

Significance
Multiple Escherichia coli genome sequences have recently been made available by advances in DNA sequencing. Analysis of these genomes has demonstrated that the fraction of genes common to all E. coli strains in the species represents a small fraction of the entire E. coli gene pool. This observation raises the question: what is a strain and what is a species? In this study, genome-scale metabolic reconstructions of multiple E. coli strains are used to reconstruct the metabolic network for an entire species and its strain-specific variants. The models are used to determine functional differences between strains and define the E. coli species based on common metabolic capabilities. Individual strains were differentiated based on niche-specific growth capabilities.
Keywords: systems biology, mathematical modeling, core and pan genome
Abstract
Genome-scale models (GEMs) of metabolism were constructed for 55 fully sequenced Escherichia coli and Shigella strains. The GEMs enable a systems approach to characterizing the pan and core metabolic capabilities of the E. coli species. The majority of pan metabolic content was found to consist of alternate catabolic pathways for unique nutrient sources. The GEMs were then used to systematically analyze growth capabilities in more than 650 different growth-supporting environments. The results show that unique strain-specific metabolic capabilities correspond to pathotypes and environmental niches. Twelve of the GEMs were used to predict growth on six differentiating nutrients, and the predictions were found to agree with 80% of experimental outcomes. Additionally, GEMs were used to predict strain-specific auxotrophies. Twelve of the strains modeled were predicted to be auxotrophic for vitamins niacin (vitamin B3), thiamin (vitamin B1), or folate (vitamin B9). Six of the strains modeled have lost biosynthetic pathways for essential amino acids methionine, tryptophan, or leucine. Genome-scale analysis of multiple strains of a species can thus be used to define the metabolic essence of a microbial species and delineate growth differences that shed light on the adaptation process to a particular microenvironment.
Over the last decade, the Escherichia coli K-12 MG1655 strain has been used extensively as a model organism for research on microbial metabolic systems biology. However, the increasing availability of genomic sequences for other E. coli strains suggests that this nonpathogenic laboratory strain’s genes are a small part of the genomic diversity in the E. coli species. For instance, the E. coli O157:H7 EDL933 strain responsible for worldwide outbreaks of hemorrhagic colitis has one million more base pairs of DNA than K-12 MG1655 (∼20% larger) (1–3). Multiple genomic sequences have defined a set of genes that is common to all E. coli strains (i.e., a “core” genome), and it has been determined that they represent a small fraction of the entire E. coli gene pool. The growing availability of whole genome sequences for E. coli strains thus brings into focus the question: what is a strain and what is a species?
A recent study of 20 E. coli strains found that a large fraction of the shared genomic elements with known function are related to metabolism (4). Therefore, it is important to characterize the genes that encode a core set of metabolic capabilities to understand their effect on cellular functions, as they constitute a common denominator that can be used to define the core metabolic potential of the E. coli species. Metabolic network reconstructions have proven to be powerful tools to probe the genomic diversity of metabolism between organisms (5–10). As useful as genome annotation is, it does not provide an understanding of the integrated function of gene products to produce phenotypic states. K-12 MG1655 was the first E. coli strain to have its genome entirely sequenced (11). A first genome-scale metabolic reconstruction was completed for this strain 3 y later (12). Since then, the reconstruction of MG1655 has undergone a series of expansions in the intervening 13 y as more information about the genome and its annotation has become available (13–15). The most recent reconstruction, iJO1366 (15), accounts for 1,366 genes (39% of functionally annotated genes on the genome) and their gene products. The genome-scale metabolic reconstruction for E. coli K-12 MG1655 is the most complete metabolic reconstruction available to date (16, 17). However, as more E. coli genomic sequences have become available, it has become clear that E. coli K-12 MG1655 only partially represents the E. coli species. Thus, it is important to construct genome-scale models (GEMs) for other E. coli strains because of this species’ importance to human health, basic microbiological science, and industrial biotechnology (18).
The goal of this study was to construct GEMs for all E. coli strains with fully sequenced genomes and thus to reconstruct the metabolic network for an entire species and its strain-specific variants. Shigella strains were included on the basis of 16S ribosomal profiling experiments that classify Shigella strains as members of the E. coli species (19), despite the historical distinction of having their own genus. Therefore, the formulated GEMs span commensal strains, as well as both intestinal and extraintestinal pathogenic strains of E. coli, allowing for a comprehensive analysis of the representative metabolic capabilities of the E. coli species.
Results
Characteristics of E. coli Core and Pan Metabolic Content.
A set of 55 E. coli genome-scale reconstructions was built and used to compare gene, reaction, and metabolite content between strains (Datasets S1 and S2). The content shared among all reconstructions thereby defines the core metabolic capabilities among all of the strains. Similarly, the metabolic capabilities of all of the strains were combined to define the full set that encompasses all models and thereby define the “pan” metabolic capabilities among all of the strains. By analogy to mathematical set theory, the core metabolic content is the intersection of the gene, reaction, and metabolite content of all 55 models, whereas the pan metabolic content is the union of these features among the models (Fig. 1A).
Fig. 1.

Core and pan metabolic capabilities of the E. coli species. The core and pan metabolic content was determined for genome-scale metabolic models (GEMs) of 55 unique E. coli strains. (A) The core content, illustrated by the intersection of the Venn diagram, is shared with all strains. The pan content consists of all content in any model and includes the core content. The Venn diagram is not to scale. (B) Classification of reactions in the core and pan reactomes by metabolic subsystem.
The size and content of the core metabolic content characterizes the metabolic foundation of E. coli as a species. The core model has 965 metabolic genes that catalyze 1,773 reactions using 1,665 metabolites. The most highly conserved subsystems were lipid metabolism, cell wall, membrane and envelope metabolism, nucleotide metabolism, and cofactor and prosthetic group metabolism. Reactions involved in lipid metabolism, cell wall/membrane/envelope metabolism, and cofactor and prosthetic group biosynthesis were highly represented (>80%) in the core reactome. Most of these reactions synthesize essential components such as vitamins and cofactors like riboflavin, Coenzyme A, and biotin, as well as quinones and isoprenoids. In contrast, only 36% of carbohydrate metabolism reactions were part of the core reactome. These reactions were comprised of central metabolism reactions including anaplerotic reactions, the citric acid cycle, glycolysis/gluconeogenesis, and the pentose phosphate pathway.
The pan metabolic capabilities are comprised of the total number of different reactions found in all strains and are thus an indicator of the full metabolic capabilities within a species. The E. coli pan reconstruction content contains 1,460 metabolic genes, 2,501 reactions, and 2,043 metabolites. About 64% of reactions in carbohydrate metabolism were part of the pan reactome, the largest group (Fig. 1B). A majority of these reactions are involved in alternate carbon source metabolism. Cell wall and membrane envelope metabolism accounted for 18% of reactions in the pan reactome. These reactions account for a major phenotypic distinction between E. coli strain’s serogroup, in particular the O antigen (20). Also, 30% of amino acid metabolism reactions are part of the pan reactome.
Ability to Catabolize Different Nutrient Sources Distinguishes Metabolic Models of E. coli Strains.
The conversion of static metabolic network reconstructions into computable mathematical models allows computation of phenotypes based on the content of each reconstruction. Thus, the 55 strain-specific reconstructed networks were converted into GEMs that allow for the simulation of phenotype (21). This set of GEMs allows for a meaningful interpretation of the content of each reconstruction and allows for the prediction of a strain’s microenvironmental and ecological niche.
Reactions belonging to the alternate carbon metabolism subsystem made up the majority of reactions in the pan reactome (Fig. 1B). Thus, it was hypothesized that these capabilities may reflect functional differences in the ability of different strains of E. coli to adapt to different nutritional environments. To test this hypothesis, growth was simulated in silico for all 55 E. coli and Shigella GEMs on minimal media in 654 growth conditions. The conditions were composed of all sole growth supporting carbon, nitrogen, phosphorous, and sulfur sources in both aerobic and anaerobic environments (Fig. 2 and Dataset S1).
Fig. 2.
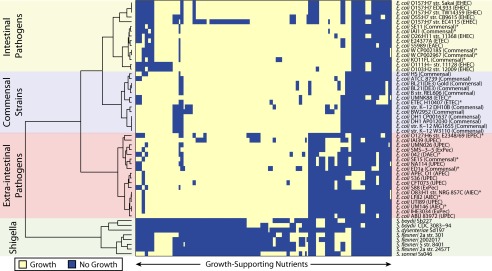
Clustering of species by unique growth-supporting conditions. Predicted metabolic phenotypes on the variable growth-supporting nutrient conditions composed of different carbon, nitrogen, phosphorous, and sulfur nutrient sources in aerobic and anaerobic conditions. Strains are clustered based on their ability to sustain growth in each different environment. Rows represent individual strains, and columns represent different nutrient conditions. In general, strains clustered into their respective pathotypes of commensal E. coli strains, intestinal pathogenic E. coli strains, extra-intestinal pathogenic E. coli strains, and Shigella strains. An asterix symbol indicates those strains that clustered outside of their respective pathotype. All growth conditions are listed in Dataset S1.
In contrast to the E. coli GEMs, the Shigella GEMs displayed a large loss of catabolic capabilities across the 654 growth conditions. This computational result supports evidence showing that Shigella strains have lost catabolic pathways for many nutrient sources (22). Models of Shigella strains completely lost the capability to sustain growth on nutrient sources for which more than 90% of E. coli models had growth capabilities. Some of these nutrients include d-alantoin, d-malate, and xanthine as carbon sources, as well as inosine as a nitrogen source. Furthermore, only one of the eight Shigella strain models (13%) was able to sustain growth on choline or l-fucose, two carbon sources that most E. coli strain models examined were predicted to catabolize.
Set of Substrates Differentiate Pathogenic Strains from Commensal (Nonpathogenic) Strains.
Based on simulated growth phenotypes, we observed a general separation of commensal strains from both extraintestinal pathogenic E. coli (ExPec) and intestinal pathogenic E. coli (InPec) strains of E. coli, suggesting that a classification schema of strains based on metabolic capabilities is possible (Fig. 3). Common laboratory strains of E. coli such as E. coli K-12 MG1655 are nonpathogenic, commensal strains. As a first step toward establishing such a schema, the separation between ExPec and commensal strain models was examined. A Fisher’s exact test was used to establish that models of ExPec strains exhibited a statistically significant capability to catabolize four unique compounds with P < 0.05 (Table 1).
Fig. 3.

Classification of E. coli pathotypes based on growth-supporting conditions. Growth-supporting nutrients were used to create a classification tree. This tree can be used to determine if an E. coli strain is commensal, an intestinal pathogen, or an extraintestinal pathogen. For example, following the tree to the right shows that 77% of E. coli strains that cannot grow on α-mannosylglycerate, fructoselysine, or taurine as sole carbon sources are expected to be extraintestinal pathogens. Thus, a small number of nutrient sources can be used to classify E. coli strains of different types.
Table 1.
Nutrients predicted to give commensal strains or extraintestinal pathogenic strains of E. coli a catabolic advantage
| Source | Commensal (%) | ExPec (%) | P value |
| ExPec strain nutrients | |||
| 3-Phospho-d-glycerate | 0 | 36 | 1.3E−2 |
| l-Arginine | 11 | 64 | 5.0E−3 |
| Cellobiose | 33 | 82 | 1.3E−2 |
| N-acetyl-d-galactosamine | 67 | 100 | 3.9E−2 |
| Commensal strain nutrients | |||
| Fructoselysine | 89 | 0 | 2.2E−6 |
| Psiscoselysine | 89 | 0 | 2.2E−6 |
| Dopamine | 89 | 0 | 2.2E−6 |
| Phenethylamine | 89 | 0 | 2.2E−6 |
| Tyramine | 89 | 0 | 2.2E−6 |
| Phenylacetaldehyde | 72 | 0 | 1.2E−4 |
| α-Mannosylglycerate | 94 | 9 | 5.7E−6 |
| 4-Hydroxyphenylacetate | 56 | 9 | 1.3E−2 |
| Cyanate | 83 | 27 | 3.8E−3 |
| Melibiose | 78 | 27 | 9.7E−3 |
| Phenylpropanoate | 72 | 27 | 2.0E−2 |
| 3-(3-Hydroxy-phenyl)propionate | 89 | 36 | 5.0E−3 |
| 3-Hydroxycinnamic acid | 89 | 36 | 5.0E−3 |
GEM predicted advantages for sole growth-supporting nitrogen and carbon sources between commensal and extraintestinal pathogenic E. coli (ExPec) strains. Percentages indicate the portion of each pathotype able to catabolize the listed nutrient source. ExPec strains had a statistically significant capability to catabolize four unique carbon sources compared to commensal strains that overrepresented 13 different sole growth-supporting carbon and nitrogen sources.
Most models of strains widely regarded as safe laboratory strains such as K-12 strains, BW2952, and DH1 were unable to grow on a unique subset of nutrients. Notably, N-acetyl-d-galactosamine supported growth in 100% of ExPec strain models compared with 67% of commensal strain models (P = 3.9 × 10−2). Additionally, several commensal strain models exhibited a statistically significant overrepresentation of catabolic pathways for 13 nutrient sources (Table 1). For example, fructoselysine and psiscoselysine share the same catabolic pathway and were not catabolizable by any of the ExPec models; however, 89% (P = 2.2 × 10−6) of the intestinal strain models could use fructoselysine or psiscoselysine as a sole carbon source. Fructoselysine is poorly digested in the human small intestine, and little is excreted, hinting that the majority of dietary fructoselysine may be digested by the intestinal microbiota (23). Further, 4-hydroxyphenylacetate, an aromatic compound, was catabolized as a sole carbon source for 55% of commensal strain models compared with only 9% of ExPec strain models (P = 1.3 × 10−2). Hydroxyphenylacetic acids are produced by bacterial fermentation of short chain peptides and amino acids in the human large intestine (24). 4-Hydroxyphenylacetate undergoes eight different enzymatic reactions before being converted to pyruvate and succinate-semialdehyde that can then be converted to succinate and enter the tricarboxylic acid cycle (25).
Next, the models of strains known to reside intestinally were compared to investigate differences between commensal and InPec strains. Models of InPec strains displayed an advantage in their ability to support growth on seven unique carbon and nitrogen sources (Table 2). Some of the substrates had unique enrichment specifically among the enterohemmorhagic (EHEC) strains, responsible for worldwide cases of diarrhea and hemolytic uremic syndrome (HUS) (26). Sucrose supported growth for 65% of the InPec strains including 100% of EHEC strains compared with only 33% of commensal strains (P = 5.0 × 10−2). Also, consistent with other reports (27), urease activity was present in EHEC strain models only. Urea supported growth as a sole nitrogen source for 47% of InPec strain models, including 100% of the EHEC models compared with 0% of commensal strain models (P = 1.0 × 10−3). Urease degrades urea into CO2 and NH4 and therefore may provide an additional source of nitrogen for cells in nitrogen-limited environments.
Table 2.
Nutrients predicted to give commensal strains or intestinal pathogenic strains of E. coli a catabolic advantage
| Source | Commensal (%) | InPec (%) | P value |
| InPec strain nutrients | |||
| Sucrose | 33 | 65 | 5.0E−2 |
| Raffinose | 28 | 65 | 2.6E−2 |
| l-Arginine | 11 | 65 | 1.3E−3 |
| d-Arabitol | 0 | 24 | 4.6E−2 |
| Ribitol | 0 | 24 | 4.5E−2 |
| Agmatine | 0 | 47 | 1.0E−3 |
| Urea | 0 | 47 | 1.0E−3 |
| Commensal strain nutrients | |||
| Galactonate | 100 | 65 | 7.6E−3 |
| α-Mannosylglycerate | 94 | 35 | 2.6E−4 |
| Dopamine | 89 | 41 | 3.5E−3 |
| Phenethylamine | 89 | 41 | 3.5E−3 |
| Tyramine | 89 | 41 | 3.5E−3 |
| 5-Dehydro-d-gluconate | 78 | 24 | 1.6E−3 |
| l-Idonate | 78 | 24 | 1.6E−3 |
| d-Allose | 78 | 41 | 2.5E−2 |
| Butyrate | 78 | 47 | 5.0E−2 |
| Acetoacetate | 78 | 47 | 5.0E-2 |
| 4-Hydroxyphenylacetate | 56 | 18 | 2.0E-2 |
GEM predicted advantages for sole growth-supporting nitrogen and carbon sources between commensal and intestinal pathogenic E. coli (InPec) strains. Percentages indicate the portion of each pathotype able to catabolize the listed nutrient source. InPec strains had a statistically significant capability to catabolize seven unique carbon and nitrogen sources compared to commensal strains that overrepresented 11 different growth-supporting carbon and nitrogen sources.
In contrast to InPec strains, commensal strains displayed an advantage in their ability to degrade 11 unique carbon and nitrogen sources (Table 2). The short chain fatty acids (SCFAs) acetoacetate and butyrate were found to support growth as sole carbon sources for 78% of commensal strain models compared with 47% of InPec models (P = 5.0 × 10−2). Notably, none of the EHEC strain models were able to catabolize either of these two compounds, and only 13% of Shigella models were able to use them as a sole source of carbon.
Metabolic Models Combined with Gap-Filling Methods Facilitate Investigation into the Genetic Basis of Strain-Specific Auxotrophies.
In addition to investigating growth-supporting nutrients, GEMs can also be used to examine the genetic bases of strain-specific auxotrophies. Twelve of the 55 reconstructed GEMs were unable to generate essential biomass components from glucose M9 minimal media without addition of growth-supporting compounds to the in silico media. The SMILEY algorithm, a method to fill gaps in metabolic networks (28), was used to examine the genetic bases of these model auxotrophies (Fig. 4). Based on this analysis, six of the eight Shigella strains exhibited an auxotrophy for niacin (vitamin B3) in silico. These simulation results are consistent with literature data indicating that many strains of Shigella, including Shigella sonnei Ss046 and Shigella boydii sb227, are unable to grow without addition of niacin to M9 minimal media with glucose (29). Gap analysis attributes this auxotrophy to the lack of l-aspartate oxidase activity, encoded by the gene nadB, in the nicotinic acid biosynthesis pathway. A bioinformatic analysis of nadB suggests that it is a pseudogene due to numerous nonsynonymous mutations compared with the sequence of nadB in E. coli K-12.
Fig. 4.

Model predicted strain-specific auxotrophies. GEM predicted minimal media conditions for each auxotrophic strain. Shigella strains lack essential vitamin biosynthesis capabilities for niacin (vitamin B3), thiamin (vitamin B1), and folate (vitamin B9). Other strains have lost biosynthetic pathways for the essential amino acids methionine, tryptophan and leucine, thus becoming auxotrophic for these compounds.
Two additional examples of identifying and confirming strain-specific auxotrophies involved models for Shigella flexneri 2a str 301, (auxotrophic for methionine) and E. coli strain DH10B (auxotrophic for leucine). Gap analysis of S. flexneri 2a str 301 suggested that the auxotrophy is due to the absence of homoserine O-transsuccinylase, encoded by metA in E. coli K-12 MG1655. A bioinformatics analysis suggested that metA is a pseudogene in S. flexneri 2a str 301 due to single base pair deletion, causing a frameshift mutation at amino acid position 212/310 and hence premature termination of the full-length protein. Both of these observations were confirmed with literature evidence. Specifically, S. flexneri strains are known to require methionine (30) in minimal media and E. coli DH10B is known to require leucine due to a deletion of the leuABCD operon (31).
Experimental Validation of Unique Nutrients Shows High Model Accuracy.
To assess the accuracy of in silico growth simulations, 12 of the 55 reconstructed strains were screened for growth on six carbon sources. Growth was estimated by optical density 48 h after inoculation. OD values of >0.08 were considered growth. The 12 strains consisted of 3 ExPec strains, 3 InPec strains, 5 commensal strains, and 1 Shigella strain, thereby spanning the pathotypes discussed above. Six carbon sources were selected based on their predicted ability to classify strains according to different pathotypes. In other words, each of these six substrates was expected to support growth of certain strains but not others. The models are validated by true-positive and true-negative results that highlight cases where the models are in agreement with experimental results. In contrast, false-positive and false-negative cases indicate potential errors or gaps in the models (17) (Fig. 5A). The experimental results showed a high level of accuracy with 80% of GEM predictions agreeing with experiments.
Fig. 5.

Comparison of GEM predictions to experimental results. Comparison of GEM predictions to experimental results revealed a high level of accuracy (80%) both for true positives (quadrant 1) and true negatives (quadrant 4). False negative cases (quadrant 2) represent missing knowledge and are an opportunity for biological discovery. False positive cases (quadrant 3) represent missing context-specific information such as transcriptional regulation. (B) A detailed breakdown of the comparisons based on the pathways (rows) and screened strains (columns).
Comparison of in silico and experimental results revealed complete agreement for two different carbon sources: acetoacetate and deoxyribose were predicted correctly with 100% accuracy (8 and 2 true positives, as well as 4 and 10 true negatives, respectively) across all 12 strains (Fig. 5B). Acetoacetate is transported into the cell via a proton symporter encoded by atoE. Growth of E. coli on SCFAs, such as acetoacetate, requires activation of the acid to its respective thioester (32). For acetoacetate, this activation is catalyzed by acetoacetyl-CoA transferase encoded by atoA and atoD that form a four unit enzymatic complex. The four strains lacking these two genes were correctly predicted not to grow on acetoacetate as a sole carbon source. Deoxyribose is the second compound predicted with 100% accuracy. It is transported into the cell via a proton symporter encoded by deoP. Deoxyribose is then phosphorylated to deoxyribose-5-phosphate by deoxyribose kinase, encoded by deoK (33). Finally, deoxyribose-5-phosphate is converted into acetaldehyde and glyceraldehyde-3-phosphate by deoxyribose-phosphate aldolase encoded by deoC. Only two of the strains tested, E. coli O42 and E. coli CFTO73, possessed these genes and were correctly predicted by the models to be able to grow on deoxyribose as a sole carbon source. These cases validate the approach and demonstrate high accuracy to discriminate between different strains’ capabilities.
A single pathway for two aromatic phenyl compounds, phenylacetaldehyde and phenylethylamine, was responsible for significant differences between model predictions and experimental results. The growth predictions for phenylacetaldehyde and phenylethylamine (tested individually) exhibited identical profiles of seven false negatives and only five true positives during in vivo growth screens (42% accuracy). These two compounds share a catabolic pathway whereby phenylethylamine is converted to phenylacetaldehyde by phenylethylamine oxidase encoded by tynA (34). Phenylacetaldehyde dehydrogenase, encoded for by feaB, then converts phenylacetaldehyde to phenylacetic acid. The acid is subsequently converted to phenylacetyl-CoA by phenylacetate-CoA ligase encoded by paaK. The genes coding for enzymes that catalyze these reactions in E. coli K-12 MG1655 had very low identity (<40%) at the amino acid level to genes in strains that proved capable of using these substrates as sole sources of carbon. Therefore, the growth experiments indicate that either these low identity enzymes are carrying out the activity or there is an alternate pathway for catabolism of these two compounds.
Discussion
GEMs of metabolism are powerful tools that can be deployed to investigate similarities and differences between strains of the same species. Unique GEMs for 55 different E. coli strains were constructed and used to (i) compare and contrast core and pan metabolic capabilities within the E. coli species; (ii) determine functional differences between strains by computing growth phenotypes on more than 650 different nutrients both aerobically and anaerobically; and (iii) explore the genetic basis behind strain-specific auxotrophies. These computational classifications and studies were fortified by performing in vitro screens of select discriminating compounds and strains resulting in a high level of accuracy (80%).
The majority of reactions found in pan metabolism fell into the metabolic subsystem of alternate carbon metabolism. It was hypothesized that these differences give different strains advantages in preferred microenvironmental niches. A clustering analysis based on computed metabolic phenotypes clearly distinguished E. coli strains from Shigella and largely separated E. coli strains known to exhibit a commensal intestinal lifestyle from those known to exhibit both intestinal and extraintestinal pathogenic lifestyles. This separation was based solely on the catabolic capabilities of different strains for unique nutrient sources. A major distinction that appeared was the capability to degrade fructoselysine and psiscoselysine, indicating that these compounds may be a defining feature of intestinal E. coli strains. This pathway is missing from all extraintestinal E. coli strains investigated. One possible mechanism that explains this feature begins with the observation that fructoselysine is poorly digested in the small intestine and absorption occurs only through diffusion (23). As a result, fructoselysine moves to the large intestine where it is present in abundance. However, excretory levels of fructoselysine are low; thus, it has been postulated that the intestinal microbiota, including E. coli, ferment almost all dietary fructoselysine. E. coli K-12 MG1655 has been shown to sustain growth on fructoselysine as a sole carbon source in anaerobic environments (23), and 88% of the intestinal strains modeled are predicted to be capable of using fructoselysine or psiscoselysine as a sole carbon source.
Another example of strain discrimination was the aromatic compound 4-hydroxyphenylacetic acid that was catabolized by 55% of commensal intestinal strain models compared with 9% of extraintestinal strain models. Hydroxyphenylacetic acids are one of several classes of aromatic compounds produced by bacterial fermentation of short chain peptides and amino acids in the human large intestine (24). Specifically, 3- and 4-hydroxyphenylacetic acid have been identified as products of tyrosine fermentation (34) by the diverse colonic microbiota. Therefore, these compounds are likely present at high levels in the intestine. Thus, utilization of 4-hydroxyphenylacetic acid as a sole carbon source may provide a competitive advantage over other strains of E. coli in the gut.
In addition to unique growth capabilities, the GEMs are also able to reliably predict strain-specific auxotrophies. This ability is important as auxotrophies often indicate cases of antagonistic pleiotrophy, whereby ancestral traits that interfere with virulence are lost to a newly evolved pathogen. Traits absent in pathogenic strains of a species but commonly expressed in commensal ancestors are strong candidates for pathoadaptive mutations. Evidence of this model of pathogen evolution was first provided by Shigella and E. coli. Widespread niacin auxotrophies in Shigella strains were identified due to disruption of nadA and nadB genes that code for the enzyme complex that converts l-aspartate to quinolate, a precursor to NAD synthesis. This finding is validated by previous literature confirming that quinolate inhibits invasion and cell-to-cell spread of Shigella flexneri 5a. Reintroduction of functional copies of nadA and nadB into this strain restored the ability to synthesize quinolate but also resulted in strong attenuation of virulence in this strain (35). Therefore, several of the additional auxotrophies identified for other vitamins such as folate and thiamin, as well as amino acids leucine, methionine, and tryptophan, may indicate further cases of antagonistic pleiotrophy. Future studies could explore the impact of these auxotrophies on virulence in each strain to potentially elucidate new pathoadaptive mutations.
Growth experiments were performed for six carbon sources tested on 12 different strains to evaluate the accuracy of the developed models. The overall accuracy of the models was 80%, a level that is in line with predecessor models (12–15). This high level of accuracy is notable because the substrates tested were selected due to their ability to differentiate among strains, making them some of the most difficult compounds to correctly predict. Three of the strains, 8739, HS, and MG1655, had 100% predictive accuracy. These strains are all safe, commensal laboratory strains, which likely contributed to them having better genome annotations and subsequently more accurate model predictions. Cases where the models are incorrect provide opportunities for biological discovery. False positives represent missing context-specific information in a GEM. These cases occur when model-predicted growth on a compound disagrees with the lack of growth observed experimentally. For example, growth on d-malate was a false positive for two models of E. coli strains. Even though both strains have a gene that has high identity to the d-malate decarboxylating oxidoreductase enzyme, encoded by yeaU, they were unable to grow on this compound. This could indicate a case where expression of this enzyme is transcriptionally repressed. Adaptive laboratory evolution of these two strains on d-malate may relieve the transcriptional repression of yeaU and lead to identification of novel regulators involved in controlling the catabolism of this compound (36).
In contrast to false positives, false negatives occur when comparing in silico and in vitro data to identify missing content in a GEM. Growth predictions for phenylacetaldehyde and phenylethylamine consisted of seven false negatives. These two compounds share the same catabolic pathway. The three genes catalyzing reactions in this pathway for E. coli K-12 MG1655 had very low identity (<40%) to genes in strains that proved capable of using these substrates as sole sources of carbon. Therefore, the growth experiments indicate that either a particular domain on these low identity enzymes is carrying out the activity or there is an alternate pathway for catabolism of these two compounds. Further characterization and studies on gene KOs for this pathway in each strain could lead to identification of a new alternate pathway for phenylacetaldehyde and phenylethylamine catabolism.
The work presented here shows that strain-specific models of E. coli can guide further studies regarding the advantages conferred by unique nutrients to E. coli strains in different niches. Additionally, the models reliably predict strain-specific auxotrophies documented in the literature, as well as novel auxotrophies that offer a strong case for future study. Taken together, this study represents a step toward the definition of a bacterial species based on common metabolic capabilities and its strains based on niche-specific growth capabilities. In addition to this fundamental advance, the niche-specific characteristics provide a basis for understanding strain and species-specific pathogenesis. Similar studies of diverse strains for species beyond E. coli will further define the concept of a species. Ultimately, this understanding can be leveraged to formulate strain- and species-specific drug development and therapeutic approaches.
Materials and Methods
The strain-specific model reconstruction procedure, gap filling algorithms, and in silico growth simulation conditions are described in SI Materials and Methods. Heatmap and phylogenetic tree and decision tree construction are described in SI Materials and Methods Carbon source and growth testing protocols are described in SI Materials and Methods. Eleven strains of E. coli and one strain of S. flexneri were tested as part of this study. The 11 E. coli strains are SMS 3–5; CFT073; HS; DH1; UMN 026; K011; Sakai; ATCC 8739; 042; EDL933; and K-12 MG1655. The S. flexneri strain was 2457T. E. coli 042 was a gift from Ian Henderson (Birmingham University, Birmingham, UK). All other strains were purchased from ATCC.
Supplementary Material
Acknowledgments
We thank Ian Henderson for the gift of E. coli strain 042. We thank Kathy Andrews and Donghyuk Kim for assistance with culture and storage of the strains used in this work. Additionally, we thank Aarash Bordbar for helpful discussions and insights. This work was funded by National Institutes of Health/National Institute of General Medical Sciences Grants 1R01GM098105 and 1R01GM057089.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. A.A.C. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1307797110/-/DCSupplemental.
References
- 1.Hayashi T, et al. Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 2001;8(1):11–22. doi: 10.1093/dnares/8.1.11. [DOI] [PubMed] [Google Scholar]
- 2.Perna NT, et al. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature. 2001;409(6819):529–533. doi: 10.1038/35054089. [DOI] [PubMed] [Google Scholar]
- 3.Canchaya C, Proux C, Fournous G, Bruttin A, Brüssow H. Prophage genomics. Microbiol Mol Biol Rev. 2003;67(2):238–276. doi: 10.1128/MMBR.67.2.238-276.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Touchon M, et al. Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet. 2009;5(1):e1000344. doi: 10.1371/journal.pgen.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liao YC, et al. An experimentally validated genome-scale metabolic reconstruction of Klebsiella pneumoniae MGH 78578, iYL1228. J Bacteriol. 2011;193(7):1710–1717. doi: 10.1128/JB.01218-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baumler DJ, Peplinski RG, Reed JL, Glasner JD, Perna NT. The evolution of metabolic networks of E. coli. BMC Syst Biol. 2011;5:182. doi: 10.1186/1752-0509-5-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Archer CT, et al. The genome sequence of E. coli W (ATCC 9637): Comparative genome analysis and an improved genome-scale reconstruction of E. coli. BMC Genomics. 2011;12:9. doi: 10.1186/1471-2164-12-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoon SH, et al. Comparative multi-omics systems analysis of Escherichia coli strains B and K-12. Genome Biol. 2012;13(5):R37. doi: 10.1186/gb-2012-13-5-r37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Charusanti P, et al. An experimentally-supported genome-scale metabolic network reconstruction for Yersinia pestis CO92. BMC Syst Biol. 2011;5:163. doi: 10.1186/1752-0509-5-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thiele I, et al. A community effort towards a knowledge-base and mathematical model of the human pathogen Salmonella Typhimurium LT2. BMC Syst Biol. 2011;5:8. doi: 10.1186/1752-0509-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blattner FR, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277(5331):1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 12.Edwards JS, Palsson BO. The Escherichia coli MG1655 in silico metabolic genotype: Its definition, characteristics, and capabilities. Proc Natl Acad Sci USA. 2000;97(10):5528–5533. doi: 10.1073/pnas.97.10.5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reed JL, Vo TD, Schilling CH, Palsson BO. An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR) Genome Biol. 2003;4(9):R54. doi: 10.1186/gb-2003-4-9-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feist AM, et al. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol Syst Biol. 2007;3:121. doi: 10.1038/msb4100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orth JD, et al. A comprehensive genome-scale reconstruction of Escherichia coli metabolism—2011. Mol Syst Biol. 2011;7:535. doi: 10.1038/msb.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCloskey D, Palsson BO, Feist AM. Basic and applied uses of genome-scale metabolic network reconstructions of Escherichia coli. Mol Syst Biol. 2013;9:661. doi: 10.1038/msb.2013.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feist AM, Palsson BO. The growing scope of applications of genome-scale metabolic reconstructions using Escherichia coli. Nat Biotechnol. 2008;26(6):659–667. doi: 10.1038/nbt1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee SY. Systems Biology and Biotechnology of Escherichia coli. Springer, New York; 2009. [Google Scholar]
- 19.Pupo GM, Lan R, Reeves PR. Multiple independent origins of Shigella clones of Escherichia coli and convergent evolution of many of their characteristics. Proc Natl Acad Sci USA. 2000;97(19):10567–10572. doi: 10.1073/pnas.180094797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DebRoy C, Roberts E, Fratamico PM. Detection of O antigens in Escherichia coli. Anim Health Res Rev. 2011;12(2):169–185. doi: 10.1017/S1466252311000193. [DOI] [PubMed] [Google Scholar]
- 21.Feist AM, Herrgård MJ, Thiele I, Reed JL, Palsson BO. Reconstruction of biochemical networks in microorganisms. Nat Rev Microbiol. 2009;7(2):129–143. doi: 10.1038/nrmicro1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bliven KA, Maurelli AT. Antivirulence genes: Insights into pathogen evolution through gene loss. Infect Immun. 2012;80(12):4061–4070. doi: 10.1128/IAI.00740-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erbersdobler HF, Faist V. Metabolic transit of Amadori products. Nahrung. 2001;45(3):177–181. doi: 10.1002/1521-3803(20010601)45:3<177::AID-FOOD177>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 24.Smith EA, Macfarlane GT. Enumeration of human colonic bacteria producing phenolic and indolic compounds: effects of pH, carbohydrate availability and retention time on dissimilatory aromatic amino acid metabolism. J Appl Bacteriol. 1996;81(3):288–302. doi: 10.1111/j.1365-2672.1996.tb04331.x. [DOI] [PubMed] [Google Scholar]
- 25.Prieto MA, Díaz E, García JL. Molecular characterization of the 4-hydroxyphenylacetate catabolic pathway of Escherichia coli W: Engineering a mobile aromatic degradative cluster. J Bacteriol. 1996;178(1):111–120. doi: 10.1128/jb.178.1.111-120.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karch H, Tarr PI, Bielaszewska M. Enterohaemorrhagic Escherichia coli in human medicine. Int J Med Microbiol. 2005;295(6-7):405–418. doi: 10.1016/j.ijmm.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 27.Nakano M, et al. Association of the urease gene with enterohemorrhagic Escherichia coli strains irrespective of their serogroups. J Clin Microbiol. 2001;39(12):4541–4543. doi: 10.1128/JCM.39.12.4541-4543.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reed JL, et al. Systems approach to refining genome annotation. Proc Natl Acad Sci USA. 2006;103(46):17480–17484. doi: 10.1073/pnas.0603364103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prunier AL, Schuch R, Fernández RE, Maurelli AT. Genetic structure of the nadA and nadB antivirulence loci in Shigella spp. J Bacteriol. 2007;189(17):6482–6486. doi: 10.1128/JB.00525-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zagaglia C, et al. Virulence plasmids of enteroinvasive Escherichia coli and Shigella flexneri integrate into a specific site on the host chromosome: Integration greatly reduces expression of plasmid-carried virulence genes. Infect Immun. 1991;59(3):792–799. doi: 10.1128/iai.59.3.792-799.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Durfee T, et al. The complete genome sequence of Escherichia coli DH10B: Insights into the biology of a laboratory workhorse. J Bacteriol. 2008;190(7):2597–2606. doi: 10.1128/JB.01695-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jenkins LS, Nunn WD. Genetic and molecular characterization of the genes involved in short-chain fatty acid degradation in Escherichia coli: The ato system. J Bacteriol. 1987;169(1):42–52. doi: 10.1128/jb.169.1.42-52.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bernier-Fébreau C, et al. Use of deoxyribose by intestinal and extraintestinal pathogenic Escherichia coli strains: A metabolic adaptation involved in competitiveness. Infect Immun. 2004;72(10):6151–6156. doi: 10.1128/IAI.72.10.6151-6156.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Díaz E, Ferrández A, Prieto MA, García JL. Biodegradation of aromatic compounds by Escherichia coli. Microbiol Mol Biol Rev. 2001;65(4):523–569. doi: 10.1128/MMBR.65.4.523-569.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prunier AL, et al. nadA and nadB of Shigella flexneri 5a are antivirulence loci responsible for the synthesis of quinolinate, a small molecule inhibitor of Shigella pathogenicity. Microbiology. 2007;153(Pt 7):2363–2372. doi: 10.1099/mic.0.2007/006916-0. [DOI] [PubMed] [Google Scholar]
- 36.Lee DH, Palsson BO. Adaptive evolution of Escherichia coli K-12 MG1655 during growth on a Nonnative carbon source, L-1,2-propanediol. Appl Environ Microbiol. 2010;76(13):4158–4168. doi: 10.1128/AEM.00373-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.