmission, perhaps via an unknown intermediate vector, in line with our working hypothesis that rodents serve as a reservoir for gammaretroviral spread among mammalian orders. Additionally, in contrast to interpretations based on analyses performed with datasets containing ERVs sparsely sampled across taxonomic diversity in a different study (27), our analyses suggest that a bottlenose dolphin (Tursiops truncatus) ERV located close to REV is not highly divergent from counterparts in other mammals, instead clustering with high support (0.99 SH) with cat and rabbit sequences at the base of the gammaretroviral clade (Fig. S1). Together, these findings highlight the use of our methodology in which ERVs across a wide taxonomic range are considered.
KoRV Transmission Route.
Gibbon ape leukemia virus (GaLV) and KoRV share high sequence similarity; however, the gibbon and koala are native to different continents, so retrovirus transmission most likely occurred via an intermediate vector (7, 8). Several Asian mice harbor viruses known to cross-hybridize with GaLV at high stringency (28), and these have been suggested to be ancestral to GaLV (29) and represent the source of KoRV (7, 18). Our results show that ERVs screened from the laboratory mouse (C57BL; Mus musculus) are phylogenetically closer to KoRV (Fig. S1, clade V, Koala; clade 3 in Fig. 3; and Fig. S2) than Asian mice retroviral sequences (Mus spretus ERV, MSEV and Mus dunni ERV, MDEV), challenging this assertion. However, for a more complete understanding of potential roles in gammaretrovirus spread, it is necessary to examine additional gamma-ERV variants from Asian mice. Our screen of 60 vertebrate genomes did not uncover additional ERVs in a closer alliance with KoRV. In line with our hypothesis that rats may have acted as spreaders of gammaretroviruses, it is not inconceivable that infected rats hitched their way to Australia in ships alongside humans during European colonization beginning in the 18th century. Hence, it may also prove informative to screen the genome of the black or “ship” rat (R. rattus) for related ERVs. Given that rats do not seem to share a common environment with koalas, which live in trees in arid environments, it is possible that an additional native vector may have been involved in the spread of gammaretroviruses to koala hosts. Consequently, it may be worthwhile to screen indigenous Australian bats and rodents for gammaretroviruses and associated ERVs to elucidate further on the possible intermediate vector of KoRV. It is also worth noting that KoRV recently was shown to exist in two subtypes (KoRV-A and -B), with different genomic structures, cell receptor use, and suggested pathology (16), which further complicates tracing of the distribution and potential transmission routes of KoRV.
Concluding Remarks
ERVs are representative of XRVs at the time of their infection (1). Consequently, the rich catalog of ERVs present in the genomes of vertebrates constitutes a valuable resource for understanding retrovirus–host evolution. For example, in addition to the role of host life history and ecology discussed above, the patterns uncovered in our analyses provide a platform for examining the importance of host genetic factors in the spread and distribution of XRVs. Host restriction factors have been shown to play a key role in host defense, as have mutations in the cell surface receptors required for retroviral infection (2). Of direct relevance here is a recent study that found that birds with a high risk of exposure to mice harboring an infective gammaretrovirus (ground-dwelling fowl and raptor species) have evolved receptor-disabling mutations, suggestive of a defensive role (30). Examination of ERV distribution patterns among host taxa provides opportunities to frame further investigations into the evolution of host genetic factors against retroviruses.
Questions relating to the evolution of retroviruses and their hosts are particularly relevant given the emergence of several prominent diseases linked with retroviral infection. These include the transmission of KoRV discussed here and the lentiviral HIV-1 and -2, which arose from multiple independent cross-species transmissions to humans from chimpanzees and sooty mangabeys, respectively (31). The occurrence of further retroviral zoonoses cannot be discounted, particularly given cases of close relationship among retroviral sequences isolated from disparate host taxa within the gammaretroviruses and associated class I ERVs. Our results demonstrate considerable phylogenetic distance between mammalian gammaretroviruses and their most closely related human ERVs (Fig. S1). Nonetheless, the phylogenies also show that previous invasions of class I retroviruses into primate genomes have led to extensive radiations of retroviral sequences.
Here, we use the potential of the genomic record to draw inferences regarding the evolutionary history of XRVs. Specifically, we adopt a specialized approach designed to reduce noise and maximize phylogenetic signal in our dataset, applicable to both XRV and ERVs. With reference to diverse studies, we demonstrate that our methodology markedly improves the ability to draw inferences regarding retroviral evolution, hypotheses of cross-species transmission, and the potential to identify reservoir hosts. Thus, broad-scale analyses such as those carried out here hold significant promise, from informing diverse fields in retrovirus biology regarding patterns of cross-species transfer to providing a platform for development of an improved retroviral taxonomy.
Materials and Methods
ERV Detection.
We used our RetroTector software (14) to screen for ERV loci in available vertebrate genomes (http://hgdownload.soe.ucsc.edu/downloads.html), because it uses conserved amino acid motifs from across the retroviral genome. RetroTector performs analyses across ERV reading frames and collects results, including positions for hits to reference motifs, into local MySQL databases for downstream analyses. Before screening of genomes consisting of small, nonassembled contigs, fragments of at least 10 kb were sorted according to decreasing size and concatenated to facilitate automated ERV detection. To increase fidelity, for each genome common nonretroviral repeats were derived from RepeatMasker and used to construct “Brooms” for the option to sweep across genomic sequences as an initial step in the automated RetroTector screening. This option is particularly useful for limiting false estimates and scoring of ERVs due to secondary nonretroviral integrations that may interfere with RetroTector accuracy. Full-length and partial ERVs scoring 300 and above were included in downstream analyses.
Sequence Alignment.
Nucleotide sequence alignments were constructed individually for a set of 28 regions sampled from across the gag (encoding the matrix, MA, capsid, CA and nucleocapsid, NC), pro (encoding the protease, PR), and pol (encoding the reverse transcriptase, RT and integrase, IN) genes of each ERV. These regions correspond to highly conserved amino acid motifs identified by the RetroTector software (14) and conserved ”spacer” sequences located between these motifs. ERV regions of lower conservation within the gag–pro–pol span were excluded, reducing the overall length of the alignment. The regions used here are referred to according to abbreviations for conserved motifs, for which locations are presented in the RetroTector publication (14), for gag (MA1, MA2, CA1, CA2, NC1, NC2), pro (PR2, PR3), and pol (RT1, RT1_2, RT2, RT2_3, RT3, RT3_4, RT4, RT4_5, RT5, RT5_6, RT6, IN1, IN1_2, IN2, IN2_3, IN3, IN3_4, IN5, IN5_6, IN6), where underscores denote intermediate spacer sequences between highly conserved amino acid motifs. Rapidly evolving regions of the retroviral genome such as the env gene were omitted to facilitate a high phylogenetic signal to noise ratio.
Nucleotide alignments were constructed for each motif and spacer region separately by using custom Perl scripts (developed by A.H.) that extract sequences from local MySQL databases containing RetroTector results. During this process, sequences are aligned using Muscle (32), followed by concatenation of the multiple alignments for downstream analyses. Alignment positions containing <10% data were trimmed by using the program trimAl (33), resulting in an alignment containing 1,553 bp. Detected ERVs may lack data, due to the absence or degradation of several motifs. Because large amounts of missing data can be problematic in phylogenetic analyses, particularly when distributed nonrandomly and for comparatively small datasets (34, 35), ERVs represented by <40% data (60% alignment gaps) were excluded from the alignment. A set of 92 reference retroviruses and ERVs, derived from GenBank, RepBase, and the literature (4, 7, 18, 36–40), were included in alignments together with the screened ERVs as used (9) to provide a phylogenetic framework from which to draw inferences.
Phylogenetic Analyses.
To reduce large numbers of ERVs in different genomes to an extent where general patterns could be inferred in evolutionary analyses, we developed a phylogenetic approach to collapse highly similar sequences. Specifically, for ERVs in each genome, maximum-likelihood phylogenies were inferred with the program ExAML (Version 1.0.0) (41) by using the general time-reversible (GTR) model of nucleotide sequence evolution, a gamma model of rate heterogeneity with four discrete rates, and a randomized parsimony starting tree estimated with RAxML (Version 7.3.6) (42). Subsequently, all nodes containing branches below a threshold level of 0.07 mean number of substitutions per site were collapsed by using a custom Perl script (developed by A.H.), which includes commands from the IO module of the Bio::Phylo package (Version 0.56) (43). More specifically, a depth-first, postorder tree traversal was performed to traverse the tree recursively, adding collapsed branch lengths to existing branches and working in from the tips until the threshold level was exceeded. The collapsed tree was parsed, and the taxon containing the highest proportion of data for each clade was output to a new alignment as the representative for that clade. The threshold level of 0.07 mean number of substitutions per site was established by performing analyses for all screened genomes, varying the cutoff for collapsing nodes from 0.01 to 0.24 in 0.01 increments. A level of 0.07 mean number of substitutions per site corresponds to a conservative threshold whereby the rate of taxon reduction decreases for the majority of genomes examined. A new alignment was constructed by using the custom alignment Perl script (above) for the set of reduced ERV taxa from the 60 host genomes together with the reference sequences. A phylogenetic tree was inferred for this alignment with FastTree2 (Version 2.1.7) (44), by using the GTR model of nucleotide sequence evolution and the CAT (category) approximation to account for variation in rates across sites. Twelve extreme long-branch taxa were excluded from the final analysis. Clade support values were estimated by using the Shimodaira–Hasegawa test (SH-like local support values) (19) as implemented in FastTree2. The resulting phylogeny (Fig. S1) was rooted by using the Caenorhabditis elegans retrotransposon Cer1 (GenBank accession no. U15406.1), which is a gypsy/Ty3 element serving as an outgroup to Retroviridae in our analyses. Three ERV taxa (oo367, dr3639, and ct1703) at the base of a clade formed by the Alpha, Beta, Delta, and Lenti clades were excised from the tree presented in Fig. S1 to improve clarity.
The phylogenetic trees in Fig. 3 and Fig. S2 (with loci details in Table S2) were estimated with MrBayes (Version 3.1.2) (45), by using two simultaneous runs of 10 million generations, each comprising one cold chain and seven heated chains, with a temperature of 0.1, a GTR model of nucleotide sequence evolution, and a gamma model with four distinct categories to account for variation in rates across sites. The analysis for Fig. 3 was initiated with a starting tree inferred with EXaML. Multiple independent runs and examination with Tracer (Version 1.5) (http://tree.bio.ed.ac.uk/software/tracer/) were used to inspect chain convergence. Trees were formatted by using FigTree (Version 1.4) (http://tree.bio.ed.ac.uk/software/figtree/).
Host Species Phylogenies.
Vertebrate host species trees were manually constructed with reference to recently published large-scale vertebrate phylogenetic analyses (46, 47).
ERV Orthology in Mouse and Rat.
Alignments between mouse (mm10) and rat (rn5) genomes were generated by using Satsuma (48). ERV insertion sites were then mapped across the genomes (source to target) by finding the closest orthologous anchors on each side, discarding positions where the target region did not contain an ERV insertion between flanking orthologous anchors. Remaining regions, including any that could not be unambiguously mapped, were examined and manually counted as orthologous or nonorthologous based on local identity dot plots.
Supplementary Material
Acknowledgments
We thank Leif Andersson for critical reading of the manuscript and Rutger Vos for feedback on the BioPerl package Bio::Phylo. Analyses were performed by using the UPPMAX computer cluster (www.uppmax.uu.se). This work was supported by the Swedish Research Council, Formas, and the Wenner–Gren Foundation.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1315419110/-/DCSupplemental.
References
- 1.Jern P, Coffin JM. Effects of retroviruses on host genome function. Annu Rev Genet. 2008;42:709–732. doi: 10.1146/annurev.genet.42.110807.091501. [DOI] [PubMed] [Google Scholar]
- 2.Stoye JP. Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nat Rev Microbiol. 2012;10(6):395–406. doi: 10.1038/nrmicro2783. [DOI] [PubMed] [Google Scholar]
- 3.Stoye JP, et al. Retroviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ, editors. Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses. San Diego: Elsevier Academic; 2012. pp. 477–495. [Google Scholar]
- 4.Jern P, Sperber GO, Blomberg J. Use of endogenous retroviral sequences (ERVs) and structural markers for retroviral phylogenetic inference and taxonomy. Retrovirology. 2005;2:50. doi: 10.1186/1742-4690-2-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feschotte C, Gilbert C. Endogenous viruses: Insights into viral evolution and impact on host biology. Nat Rev Genet. 2012;13(4):283–296. doi: 10.1038/nrg3199. [DOI] [PubMed] [Google Scholar]
- 6.Rosenberg N, Jolicoeur P. Retroviral pathogenesis. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor, NY: Cold Spring Harbor Lab Press; 1997. pp. 475–586. [PubMed] [Google Scholar]
- 7.Martin J, Herniou E, Cook J, O’Neill RW, Tristem M. Interclass transmission and phyletic host tracking in murine leukemia virus-related retroviruses. J Virol. 1999;73(3):2442–2449. doi: 10.1128/jvi.73.3.2442-2449.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanger JJ, Bromham LD, McKee JJ, O’Brien TM, Robinson WF. The nucleotide sequence of koala (Phascolarctos cinereus) retrovirus: A novel type C endogenous virus related to Gibbon ape leukemia virus. J Virol. 2000;74(9):4264–4272. doi: 10.1128/jvi.74.9.4264-4272.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Groenen MAM, et al. Analyses of pig genomes provide insight into porcine demography and evolution. Nature. 2012;491(7424):393–398. doi: 10.1038/nature11622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cingöz O, et al. Characterization, mapping, and distribution of the two XMRV parental proviruses. J Virol. 2012;86(1):328–338. doi: 10.1128/JVI.06022-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fiebig U, Hartmann MG, Bannert N, Kurth R, Denner J. Transspecies transmission of the endogenous koala retrovirus. J Virol. 2006;80(11):5651–5654. doi: 10.1128/JVI.02597-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le Tissier P, Stoye JP, Takeuchi Y, Patience C, Weiss RA. Two sets of human-tropic pig retrovirus. Nature. 1997;389(6652):681–682. doi: 10.1038/39489. [DOI] [PubMed] [Google Scholar]
- 13.Patience C, Takeuchi Y, Weiss RA. Infection of human cells by an endogenous retrovirus of pigs. Nat Med. 1997;3(3):282–286. doi: 10.1038/nm0397-282. [DOI] [PubMed] [Google Scholar]
- 14.Sperber GO, Airola T, Jern P, Blomberg J. Automated recognition of retroviral sequences in genomic data—RetroTector. Nucleic Acids Res. 2007;35(15):4964–4976. doi: 10.1093/nar/gkm515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duffy S, Shackelton LA, Holmes EC. Rates of evolutionary change in viruses: Patterns and determinants. Nat Rev Genet. 2008;9(4):267–276. doi: 10.1038/nrg2323. [DOI] [PubMed] [Google Scholar]
- 16.Xu W, et al. An exogenous retrovirus isolated from koalas with malignant neoplasias in a US zoo. Proc Natl Acad Sci USA. 2013;110(28):11547–11552. doi: 10.1073/pnas.1304704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tarlinton RE, Meers J, Young PR. Retroviral invasion of the koala genome. Nature. 2006;442(7098):79–81. doi: 10.1038/nature04841. [DOI] [PubMed] [Google Scholar]
- 18.Ávila-Arcos MC, et al. One hundred twenty years of koala retrovirus evolution determined from museum skins. Mol Biol Evol. 2013;30(2):299–304. doi: 10.1093/molbev/mss223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimodaira H, Hasegawa M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol. 1999;16(8):1114–1116. [Google Scholar]
- 20.Niewiadomska AM, Gifford RJ. The extraordinary evolutionary history of the reticuloendotheliosis viruses. PLoS Biol. 2013;11(8):e1001642. doi: 10.1371/journal.pbio.1001642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cui J, et al. Identification of diverse groups of endogenous gammaretroviruses in mega- and microbats. J Gen Virol. 2012;93(Pt 9):2037–2045. doi: 10.1099/vir.0.043760-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hardus ME, et al. Behavioral, ecological, and evolutionary aspects of meat-eating by Sumatran orangutans (Pongo abelii) Int J Primatol. 2012;33(2):287–304. doi: 10.1007/s10764-011-9574-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hofreiter M, Kreuz E, Eriksson J, Schubert G, Hohmann G. Vertebrate DNA in fecal samples from bonobos and gorillas: Evidence for meat consumption or artefact? PLoS ONE. 2010;5(2):e9419. doi: 10.1371/journal.pone.0009419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Musser G, Carleton M. Superfamily Muroidea. In: Wilson DE, Reeder DM, editors. Mammal Species of the World: A Taxonomic and Geographic Reference. 3rd Ed. Vol 2. Baltimore: Johns Hopkins Univ Press; 2005. pp. 894–1531. [Google Scholar]
- 25.Aplin KP, et al. Multiple geographic origins of commensalism and complex dispersal history of Black Rats. PLoS ONE. 2011;6(11):e26357. doi: 10.1371/journal.pone.0026357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lamere SA, et al. Molecular characterization of a novel gammaretrovirus in killer whales (Orcinus orca) J Virol. 2009;83(24):12956–12967. doi: 10.1128/JVI.01354-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang L, et al. Ancient invasion of an extinct gammaretrovirus in cetaceans. Virology. 2013;441(1):66–69. doi: 10.1016/j.virol.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 28.Lieber MM, et al. Isolation from the Asian mouse Mus caroli of an endogenous type C virus related to infectious primate type C viruses. Proc Natl Acad Sci USA. 1975;72(6):2315–2319. doi: 10.1073/pnas.72.6.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weiss RA. On the concept and elucidation of endogenous retroviruses. Philos Trans R Soc Lond B Biol Sci. 2013;368(1626):20120494. doi: 10.1098/rstb.2012.0494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin C, Buckler-White A, Wollenberg K, Kozak CA. The avian XPR1 gammaretrovirus receptor is under positive selection and is disabled in bird species in contact with virus-infected wild mice. J Virol. 2013;87(18):10094–10104. doi: 10.1128/JVI.01327-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharp PM, Hahn BH. The evolution of HIV-1 and the origin of AIDS. Philos Trans R Soc Lond B Biol Sci. 2010;365(1552):2487–2494. doi: 10.1098/rstb.2010.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edgar RC. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25(15):1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wiens JJ, Morrill MC. Missing data in phylogenetic analysis: Reconciling results from simulations and empirical data. Syst Biol. 2011;60(5):719–731. doi: 10.1093/sysbio/syr025. [DOI] [PubMed] [Google Scholar]
- 35.Simmons MP. Misleading results of likelihood-based phylogenetic analyses in the presence of missing data. Cladistics. 2012;28(2):208–222. doi: 10.1111/j.1096-0031.2011.00375.x. [DOI] [PubMed] [Google Scholar]
- 36.Dewannieux M, et al. Identification of an infectious progenitor for the multiple-copy HERV-K human endogenous retroelements. Genome Res. 2006;16(12):1548–1556. doi: 10.1101/gr.5565706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jern P, Sperber GO, Ahlsén G, Blomberg J. Sequence variability, gene structure, and expression of full-length human endogenous retrovirus H. J Virol. 2005;79(10):6325–6337. doi: 10.1128/JVI.79.10.6325-6337.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katzourakis A, Gifford RJ, Tristem M, Gilbert MT, Pybus OG. Macroevolution of complex retroviruses. Science. 2009;325(5947):1512. doi: 10.1126/science.1174149. [DOI] [PubMed] [Google Scholar]
- 39.Keckesova Z, Ylinen LM, Towers GJ, Gifford RJ, Katzourakis A. Identification of a RELIK orthologue in the European hare (Lepus europaeus) reveals a minimum age of 12 million years for the lagomorph lentiviruses. Virology. 2009;384(1):7–11. doi: 10.1016/j.virol.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jern P, Stoye JP, Coffin JM. Role of APOBEC3 in genetic diversity among endogenous murine leukemia viruses. PLoS Genet. 2007;3(10):2014–2022. doi: 10.1371/journal.pgen.0030183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stamatakis A, Aberer J (2013) Novel parallelization schemes for large-scale likelihood-based phylogenetic inference. 2013 IEEE International Parallel & Distributed Processing Symposium (IPDPS), May 20–24, Boston (IEEE Computer Society, New York), pp 1195–1204.
- 42.Stamatakis A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 43.Vos RA, Caravas J, Hartmann K, Jensen MA, Miller C. BIO:Phylo-phyloinformatic analysis using perl. BMC Bioinformatics. 2011;12:63. doi: 10.1186/1471-2105-12-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Price MN, Dehal PS, Arkin AP. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5(3):e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19(12):1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 46.Crawford NG, et al. More than 1000 ultraconserved elements provide evidence that turtles are the sister group of archosaurs. Biol Lett. 2012;8(5):783–786. doi: 10.1098/rsbl.2012.0331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meredith RW, et al. Impacts of the Cretaceous Terrestrial Revolution and KPg extinction on mammal diversification. Science. 2011;334(6055):521–524. doi: 10.1126/science.1211028. [DOI] [PubMed] [Google Scholar]
- 48.Grabherr MG, et al. Genome-wide synteny through highly sensitive sequence alignment: Satsuma. Bioinformatics. 2010;26(9):1145–1151. doi: 10.1093/bioinformatics/btq102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.