

Significance
This study determined that two histological phenotypes of cancer can arise from a common target cell. Whereas luminal cells are not efficient cells of origin, luminal-like tumor cells isolated from human prostate adenocarcinoma can serially propagate advanced disease.
Abstract
The relationship between the cells that initiate cancer and the cancer stem-like cells that propagate tumors has been poorly defined. In a human prostate tissue transformation model, basal cells expressing the oncogenes Myc and myristoylated AKT can initiate heterogeneous tumors. Tumors contain features of acinar-type adenocarcinoma with elevated eIF4E-driven protein translation and squamous cell carcinoma marked by activated beta-catenin. Lentiviral integration site analysis revealed that alternative histological phenotypes can be clonally derived from a common cell of origin. In advanced disease, adenocarcinoma can be propagated by self-renewing tumor cells with an androgen receptor-low immature luminal phenotype in the absence of basal-like cells. These data indicate that advanced prostate adenocarcinoma initiated in basal cells can be maintained by luminal-like tumor-propagating cells. Determining the cells that maintain human prostate adenocarcinoma and the signaling pathways characterizing these tumor-propagating cells is critical for developing effective therapeutic strategies against this population.
Tumors that arise from a given tissue in the body exhibit heterogeneity with respect to their molecular alterations, biological behavior, and response to therapy (1). Such variation presents a serious challenge for clinical cancer management. In many organ sites, tumors have been classified into subtypes based on their molecular and histological features (2). Subtypes of cancer can reflect distinct states of differentiation within a given tissue, leading Visvader and coworkers to propose that different epithelial tumor subtypes can arise from transformation of distinct cells of origin with different developmental potential (3). Functional studies in the mouse mammary gland and mouse lung support this model (4, 5). However, there is limited functional evidence for such a mechanism in human epithelial cancer.
Several recent studies using mouse models have revealed that the same phenotypic cell that initiates cancer can be responsible for tumor maintenance or propagation. Lgr5+ intestinal stem cells can initiate and maintain murine intestinal adenomas (6, 7). In mouse models of skin cancer, hair follicle bulge stem cells can serve as target cells for transformation (8) and CD34+ cells resembling their normal bulge stem cell counterpart are capable of propagating the disease as a cancer stem cell population (9). Mouse models of breast cancer demonstrate that tumors can arise from the transformation of luminal cells (4), and recent studies using human tumor samples indicate that breast cancer can also be propagated by luminal-like cells (10). In most human epithelial cancers it has not been determined whether the cell types that give rise to cancer are also capable of maintaining advanced disease.
The predominant histological subtype of prostate cancer is acinar-type adenocarcinoma (11), with features of luminal secretory cells, rare neuroendocrine cells, and an absence of basal cells. A number of less common histological variants are found in prostate cancer, including small cell carcinoma and squamous cell carcinoma. Both of these variants are associated with poor prognosis, aggressive disease, and resistance to hormonal therapy (androgen deprivation and/or androgen receptor blockade) (11). Small-cell carcinoma is characterized by proliferating neuroendocrine cells and loss of p53 (12). Squamous cancers have features of basal cells and can occur either in the context of adenocarcinoma or alone as squamous cell carcinoma (11, 13, 14). Based on their different phenotypes and response to hormonal therapy, different histological variants of prostate cancer are predicted to arise from distinct cells of origin (13).
The relationship between the cells that initiate and maintain human prostate adenocarcinoma is not known. Naïve human prostate basal cells can initiate acinar-type adenocarcinoma in response to oncogenic stimulation (15). Consistent with these findings, basal cells from the BPH-1 human prostate cell line can initiate human prostate cancer in response to combined estrogen and testosterone treatment (16). These collective data suggest that human prostate tumors may set aside a subset of basal cells within the tumor to ensure continuous production of malignant luminal-like cancer cells.
Human prostate cancer cells with a basal phenotype have been reported to produce luminal cancer progeny in vitro (17). Using cell lines that were originally derived from human prostate tumors, it was shown that basal cell marker CD44 enriched for tumor-propagating cells in the absence of differentiated luminal cell markers (18). A recent study demonstrates that advanced chemotherapy-resistant prostate cancer is maintained by cells lacking basal or luminal cytokeratins (19). No study has defined the role of basal or luminal-like cells isolated directly from primary human prostate cancer in tumor propagation.
In the present study, we use a tissue-regeneration model of human prostate cancer to determine whether the cells at the origin of prostate cancer are continually required to maintain the disease as tumor-propagating cells. Benign cell populations isolated from primary human prostate tissue were first tested for their susceptibility to transformation by defined oncogenes. In the resulting tumors, cancer cell populations were further transplanted to define the cells capable of propagating the disease. Tumors driven by expression of oncogenes Myc and myristoylated/activated AKT (myrAKT) initiating in basal cells exhibit features of both adenocarcinoma and squamous cell carcinoma with different signaling pathways characteristic of each histological pattern. eIF4E-driven protein translation pathway is elevated in adenocarcinoma, whereas activation of beta-catenin is associated with squamous differentiation in experimental and clinical human prostate cancer. Using lentiviral integration site analysis, we determined that alternative histological phenotypes of prostate cancer can arise from a clonal cell of origin. Adenocarcinoma can be serially propagated by cells with a luminal phenotype. Our results indicate that cancer initiated in basal cells can evolve to adenocarcinoma maintained by luminal-like cells.
Results
Basal Cells Initiate Heterogeneous Human Prostate Cancer.
Two of the most common alterations identified in human prostate tumors are increased expression of Myc (20) and activation of AKT, typically via loss of PTEN (21). Coexpression of Myc and phosphorylated AKT are rarely observed in primary localized prostate cancer, but are commonly found in advanced metastatic prostate tumors (Fig. 1A). The oncogenes Myc, myrAKT, or both Myc and myrAKT were introduced via lentivirus into highly enriched populations of human prostate basal (CD45−Trop2+CD49fhiCD26−) and luminal (CD45−Trop2+CD49floCD26+) cells isolated by FACS. Basal and luminal epithelial cells were purified from preparations of dissociated, freshly isolated prostate tissue from six patients undergoing radical prostatectomy. Transduced human prostate epithelial cells were combined with inductive murine urogenital sinus mesenchyme (UGSM) with Matrigel and implanted s.c. into immune-deficient NOD-SCID-IL2Rγnull mice (Fig. S1A). Consistent with previous findings (15), infection of human prostate luminal-enriched cells did not result in any detectable epithelial structures after 12 wk in vivo, regardless of whether cells were transduced with a single oncogene or the combination of Myc and myrAKT. Even implantation of 100,000 luminal cells was insufficient to generate primary tumors (Fig. S1B).
Fig. 1.

Myc and myrAKT are coexpressed in advanced prostate cancer and synergize to initiate human prostate cancer. (A) Localized (black line) and metastatic (red line) human prostate cancer specimens were subjected to Western blot analysis with antibodies against phosphorylated AKT (pAKT), Myc, and Erk as a loading control. (B) Naïve benign basal or luminal cells were transduced with Myc, myrAKT, or both Myc and myrAKT at varying cell doses. Representative tumors initiated from basal cells expressing Myc, myrAKT, or both after 8 wk of regeneration. (Scale bar, 1 cm.) (C) Histology of representative lesions initiated from basal cells expressing Myc, myrAKT, or both stained for H&E, AR, and p63. (Scale bars, 100 μm.)
When expressed in naïve human prostate basal-enriched cells, Myc or myrAKT alone gave rise to benign glands or low-grade prostatic intraepithelial neoplasia lesions with distinct p63+ basal and androgen receptor-positive (AR+) luminal layers (Fig. 1 B and C). The two oncogenes dramatically synergized in human prostate basal cells to consistently generate large tumors in as little as 6 wk (Fig. 1B). Transduced basal cells were implanted in limiting numbers (105, 5 × 104, 104, and 103) in vivo to determine the number of target cells required to respond to oncogenic stimulation. As few as 10,000 basal cells were sufficient to initiate tumors in 12 wk (Fig. S1B). Metastasis was not observed in tumor-bearing mice, indicating that further genetic alterations or a longer time period may be required to observe metastatic lesions.
Regenerated tumors contained both acinar-type adenocarcinoma and squamous features (Fig. 2A). Adenocarcinoma regions were defined by high levels of expression of the luminal markers Keratin 8 (K8) and CD26, heterogeneous expression of AR, scattered chromogranin A+ neuroendocrine-like (NE) cells and an absence of basal markers Keratin 14 (K14), p63, and Keratin 5 (K5) (Fig. 2B). Conversely, squamous regions expressed basal cell markers K14, p63, and K5 and lacked cells expressing K8, CD26, AR, or chromogranin A. All tumors were confirmed to express oncogenes Myc and myrAKT and have a human origin based on staining with a pan-HLA antibody (Fig. 2B and Fig. S2). This heterogeneous tumor provides a model to investigate the relationship between different histological phenotypes within an individual cancer. In clinical prostate cancer, the squamous phenotype is rare in primary tumors. Squamous differentiation is predominantly observed in aggressive metastatic tumors that are resistant to androgen-deprivation therapy (22).
Fig. 2.

Heterogeneous human prostate tumors containing squamous and acinar-type adenocarcinoma phenotypes. (A) H&E stains demonstrate heterogeneous tumors generated from the transformation of naïve benign basal cells from six distinct individuals. Dotted lines represent the borders of adenocarcinoma and squamous phenotypes. (Scale bars, 100 μm.) (B) Representative adenocarcinoma and squamous regions are identified based on staining for H&E and antibodies against luminal markers K8, CD26, and AR, the neuroendocrine marker chromogranin A, and basal/squamous markers K14, p63, and K5, and oncogenes Myc and myrAKT/pAKT. (Scale bars, 100 μm.)
Distinct Histological Phenotypes Share a Clonal Origin.
Human prostate cancer often presents as a multifocal disease where distinct cancerous lesions are present within an individual patient’s tissue. Multiple tumor foci can regularly be identified within regenerated tumors using the in vivo tissue-regeneration assay (15). We identified certain regions containing cells with adjacent squamous (K14/p63/K5) and luminal (K8/CD26) phenotypes in close proximity without defined borders (Fig. 3 A and B). These mixed regions made up an average of six foci per tumor, covering ∼18% of the total tumor area (Fig. S3). These regions containing both histological phenotypes originate either from the merging of two neighboring glands of distinct histological phenotypes or from a single gland capable of multilineage differentiation. The most definitive method to distinguish these possible outcomes is lentiviral integration site analysis (23).
Fig. 3.

Distinct histological variants in heterogeneous tumors can share a clonal origin. (A) Schematic of two different heterogeneous tumors containing adenocarcinoma, squamous, or both phenotypes. Regions X, Y, and Z. (B) Representative regions X, Y, and Z are shown with serial tissue sections stained with K8 to highlight adenocarcinoma and either p63 or K5 to highlight squamous regions. Dotted lines indicate region excised using laser capture microdissection. (Scale bars, 100 μm.) (C) Schematic of lentiviral integration site analysis. LTR, long terminal repeat (viral DNA). (D) Venn diagrams depict shared lentiviral integration sites in DNA isolated and amplified from neighboring adenocarcinoma (red) and squamous (green) phenotypes (region X), distinct adenocarcinoma gland (region Y), and additional neighboring adenocarcinoma and squamous phenotypes (region Z). (E) Table lists all unique integration sites (IS) with genomic location identifiers (chromosome, orientation, and nucleotide position) representing at least 1% of total reads (indicated by +) in each sample. Rows in yellow represent shared IS between distinct histological phenotypes in the same region, rows in red indicate IS unique to adenocarcinoma, and green represent IS unique to squamous.
Tumors were initiated from naïve human prostate basal cells transduced with lentivirus, resulting in the integration of viral DNA into the genome of the target cell and all of its progeny. If both adenocarcinoma and squamous phenotypes within an individual region arise from the same transformed basal cell, they should share a set of common lentiviral integration sites. However, if distinct histological phenotypes in an individual region arise from distinct cells of origin, they should not share any common integration sites.
Laser capture microdissection was performed on neighboring adenocarcinoma and squamous phenotypes within an individual lesion (region X) and DNA was isolated separately from adenocarcinoma and squamous cells for lentiviral integration site analysis (Fig. 3B). DNA was also isolated from an individual lesion containing only the adenocarcinoma phenotype (region Y). In a distinct tumor, DNA was collected from neighboring adenocarcinoma and squamous phenotypes in close proximity (region Z). PCR primers specific for the viral DNA were used to extend into the genomic DNA (Fig. 3C). Illumina sequencing was performed and reads were aligned to the genome to map integration sites (Fig. 3C). Neighboring adenocarcinoma and squamous cells (region X) shared common integration sites (Fig. 3 D and E), showing that they are derived from a clonal origin. In contrast, tissue taken from a distinct adenocarcinoma lesion (region Y) had entirely unique integration sites (Fig. 3 D and E). Common integration sites were also shared between neighboring adenocarcinoma and squamous phenotypes taken from a separate tumor (Region Z, Fig. 3 D and E). No overlapping integration sites were observed between different regions, as would be expected.
Beta-catenin signaling has been implicated in squamous differentiation and tumorigenesis in the prostate, mammary gland, and skin (9, 24, 25). In human regenerated prostate tumors, the expression of total and activated forms of beta-catenin was evaluated in both adenocarcinoma and squamous tissues. In contrast to total beta-catenin, which is expressed in both histological variants, active beta-catenin is highly expressed in squamous areas (Fig. S4). Elevated Wnt signaling in the prostate microenvironment can transform adjacent naïve benign epithelium (26). Wnt ligands are elevated in prostate cancer stromal cells following treatment, leading to beta-catenin activation in the adjacent tumor cells (27). This is consistent with the presence of squamous differentiation in prostate cancer, which is rare in primary tumors but more commonly observed in late-stage prostate cancer following treatment (14, 22). High levels of activated beta-catenin could also be detected in clinical metastatic castration-resistant prostate cancer with squamous differentiation (Fig. S4).
Luminal-Like Cancer Cells Serially Propagate Adenocarcinoma in the Absence of Basal-Like Cells.
Previous studies have established optimal conditions for tumorigenicity of dissociated human tumor cell preparations (28). Regenerated primary human prostate tumor cells initiated from the transformation of five distinct benign patient samples were dissociated to single cells. Human tumor cells were identified by FACS based on staining of a pan-HLA antibody (Fig. 4A). Luminal-like tumor cells were then isolated on the basis of low expression of CD49f and transplanted into secondary NSG mice.
Fig. 4.

Luminal-like cells isolated from primary tumors propagate adenocarcinoma. (A) Tumors initiated from CD49fhi cells expressing Myc and myrAKT are dissociated to single cells and gated based on HLA+ and CD49f. Isolated HLA+ CD49flo cells are transplanted back into recipient mice and harvested 6–12 wk later. (Scale bar, 1 cm.) (B) H&E-stained overview of a representative secondary tumor generated from 10,000 isolated CD49flo tumor cells. Only the adenocarcinoma phenotype is observed as evidenced by stains for H&E, K8, CD26, K14, and p63. (Scale bars, 50 μm.)
Upon transplantation, isolated CD49flo tumor cells were capable of generating secondary tumors in recipient mice. Immunohistochemical staining on tumor-derived tissue sections identified a phenotype consistent with luminal/acinar-type adenocarcinoma (29) with a predominance of K8+ CD26+ luminal-like cells and an absence of K14+ p63+ basal-like cells (Fig. 4B). CD49flo tumor cells failed to regenerate the squamous phenotype. Secondary tumors derived from CD49flo cancer cells were almost entirely composed of a CD49flo Keratin 18+ luminal-like phenotype as analyzed by flow cytometry (Fig. 5A) and expressed oncogenes Myc and myrAKT (Fig. S5). As few as 100 CD49flo cells taken from secondary tumors could generate tertiary tumors with a strictly adenocarcinoma phenotype (Fig. 5B). These data suggest that CD49flo tumor cells can self-renew in a unipotent manner in vivo.
Fig. 5.

Luminal-like tumor-propagating cells maintain human prostate adenocarcinoma in the absence of basal-like cells. (A) Flow cytometry characterization of secondary tumors derived from CD49flo tumor cells. Secondary tumors derived from CD49flo tumor cells were dissociated to single cells and stained for surface levels of CD49f or subjected to intracellular flow cytometry and stained for K18. (B) Table indicates number of tumors formed per transplantation of CD49flo cells (taken from either primary or secondary tumors) at varying cell doses. (C) CD49flo secondary tumor cells can generate tertiary tumors upon transplantation. Representative tertiary tumor sections are shown, stained for H&E, the pan-human HLA-A/B/C antibody, and markers of luminal adenocarcinoma (K8 and CD26) and basal cells (K14 and p63). (Scale bars, 100 μm.)
CD49fhi tumor cells were also tested in transplantation assays. We found that CD49fhi tumor cells could regenerate secondary lesions exhibiting a mixture of squamous (K14+ p63+) and adenocarcinoma (K8+ CD26+) areas (Fig. S6) that both expressed Myc and myrAKT (Fig. S7). These data suggest the presence of multipotent cells in the CD49fhi fraction capable of propagating both squamous and adenocarcinoma phenotypes.
In numerous tissues, high expression of Myc is associated with a block in differentiation and reprogramming to pluripotency (30). In prostate epithelium, Myc overexpression has been shown to reduce levels of AR (31) and its target prostate-specific antigen (PSA) (32). We hypothesized that high levels of Myc in Myc/myrAKT-driven tumors might cause a block in differentiation, resulting in reduced levels of AR and PSA. In primary regenerated tumors, we found low and heterogeneous expression of AR with only rare PSA+ glands (Fig. 1C and Fig. S8). Levels of AR and PSA were low in secondary and tertiary tumors upon serial transplantation (Fig. 5C and Fig. S8). These results are consistent with recent studies demonstrating that low or negative levels of AR and PSA are associated with tumor-propagating cells in prostate cancer xenografts (18, 19, 33, 34). Low levels of AR and PSA are also characteristic of DU145 (35) and PC3 (36) aggressive metastatic prostate cancer cells. In fact, expression of PSA, characteristic of clinical acinar-type adenocarcinoma, is inversely correlated with patient survival (18).
Luminal-Like Cancer Cells Exhibit Elevated eIF4E-Driven Protein Translation.
Our findings demonstrate that whereas naïve benign luminal cells are not efficient cells of origin for prostate cancer, CD49flo tumor cells with a luminal phenotype can self-renew and maintain human prostate adenocarcinoma in the absence of CD49fhi or K14+ p63+ basal-like cells. Signaling pathways that are absent in benign luminal cells may become expressed in malignant luminal cells and contribute to their capacity to self-renew and initiate tumorigenesis. Two pathways implicated in prostate tumorigenesis are the MAPK pathway (37) and the JAK/STAT pathway (38). Myc and myrAKT-driven tumor cells exhibited low levels of phosphorylated Erk1/2, downstream of the MAPK pathway, and weak staining for phosphorylated STAT3, a readout of the JAK/STAT pathway (Fig. S9).
The protein translation factor eIF4E is downstream of the PI3K pathway and has been shown to cooperate with Myc in promoting cell growth and tumorigenesis (39). eIF4E is unable to promote translation when bound by 4EBP1. mTOR-mediated phosphorylation of 4EBP1 (p4EBP1) relieves this interaction and allows eIF4E-driven translation initiation. High levels of eIF4E and p4EBP1 are associated with poor prognosis in prostate cancer (40) and have been shown to drive tumor invasion (41).
Elevated levels of eIF4E, 4EBP1 and p4EBP1 were found in luminal-like tumor cells isolated from Myc and myrAKT-driven tumors but not in benign luminal cells or in neighboring squamous cells (Fig. 6 A and B and Fig. S10). Luminal-like tumor cells also express MTA1 and Sox2 (Fig. 6 A and B), two targets of eIF4E translation associated with prostate cancer progression. The self-renewal regulator Sox2 has been previously demonstrated to promote the proliferation of prostate cancer cells (42). eIF4E, p4EBP1, MTA1, and Sox2 were identified in subsets of benign basal cells but not benign luminal cells (Fig. 6A). These findings suggest that luminal-like tumor cells may acquire a limited set of self-renewal and cell-survival factors normally associated with stem-like basal cells to promote tumor propagation.
Fig. 6.

Luminal-like tumor-propagating cells exhibit elevated eIF4E-driven protein translation. (A) Immunohistochemical analysis of benign human prostate and primary tumors initiated by Myc and myrAKT stained for antibodies against eIF4E, 4EBP1, phosphorylated 4EBP1 (Thr37/46), and the eIF4E target MTA1. (Scale bars, 50 μm.) (B) Western blots of FACS-purified benign basal and luminal cells compared with luminal-like tumor cells stained with antibodies aginst eIF4E, phosphorylated 4EBP1 (Thr37/46), 4EBP1, MTA1, Sox2, the basal marker p63, and loading control Histone H3. (C) Luminal-like cells isolated from Myc and myrAKT-driven tumors were treated with DMSO, Dasatinib, Rapamycin, and PP242 for 18 h followed by an additional treatment 1 h before harvesting. Lysates were subjected to Western blot for phosphorylated 4EBP1 (Thr37/46), Sox2, MTA1, total and phosphorylated Src (Y416), and Erk2 as a loading control. (D) Luminal-like cells isolated from Myc- and myrAKT-driven tumors were plated into Matrigel and grown in a sphere assay. Cells were treated with DMSO, Dasatinib, Rapamycin, and PP242 every 48 h and sphere number and size was quantified after 10 d in vitro. Sphere number is presented as a percentage normalized to the DMSO control. (E) Sphere size is presented as the diameter in microns. Error bars represent SEM. Statistical analysis was performed using ANOVA and Newman–Keuls multiple comparison test. *P < 0.05, ***P < 0.0005. n.s., not statistically significant.
Luminal-like cancer cells were grown in vitro and treated with pharmacological inhibitors previously demonstrated to interfere with eIF4E-driven protein translation. mTOR-mediated phosphorylation of 4EBP1 can be prevented by treatment with Rapamycin, allowing nonphosphorylated 4EBP1 to bind and inhibit eIF4E. PP242 inhibits both mTORC1 and mTORC2 and has been previously demonstrated to deplete eIF4E-driven protein translation (43). Dasatinib is a Src family kinase inhibitor that does not alter 4EBP1 phosphorylation. We tested the effect of eIF4E-driven protein translation inhibition on luminal-like cancer cells isolated from Myc and myrAKT-driven tumors. Treatment with rapamycin and PP242, but not with Dasatinib, caused a reduction in MTA1 and Sox2 protein levels and depleted sphere number and size in vitro (Fig. 6 C–E).
Discussion
Epithelial cancers often exhibit significant heterogeneity at the histological level (1). By engineering expression of oncogenes or loss of tumor suppressors in specific lineages via cre-lox technology, it has been reported that different histological variants of mouse breast and lung cancer can arise from distinct cells of origin (4, 5). We have used a tissue-regeneration model to determine the origins of prostate cancer heterogeneity. Based on their distinct phenotypes and biological behaviors, histological variants of prostate cancer have been proposed to arise from different cells of origin (13). Myc- and myrAKT-initiated tumors exhibit features of both acinar-type adenocarcinoma and squamous cell carcinoma. Using lentiviral integration site analysis, we demonstrate that alternative human epithelial cancer phenotypes can arise from a common clonal target cell. Activated beta-catenin is elevated in areas with squamous differentiation both in experimental and clinical prostate cancer, suggesting a role for the beta-catenin pathway either in promoting or maintaining the squamous phenotype.
Our findings that basal cells are efficient cells of origin for human prostate cancer are consistent with previous findings by our group and others (15, 16). These studies on the human disease have all used a tissue-regeneration approach to model development concomitant with tumorigenesis. Several different groups have investigated the origins of murine prostate cancer using genetically engineered mouse models in which the tumor suppressor Pten is deleted in a subset of basal or luminal cells from a young age. Depending on the genetic background of the mouse and the frequency of Pten deletion by the promoters used, there is considerable disagreement on whether basal or luminal cells generate a more proliferative, aggressive tumor that can model lethal human prostate cancer (44–46). Regardless of the assay system, studies using both mouse and human tissue confirm that transformed basal cells can generate malignant luminal progeny in vivo.
In the present study, we find that human prostate adenocarcinoma initiated by transformed basal cells can be propagated by phenotypically luminal cancer cells (Fig. 7). Such a model resembles chronic myelogenous leukemia (CML). CML is initiated in transformed hematopoietic stem cells (47, 48). Advanced disease can be maintained by granulocyte-macrophage progenitor-like cells that have gained self-renewal (49–51).
Fig. 7.
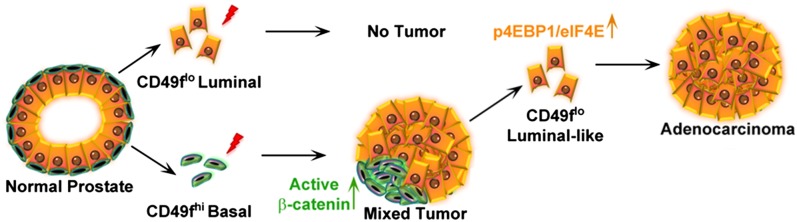
Model of human prostate cancer initiation and propagation by distinct phenotypic cell populations.
Several regulators of growth and self-renewal normally restricted to the stem-like basal cell compartment are likely to be expressed in malignant luminal cells in human prostate cancer and cooperate to promote their tumorigenic activity. In Myc- and myrAKT-driven human prostate tumors, luminal-like cancer cells exhibit elevated levels of p4EBP1 and eIF4E. Luminal-like tumor cells express MTA1 and Sox2, two targets of eIF4E-driven protein translation that are normally expressed in self-renewing basal cells but not benign luminal cells. These findings suggest that eIF4E-driven protein translation may contribute to luminal-like tumor-propagating cell survival and self-renewal. In fact, activation of eIF4E downstream of Pten deletion in murine models of prostate cancer may play an important role in luminal cell transformation (44, 46).
Methods
Full methods, including antibodies, lentiviral vectors, laser capture microdissection, DNA isolation, and nonrestrictive linear amplification-mediated (nrLAM) PCR are found in SI Methods.
Human Tissue.
Acquisition and processing of human tissue, dissociation and isolation of distinct epithelial subsets, lentiviral transduction, and in vivo implantation have all been described in detail (52). Patient tissue is provided in a de-identified manner and is exempt from institutional review board approval. Immunohistochemistry, Western blotting, and intracellular flow cytometry were performed as previously described (15).
Sequential Tumor Transplantation.
Primary tumors were minced into small pieces, dissociated in 1 mg/mL type I collagenase (Invitrogen), 1mg/mL Dispase (Invitrogen) and 50 μg/mL DNase I (Roche) at 37 °C on an Adams Nutator Mixed (BD Biosciences) for 2–4 h and further digested with 0.05% trypsin/EDTA (Invitrogen) for 5 min at 37 °C if necessary before sequential passing through 100-, 70-, and 40-μm cell strainers (BD Biosciences). Cells were stained with antibodies at 4 °C for 15–30 min and tumor cell populations were sorted on a BD FACS ARIA II into media with 50% (vol/vol) FBS (Omega Scientific). Isolated cells were counted by hemacytometer using trypan blue stain (Invitrogen), resuspended in 30 μL Matrigel (BD Biosciences), and implanted s.c. into NOD-SCID-IL2Rγnull (NSG) mice with or without UGSM cells.
Animal Work.
All primary, secondary, and tertiary tumors were transplanted s.c. into NOD-SCID-IL2Rγnull (NSG) mice. NSG mice were originally purchased from the Jackson Laboratories and were housed and bred under the care of the Division of Laboratory Animal Medicine at the University of California, Los Angeles (UCLA). Surgical castration (orchiectomy) of tumor-bearing mice was performed according to protocols approved by UCLA's Animal Research Committee.
Supplementary Material
Acknowledgments
We thank D. Cheng for cell sorting and the Tissue Procurement Core Laboratories for tissue preparation. Laser capture microdissection was performed at the California NanoSystems Institute Advanced Light Microscopy/Spectroscopy Shared Resource Facility at University of California, Los Angeles (UCLA), supported with funding from the Biosciences Core Facilities program at UCLA. T.S., J.M.D., and J.H. are supported by the Department of Defense Prostate Cancer Research Program. A.R.C. is supported by Ruth L. Kirschstein National Research Service Award GM007185. J.H. is supported by the UCLA Specialized Program of Research Excellence (SPORE) in prostate cancer (Prinicipal Investigator R. Reiter), a creativity award from the Prostate Cancer Foundation (PCF) (Principal Investigator M. Rettig) and National Institutes of Health Grant 1R01CA158627 (Principal Investigator L. Marks). A.S.G. is supported by the SPORE in Prostate Cancer (Principal Investigator R. Reiter) and a PCF Young Investigator Award. O.N.W. is an Investigator of the Howard Hughes Medical Institute and is supported by the Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research. A.S.G and O.N.W. are supported by a PCF Creativity Award (Principal Investigator O.N.W.). A.S.G, J.H., and O.N.W. are supported by a PCF Challenge Award (Principal Investigator O.N.W.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1320565110/-/DCSupplemental.
References
- 1.Visvader JE. Cells of origin in cancer. Nature. 2011;469(7330):314–322. doi: 10.1038/nature09781. [DOI] [PubMed] [Google Scholar]
- 2.Perou CM, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 3.Lim E, et al. kConFab Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15(8):907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 4.Molyneux G, et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell. 2010;7(3):403–417. doi: 10.1016/j.stem.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 5.Sutherland KD, et al. Cell of origin of small cell lung cancer: Inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell. 2011;19(6):754–764. doi: 10.1016/j.ccr.2011.04.019. [DOI] [PubMed] [Google Scholar]
- 6.Barker N, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457(7229):608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- 7.Schepers AG, et al. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science. 2012;337(6095):730–735. doi: 10.1126/science.1224676. [DOI] [PubMed] [Google Scholar]
- 8.Lapouge G, et al. Identifying the cellular origin of squamous skin tumors. Proc Natl Acad Sci USA. 2011;108(18):7431–7436. doi: 10.1073/pnas.1012720108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malanchi I, et al. Cutaneous cancer stem cell maintenance is dependent on beta-catenin signalling. Nature. 2008;452(7187):650–653. doi: 10.1038/nature06835. [DOI] [PubMed] [Google Scholar]
- 10.Kim J, et al. Tumor initiating but differentiated luminal-like breast cancer cells are highly invasive in the absence of basal-like activity. Proc Natl Acad Sci USA. 2012;109(16):6124–6129. doi: 10.1073/pnas.1203203109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Humphrey PA. Histological variants of prostatic carcinoma and their significance. Histopathology. 2012;60(1):59–74. doi: 10.1111/j.1365-2559.2011.04039.x. [DOI] [PubMed] [Google Scholar]
- 12.Chen H, et al. Pathogenesis of prostatic small cell carcinoma involves the inactivation of the P53 pathway. Endocr Relat Cancer. 2012;19(3):321–331. doi: 10.1530/ERC-11-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mott LJ. Squamous cell carcinoma of the prostate: Report of 2 cases and review of the literature. J Urol. 1979;121(6):833–835. doi: 10.1016/s0022-5347(17)57013-5. [DOI] [PubMed] [Google Scholar]
- 14.Parwani AV, et al. Prostate carcinoma with squamous differentiation: an analysis of 33 cases. Am J Surg Pathol. 2004;28(5):651–657. doi: 10.1097/00000478-200405000-00014. [DOI] [PubMed] [Google Scholar]
- 15.Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON. Identification of a cell of origin for human prostate cancer. Science. 2010;329(5991):568–571. doi: 10.1126/science.1189992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taylor RA, et al. Australian Prostate Cancer Bioresource Human epithelial basal cells are cells of origin of prostate cancer, independent of CD133 status. Stem Cells. 2012;30(6):1087–1096. doi: 10.1002/stem.1094. [DOI] [PubMed] [Google Scholar]
- 17.Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65(23):10946–10951. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- 18.Qin J, et al. The PSA(-/lo) prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell Stem Cell. 2012;10(5):556–569. doi: 10.1016/j.stem.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Domingo-Domenech J, et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell. 2012;22(3):373–388. doi: 10.1016/j.ccr.2012.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gurel B, et al. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod Pathol. 2008;21(9):1156–1167. doi: 10.1038/modpathol.2008.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang S, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4(3):209–221. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 22.Rubin MA, et al. Effects of long-term finasteride treatment on prostate cancer morphology and clinical outcome. Urology. 2005;66(5):930–934. doi: 10.1016/j.urology.2005.07.036. [DOI] [PubMed] [Google Scholar]
- 23.Paruzynski A, et al. Genome-wide high-throughput integrome analyses by nrLAM-PCR and next-generation sequencing. Nat Protoc. 2010;5(8):1379–1395. doi: 10.1038/nprot.2010.87. [DOI] [PubMed] [Google Scholar]
- 24.Bierie B, et al. Activation of beta-catenin in prostate epithelium induces hyperplasias and squamous transdifferentiation. Oncogene. 2003;22(25):3875–3887. doi: 10.1038/sj.onc.1206426. [DOI] [PubMed] [Google Scholar]
- 25.Miyoshi K, et al. Activation of beta -catenin signaling in differentiated mammary secretory cells induces transdifferentiation into epidermis and squamous metaplasias. Proc Natl Acad Sci USA. 2002;99(1):219–224. doi: 10.1073/pnas.012414099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zong Y, et al. Stromal epigenetic dysregulation is sufficient to initiate mouse prostate cancer via paracrine Wnt signaling. Proc Natl Acad Sci USA. 2012;109(50):E3395–E3404. doi: 10.1073/pnas.1217982109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun Y, et al. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat Med. 2012;18(9):1359–1368. doi: 10.1038/nm.2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quintana E, et al. Efficient tumour formation by single human melanoma cells. Nature. 2008;456(7222):593–598. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Leenders GJ, Aalders TW, Hulsbergen-van de Kaa CA, Ruiter DJ, Schalken JA. Expression of basal cell keratins in human prostate cancer metastases and cell lines. J Pathol. 2001;195(5):563–570. doi: 10.1002/path.993. [DOI] [PubMed] [Google Scholar]
- 30.Leon J, Ferrandiz N, Acosta JC, Delgado MD. Inhibition of cell differentiation: A critical mechanism for MYC-mediated carcinogenesis? Cell Cycle. 2009;8(8):1148–1157. doi: 10.4161/cc.8.8.8126. [DOI] [PubMed] [Google Scholar]
- 31.Ju X, et al. Novel oncogene induced metastatic prostate cancer cell lines define human prostate cancer progression signatures. Cancer Res. 2012;73(2):978–989. doi: 10.1158/0008-5472.CAN-12-2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernard D, Pourtier-Manzanedo A, Gil J, Beach DH. Myc confers androgen-independent prostate cancer cell growth. J Clin Invest. 2003;112(11):1724–1731. doi: 10.1172/JCI19035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maitland NJ, Frame FM, Polson ES, Lewis JL, Collins AT. Prostate cancer stem cells: Do they have a basal or luminal phenotype? Horm Cancer. 2011;2(1):47–61. doi: 10.1007/s12672-010-0058-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rajasekhar VK, Studer L, Gerald W, Socci ND, Scher HI. Tumour-initiating stem-like cells in human prostate cancer exhibit increased NF-kappaB signalling. Nat Commun. 2011;2:162. doi: 10.1038/ncomms1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chlenski A, Nakashiro K, Ketels KV, Korovaitseva GI, Oyasu R. Androgen receptor expression in androgen-independent prostate cancer cell lines. Prostate. 2001;47(1):66–75. doi: 10.1002/pros.1048. [DOI] [PubMed] [Google Scholar]
- 36.Kaighn ME, Narayan KS, Ohnuki Y, Lechner JF, Jones LW. Establishment and characterization of a human prostatic carcinoma cell line (PC-3) Invest Urol. 1979;17(1):16–23. [PubMed] [Google Scholar]
- 37.Mulholland DJ, et al. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res. 2012;72(7):1878–1889. doi: 10.1158/0008-5472.CAN-11-3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tam L, et al. Expression levels of the JAK/STAT pathway in the transition from hormone-sensitive to hormone-refractory prostate cancer. Br J Cancer. 2007;97(3):378–383. doi: 10.1038/sj.bjc.6603871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruggero D, et al. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat Med. 2004;10(5):484–486. doi: 10.1038/nm1042. [DOI] [PubMed] [Google Scholar]
- 40.Graff JR, et al. eIF4E activation is commonly elevated in advanced human prostate cancers and significantly related to reduced patient survival. Cancer Res. 2009;69(9):3866–3873. doi: 10.1158/0008-5472.CAN-08-3472. [DOI] [PubMed] [Google Scholar]
- 41.Hsieh AC, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485(7396):55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin F, et al. Sox2 targets cyclinE, p27 and survivin to regulate androgen-independent human prostate cancer cell proliferation and apoptosis. Cell Prolif. 2012;45(3):207–216. doi: 10.1111/j.1365-2184.2012.00812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feldman ME, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7(2):e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu TL, et al. Conditionally ablated Pten in prostate basal cells promotes basal-to-luminal differentiation and causes invasive prostate cancer in mice. Am J Pathol. 2013;182(3):975–991. doi: 10.1016/j.ajpath.2012.11.025. [DOI] [PubMed] [Google Scholar]
- 45.Wang ZA, et al. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nat Cell Biol. 2013;15(3):274–283. doi: 10.1038/ncb2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi N, Zhang B, Zhang L, Ittmann M, Xin L. Adult murine prostate basal and luminal cells are self-sustained lineages that can both serve as targets for prostate cancer initiation. Cancer Cell. 2012;21(2):253–265. doi: 10.1016/j.ccr.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huntly BJ, et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell. 2004;6(6):587–596. doi: 10.1016/j.ccr.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 48.Passegué E, Wagner EF, Weissman IL. JunB deficiency leads to a myeloproliferative disorder arising from hematopoietic stem cells. Cell. 2004;119(3):431–443. doi: 10.1016/j.cell.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 49.Krivtsov AV, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442(7104):818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- 50.Jamieson CH, Weissman IL, Passegué E. Chronic versus acute myelogenous leukemia: A question of self-renewal. Cancer Cell. 2004;6(6):531–533. doi: 10.1016/j.ccr.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 51.Jamieson CH, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351(7):657–667. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- 52.Goldstein AS, et al. Purification and direct transformation of epithelial progenitor cells from primary human prostate. Nat Protoc. 2011;6(5):656–667. doi: 10.1038/nprot.2011.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.