

Significance
Transport of gases between the ocean and the atmosphere has profound implications for our environment and the Earth’s climate. An example of this transport is the oceanic uptake of carbon dioxide, which has buffered us from a higher concentration of this greenhouse gas in the atmosphere while also causing ocean acidification. Here we describe the first direct measurements of air–sea methanol transfer. Atmospheric methanol, a ubiquitous and abundant organic gas of primarily terrestrial origin, is observed to be transported over thousands of kilometers and deposited over the ocean, where it is likely consumed by marine microbes. We quantify the rate of methanol deposition and examine the governing processes near the air–sea interface.
Keywords: trace gas cycling, air–sea exchange, eddy covariance, environmental chemistry, marine micrometeorology
Abstract
In the troposphere, methanol (CH3OH) is present ubiquitously and second in abundance among organic gases after methane. In the surface ocean, methanol represents a supply of energy and carbon for marine microbes. Here we report direct measurements of air–sea methanol transfer along a ∼10,000-km north–south transect of the Atlantic. The flux of methanol was consistently from the atmosphere to the ocean. Constrained by the aerodynamic limit and measured rate of air–sea sensible heat exchange, methanol transfer resembles a one-way depositional process, which suggests dissolved methanol concentrations near the water surface that are lower than what were measured at ∼5 m depth, for reasons currently unknown. We estimate the global oceanic uptake of methanol and examine the lifetimes of this compound in the lower atmosphere and upper ocean with respect to gas exchange. We also constrain the molecular diffusional resistance above the ocean surface—an important term for improving air–sea gas exchange models.
Background
Atmospheric methanol affects tropospheric oxidative capacity and air pollution by participating in the cycling of ozone and the hydroxyl radical (OH). Methanol is primarily released to air from terrestrial plants (during growth and decay); other identified sources include industrial emissions, biomass and biofuel burning, and atmospheric production (1–5). Methanol reacts with OH in the troposphere with a photochemical lifetime of ∼10 d, leading to formaldehyde (6) and carbon monoxide (7), among other products. Observations suggest that methanol can be further removed from air via deposition to land (8) and to the sea surface (9, 10). In the upper ocean, methanol supports the growth of methylotrophic bacteria (11) and has recently been found to be consumed by SAR11 alphaprotoeobacteria, the most abundant marine heterotrophs (12). The turnover time of seawater methanol is thus quite short, on the order of a few days (13, 14). However, significant oceanic concentrations of methanol have been detected in the range of 50∼400 nM (9, 15–17), leading to questions about its source.
To understand the global cycling of methanol, it is imperative to quantify its transport between the ocean and the atmosphere. Heikes et al. (3) modeled a gross air-to-sea depositional loss of −80 Tg⋅y−1 and also argued for an oceanic source of 30 Tg⋅y−1 to sustain an observed concentration of 0.9 ppb in the marine atmospheric boundary layer (MABL) of the Pacific and Atlantic. Based on aircraft measurements over the Pacific, Singh et al. (18) estimated a loss of −8 Tg⋅y−1 to the surface ocean with no appreciable oceanic source, which was later modified to −10 Tg⋅y−1 by Jacob et al. (4). Millet et al. (5) modeled a gross deposition of −101 Tg⋅y−1 to the ocean—a sink largely offset by an oceanic production of 85 Tg⋅y−1. From in situ seawater concentration measurement and modeled atmospheric distribution over the Atlantic, Beale et al. (17) recently calculated a net oceanic emission of 12 Tg⋅y−1, but saw evidence for both oceanic production and uptake. Amid these large discrepancies is the fact that the air–sea methanol flux has never been measured directly (e.g., with eddy covariance)—a void we address with this report.
Due to challenges in direct quantification, the flux of a gas across the air–sea interface is often approximated as the product of the gas transfer velocity and the air–sea concentration difference using the two-layer model (19):
Here, Cw and Ca are the bulk concentrations of the gas in water and atmosphere. H is the dimensionless Henry’s solubility expressed as the ratio of liquid-to-gas concentrations at equilibrium. Cw/H denotes the concentration on the airside of the interface that would be equilibrated with the waterside. When Cw/H is less than Ca, surface water is undersaturated relative to the atmosphere and the flux is from air to sea. Ka is the total gas transfer velocity from the perspective of atmospheric concentrations. Governed by molecular and turbulent transfer in both phases, Ka encompasses the kinetic forcing in gas exchange.
Molecular sublayers exist on both sides of the air–sea interface, where turbulent transport diminishes and molecular diffusion dominates. Conceptualizing the system as two resistors in series, Ka can be partitioned to individual transfer velocities in air and water (ka and kw, respectively):
For sparingly soluble gases (low H), transport through the aqueous molecular sublayer is the rate-limiting step (i.e., Ka ∼ Hkw). Such waterside controlled gases, including carbon dioxide (CO2) and sulfur hexafluoride (SF6), have been the subjects of decades of research (20). In contrast, transfer of very soluble (high H) and/or surface reactive gases is limited on the airside (i.e., Ka ∼ ka). For the highly soluble methanol with H of ∼5,000 at 25 °C (21), the second term in Eq. 2 contributes at most a few percent to Ka.
The airside transfer velocity is dictated by resistances from aerodynamic transport in the turbulent atmosphere (Rt) and diffusion in the airside molecular sublayer (Rm):
Our knowledge of ka stems mostly from studies of latent heat (water vapor) and sensible heat (conduction due to the air–sea temperature difference). Resistance-based models (22, 23) and, more recently, the Center for Coupled Ocean-Atmosphere Response Experiment (COARE) gas transfer model (24) suggest that at a height well above the sea surface (e.g., 10 m), Rt substantially exceeds Rm. The predominance of turbulent transport might be one reason why rates of water vapor transfer measured over the ocean are significantly lower than those observed in laboratories (25, 26), where dynamics are different.
To relate ka of water vapor or sensible heat to other gases, Rm is assumed to be proportional to Sca1/2∼2/3, where Sca is the airside Schmidt number (ratio of kinematic viscosity to molecular diffusivity in air). However, limited open-ocean observations of airside-controlled trace gases have demonstrated diverging behaviors from water vapor, which are so far unexplained. Eddy covariance measurements of the very soluble acetone resulted in air–sea flux at times opposite in direction to the prediction from the two-layer model (27). In the case of the surface reactive sulfur dioxide, aircraft flux measurements yielded ka values ∼30% lower than expected (28). Thus, flux observation of another gas with predominantly airside control, such as methanol, has the potential to reduce the uncertainty in ka and ultimately improve flux estimations based on Eq. 1.
Results
On the 22nd Atlantic Meridional Transect (AMT-22) cruise on the Royal Research Ship James Cook (October∼November 2012) from Southampton, United Kingdom, to Punta Arenas, Chile, we measured the air–sea flux of methanol directly with the eddy covariance method. Quantified by a proton transfer reaction mass spectrometer (PTR-MS) with an isotopically labeled standard, atmospheric methanol concentration (Ca) was correlated with motion-corrected vertical wind velocity (w) to yield its net vertical transport. We also measured the dissolved concentration of methanol (Cw) at ∼5 m depth from hydrocasts with the same PTR-MS coupled to a membrane inlet (16).
Fig. 1 shows the cruise track of AMT-22, color-coded by the atmospheric methanol concentration. To illustrate where sampled air masses resided previously, we overlay 5-d back-trajectories from the Hybrid Single-Particle Lagrangian Integrated Trajectory (HYSPLIT) model (29). Ca was higher in the Northern Hemisphere, as expected from the greater landmass and anthropogenic activity. At the same latitudes, our Ca values are comparable to previous maritime measurements at Cape Verde (30) and near the tropics (9). From north to south across the Intertropical Convergence Zone at ∼3°N, Ca decreased rapidly from ∼0.6 to ∼0.3 ppb. Plumes of higher Ca can be seen in continental outflow regions (e.g., off Northern Africa and North America), whereas lower values are observed in air masses that had not been in contact with land for several days. Sudden depletion in Ca often coincided with precipitation (e.g., October 11, October 14, November 13), likely in part due to removal by wet deposition and heterogeneous chemistry (3).
Fig. 1.

Cruise track of AMT-22 color-coded by the hourly atmospheric methanol concentration (n = 734) and overlaid with 5-d back-trajectories (initiated from the MABL and marked on daily intervals) for selected days. Methanol concentration was higher in the Northern Hemisphere than in the Southern, and particularly elevated in continental outflow regions (e.g., Northern Africa and North America). In contrast, depleted concentrations were observed for air masses that had not been in recent contact with land and during precipitation. Given its atmospheric lifetime of several days, methanol may be considered a tracer for terrestrial emissions, but is unlikely to undergo interhemispheric transport, which has a timescale of ∼1 y.
Latitudinal distributions of atmospheric and seawater methanol concentrations are shown in Fig. 2A. Compared with previous measurements (9, 15–17), Cw was considerably lower during AMT-22, with a mean (range) of 29 (15∼62) nM. Cw correlated weakly with Ca (r2 = 0.11, P = 0.003, two-tailed) and demonstrated no clear hemispheric trend. Surface water was undersaturated in methanol with respect to the atmosphere (Fig. 2B), consistent with rapid oceanic destruction. Saturation level was lower on average in the Northern Hemisphere (24%) than in the Southern (34%), correlating weakly with wind speed (r2 = 0.10, P = 0.007, two-tailed). Measured air–sea methanol flux (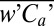 ) averaged to latitude bins is shown in Fig. 2C. Greater air-to-sea flux occurred in regions of high Ca and strong winds, with the largest oceanic uptake found in the subtropical and tropical North Atlantic.
) averaged to latitude bins is shown in Fig. 2C. Greater air-to-sea flux occurred in regions of high Ca and strong winds, with the largest oceanic uptake found in the subtropical and tropical North Atlantic.
Fig. 2.
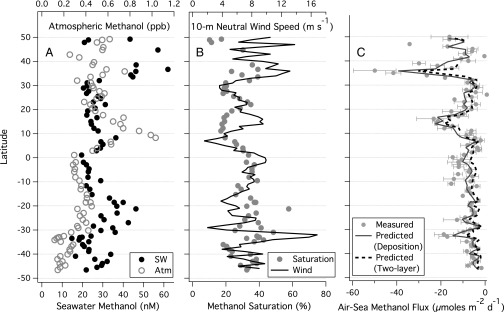
(A) Latitudinal distributions of atmospheric and seawater methanol concentrations; (B) saturation level of methanol and wind speed; (C) air–sea methanol flux measured by eddy covariance and predicted by a two-layer model and a purely deposition model based on observed concentrations (n = 73). Error bars on flux represent SE. Seawater concentration did not demonstrate any hemispheric trend, and was significantly undersaturated with respect to the atmosphere, implying rapid oceanic degradation. Methanol flux was consistently from air to sea, peaking in regions of high atmospheric concentration and strong wind. Flux averaged −14 μmol⋅m−2⋅d−1 in the subtropical and tropical Atlantic, and was as much as −50 μmol⋅m−2⋅d−1. In the South Atlantic, flux was lower in magnitude, with a mean of −8 μmol⋅m−2⋅d−1.
Two approaches of predicting bulk air–sea methanol flux based on observed concentrations are shown in Fig. 2C: the first from the two-layer model (Eq. 1) with ka from Mackay and Yeun (25) and kw from the COARE (24), and the second as purely deposition (−ka Ca) with ka from ref. 24. Though both approaches yield reasonable fits to measured flux, the agreement is somewhat fortuitous. Based on volatilization experiments in a wind-wave tank, ka from ref. 25 overestimates water transfer relative to observed rate over the ocean, which is better represented by ref. 24. However, the formulation, ka Ca, specifies a unidirectional transfer of methanol from air to sea and no return flux. Using ka from ref. 24 in the two-layer model or ka from ref. 25 in the purely deposition model results in significant underestimation and overestimation, respectively.
Discussion
We first examine the influence of air–sea exchange on the atmospheric and oceanic methanol budgets. The vertical gradient in Ca within the atmospheric surface layer (the lowest ∼10% of the MABL) can be approximated from similarity theory as −Flux/(κ u* z), with κ being the von Karmon constant and z the sampling height. In this case, Ca is estimated to increase with height at an average rate of ∼0.002 ppb⋅m−1. For a 1-km-high MABL with a mean mixing ratio of 1 ppb, deposition to the ocean removes methanol from air with a timescale of ∼4 d. Crudely assuming the global ocean to have the same methanol and wind speed distributions as during our cruise, an average methanol flux of −10 μmol⋅ m−2⋅ d−1 extrapolates to a net air–sea transport of −42 Tg⋅y−1. Substituting this flux into previous global budgets (3–5), it is evident that air–sea exchange accounts for 18∼23% of the total removal of atmospheric methanol.
The atmosphere does not appear to be the sole source of seawater methanol, however. Assuming a 50-m-deep oceanic mixed layer with a dissolved methanol concentration of 29 nM, at a mean flux of −10 μmol⋅m−2⋅d−1, the replacement time for seawater methanol is 140 d with respect to gas exchange, approximately two orders of magnitude longer than the typical turnover time due to biological consumption (13, 14). Thus, a suggested “missing” source of seawater methanol (3, 5, 13) seems justified for mass balance. Furthermore, we found methanol concentration at ∼500 m depth to be 60∼80% of the 5-m value, proportionally similar to depth profiles observed previously (9, 17). Given the measurable biological consumption of methanol at depth (14), the presence of significant concentration there suggests that its production is not limited to the near surface. A recent work shows that methanol may be produced by the marine proteobacteria Alteromonadales (31).
Now we turn our attention to the process of air–sea methanol transfer. We calculate Ka from measured flux using observed Cw (Fig. 3A) and by setting Cw to zero (Fig. 3B). To account for buoyancy effects, Ka is adjusted to neutral atmospheric stability based on similarity theory (32) and plotted against the measured friction velocity (u*, related to wind stress) as well as the approximate 10-m neutral wind speed. Also shown are parameterizations from Mackay and Yeun (25), Liss (26) adjusted for molecular weight (19), and COARE (24). The aerodynamic limit from COARE (1/Rt) defines the theoretical rate of atmospheric turbulent transfer. In addition, we show the in situ transfer velocity of sensible heat 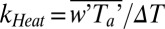 , where Ta is the air temperature from the sonic anemometer corrected for humidity, and ∆T the air–sea temperature difference.
, where Ta is the air temperature from the sonic anemometer corrected for humidity, and ∆T the air–sea temperature difference.
Fig. 3.

(A) Methanol transfer velocity calculated using measured Cw; (B) calculated using Cw = 0 (n = 73). Measured friction velocity and the approximate wind speed are shown on the abscissae. Using measured Cw, calculated methanol transfer velocity sometimes exceeds the aerodynamic limit, particularly in high winds. In contrast, using Cw = 0 leads to more reasonable Ka, implying low dissolved methanol concentrations close to the air–water interface. KHeat adjusted to neutral stability is shown as averages in u* bins. Error bars on Ka, KHeat, and u* correspond to the respective SEs.
With the two-layer approach using observed Cw (Fig. 3A), the polynomial fit 11,766 u* + 13,804 u*2 (R2 = 0.87) describes the nonlinear relationship between Ka and u*. Ka is similar to kHeat and the aerodynamic limit at low to moderate winds (u* < 0.4 m⋅s−1), which confirms the expectation that methanol is airside controlled and has minimal waterside resistance. At u* > 0.4 m⋅s−1, Ka trends ∼15% higher than the aerodynamic limit, and significantly exceeds kHeat by ∼20% (χ2 test at 95% confidence), which is inconsistent with physical theory. Uncertainties in Ka amplify in high winds due to the small sample size as well as greater measurement errors (Methods). Nevertheless, based on Eq. 3, Ka for methanol should be ∼10% lower than kHeat because of the higher Sca for methanol (1.09) (33) relative to heat (Sca = 0.64), which is not reflected in Fig. 3A.
Transfer velocity calculated with Cw = 0 equates to a one-way deposition velocity (Fig. 3B). By specifying the maximum air–sea concentration difference, the deposition velocity represents the lower limit of ka. The mean deposition velocity of 2,444 cm⋅h−1 converts to 0.68 cm⋅s−1, which is several times higher than previous estimates based on temporal trends in the atmospheric methanol concentration (10) and vertical profiles from the MABL to the free troposphere (18). We note that our measurements by eddy covariance are the most direct and do not require assumptions about the seawater saturation or atmospheric chemistry of methanol.
With Cw = 0, Ka demonstrates a near linear relationship with u*, and may be fitted by 8,814 u* + 6,810 u*2 (R2 = 0.89), which is lower than the aerodynamic limit as well as measured kHeat, and lies between laboratory results (25, 26) and the resistance-based parameterization (24). Compared with Fig. 3A, as expected, the periods with the highest saturation values had the largest reductions in Ka. We further solve for resistance in the airside molecular diffusion sublayer above the ocean surface by taking the difference between Rt and 1/Ka, which is illustrated in Fig. 4. The derived Rm is between the parameterization of 5 Sca2/3/ u* from Hicks et al. (22) and 13.3 Sca1/2/ u* from COARE (24). Because using Cw = 0 yields the minimum Ka and so maximum airside resistance, our results suggest that Rm may be overestimated in the COARE model.
Fig. 4.

Resistance in the molecular diffusion sublayer above the ocean surface (Rm), calculated as the difference between aerodynamic resistance (Rt) and 1/Ka of methanol (with Cw = 0). Rm estimated from methanol transfer lies between the parameterizations from Hicks et al. (22) and COARE (24). In all cases, Rt at a height of 18 m is several times greater than Rm.
It is surprising that using Cw = 0 yields a more physically consistent Ka than using the measured Cw. For Ka in Fig. 3A to be ∼15% lower (i.e., to approach the aerodynamic limit), Cw needs to be reduced by ∼50%. We examine the possibility of a near surface gradient in Cw. Microorganisms and dissolved organic matter tend to be enriched in the ∼0.1-mm-thick aqueous molecular sublayer (34, 35). This microlayer covers both the productive regions and the oligotrophic waters and at wind speeds of up to ∼10 m⋅s−1 (36). Breaking waves temporally disrupt the surface, but a coherent microlayer appears to reform within seconds, in part due to efficient scavenging of surface active organic materials from bulk water by rising bubbles (37). Considering the methanol budget in the microlayer, the air-to-sea transport in our study adds 10 μmol⋅m−2⋅d−1. If the concentration in the microlayer were maintained at 50% lower than in the bulk water, 26 μmol⋅m−2⋅d−1 of methanol would be diffusing into the microlayer from below at steady state (with kw = 11 cm⋅h−1 from COARE). The total methanol input into the microlayer (36 μmol⋅m−2⋅d−1) divided over a thickness of 0.1 mm would yield a concentration increase of 4 nM⋅s−1. A methanol depletion of the same rate is required for mass balance (without any in situ production), which would be at least three orders of magnitude faster than any observed biological consumption (13, 14).
The mixing time between the sea surface and 5 m depth, dependent on the turbulent diffusivity, is typically on the order of a few minutes (38). Thus, enhanced consumption in the top meters of the ocean with a timescale of a few nM per minute could result in a vertical gradient in bulk Cw. Photochemically mediated destruction of methanol by OH radical in water is fast, with a rate constant of 1 × 109 M−1⋅s−1 (39). However, the OH concentration in the surface ocean is only 1∼10 × 10−18 M (40) and therefore too low to be a significant sink for dissolved methanol. A pronounced photochemical effect would also imply a greater Ka during the day than at night, which was not observed during this cruise. In sum, known methanol sinks do not appear to be rapid enough to cause a substantially lower dissolved concentration at the interface relative to the bulk seawater. Interestingly, in an earlier measurement of acetone flux (8), a lower dissolved concentration at the surface would also to help reconcile the difference between observed uptake and predicted emission in the tropical Pacific. Along with previously measured SO2 deposition velocities that are lower than expected (28), these results allude to potential processes not well understood in the transfer of airside controlled trace gases.
Conclusion
In this study, we report direct measurements of air–sea methanol transfer by eddy covariance. The surface ocean consistently took up methanol from the atmosphere, with enhanced influx in continental outflow regions and during high winds. The low saturation of methanol in the surface seawater implies rapid oceanic destruction of this compound. Methanol transfer resembles a one-way depositional process, suggesting that methanol concentrations at the water surface may be even lower than what were measured at ∼5 m depth due to processes currently unknown. Further field measurements along with other airside-controlled compounds (e.g., water vapor, ethanol), as well as laboratory experiments of methanol uptake with and without biology would help to determine whether the deposition model always holds for highly soluble gases.
Methods
Atmospheric Measurements.
During AMT-22, atmospheric and seawater methanol concentrations were alternately quantified by a high-resolution PTR-MS (Ionicon), which was housed in the meteorological laboratory near the foredeck of the ship. Acetone and acetaldehyde were also measured and will be described elsewhere. For ∼19 h of a day, the PTR-MS operated under atmospheric mode and continuously measured at ∼2.1 Hz. Air was drawn in from an intake on the starboard side of the ship’s foremast (∼18 m above mean sea level) via ∼25 m of 6.4 mm (inner diameter) perfluoroalkoxy tubing by a vacuum pump at a flow rate of ∼23 standard liters per minute, as monitored by a digital thermal mass flow meter. A triply deuterated methanol gas standard (2.0 ± 0.1 ppm of methanol-d3; Scientific and Technical Gases Ltd.) was injected continuously into the inlet line at 30(±0.3) standard cubic centimeter per minute, as regulated by a digital thermal mass flow controller; this allows Ca to be calculated from the ratio between the ambient and deuterated signals. The use of the isotopic standard minimizes uncertainties due to instrumental drift and variable efficiencies. Background values were taken by directing ambient air through a platinum catalytic converter (350 °C) for 2 min every hour. The detection limit for mean atmospheric concentration (minutely averaged) and the noise level at ∼2.1 Hz were 0.048 and 0.21 ppb, respectively. The standard injection system was initially designed and the instrument performance characterized in detail at a coastal site (41).
In eddy covariance (EC), Ca is correlated with concurrent vertical wind velocity (w) and averaged over time to yield the vertical flux (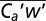 , where primes denote deviations from the respective means and the overbar signals averaging over nominally ∼1 h). Wind measurements on a ship are influenced by the ship’s movement, necessitating a motion correction. Mounted ∼40 cm from the gas intake, a sonic anemometer (WindMaster; Gill Instruments) and a motion sensor (Motionpak II; Systron Donner) measured 3D wind velocities, linear accelerations, and rotational rates at 10 Hz. Observed winds were corrected for ship’s motion (42), and further sequentially decorrelated with ship velocities and accelerations to yield true winds (24). The EC friction velocity (derived from
, where primes denote deviations from the respective means and the overbar signals averaging over nominally ∼1 h). Wind measurements on a ship are influenced by the ship’s movement, necessitating a motion correction. Mounted ∼40 cm from the gas intake, a sonic anemometer (WindMaster; Gill Instruments) and a motion sensor (Motionpak II; Systron Donner) measured 3D wind velocities, linear accelerations, and rotational rates at 10 Hz. Observed winds were corrected for ship’s motion (42), and further sequentially decorrelated with ship velocities and accelerations to yield true winds (24). The EC friction velocity (derived from 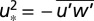 , where u is the wind velocity along the mean wind direction) closely agrees with modeled u* (24) as a function of wind speed, validating the motion correction (Fig. 5).
, where u is the wind velocity along the mean wind direction) closely agrees with modeled u* (24) as a function of wind speed, validating the motion correction (Fig. 5).
Fig. 5.

Friction velocity (u*) as a function of 10-m neutral wind speed. Measured u* by eddy covariance (n = 584) agrees well with prediction from the COARE model (24), validating the motion correction on observed winds.
Methanol flux is computed as the integral of the Ca:w cospectrum from 0.002∼1 Hz, omitting low-frequency contributions possibly related to horizontal heterogeneity. Only the wind sector from −50 to 110 degrees is considered for flux, excluding periods of contamination from the ship’s exhaust and distortion of ambient wind fields due to the ship’s superstructure. A total of 484 h of valid methanol flux observations were made, of which 29 h were during high wind conditions (u* > 0.4 m⋅s−1). As expected, correlating the methanol-d3 signal with w resulted in “null” fluxes scattered around zero. After dividing by u*, methanol flux also does not correlate with measured sensible heat flux or computed latent heat flux, implying minimal sensitivity in the instrument response to ambient fluctuations in temperature and humidity. However, in heavy swells, Ca exhibited some spurious correlations with the vertical platform acceleration and displacement at the frequency of ship’s motion (∼0.1 Hz). The former artifact was likely due to motion-induced variability in the water vapor source flow of the PTR-MS, and the latter from heaving of the ship vertically across the Ca gradient. Applying a similar decorrelation algorithm as described above to Ca removes the erroneous spike at ∼0.1 Hz and also reduces the magnitude of methanol flux by an average of 24%.
Mean methanol and sensible heat cospectra over 10 h on October 17 are shown in Fig. 6, which are well described by the expected spectral shape for atmospheric turbulent transport (43). Based on an empirical filter function (44) with a response time of 0.5 s and the shape of the theoretical spectrum at frequencies above the Nyquist (∼1 Hz), a correction for high-frequency attenuation is applied to the measured methanol flux, which is on average 17% and increases with wind speed, consistent with estimates from an ogive approach (41). Fluxes are processed hourly and averaged to 1° latitude bins. At a nominal ship velocity of 18 km⋅h−1, each latitude bin corresponds to ∼6 h. Random uncertainty in methanol flux is ∼20% for the bin average given a sampling error of ∼50% for hourly measurements (45).
Fig. 6.

Normalized cospectra of sensible heat and methanol over 10 h on October 17, a day with high winds and large methanol flux. Both cospectra are well described by the theoretical spectral shape characteristic of atmospheric turbulent transport (43). Attenuated flux at high frequency is corrected following a filter-function approach (44).
Seawater Measurements and Computation of Ka.
Discrete seawater samples (triplicates) were taken primarily from predawn and noontime conductivity, temperature, salinity (CTD) hydrocasts daily. Unfiltered water was transported from the 5-m Niskin bottle via a short piece of Tygon tubing into opaque glass bottles (∼300 mL). Contact with air was avoided by sampling first from the Niskin and overfilling the glass bottles before capping. An additional sample from the deepest Niskin (nominally at 500 m depth) was collected at noon. Several water samples were also obtained from the ship’s nontoxic underway water supply on November 13, when no CTD was commenced during a storm, and on November 20, after the completion of CTD work. An intercomparison earlier during the cruise yielded no significant difference in Cw between the 5-m CTD and the water collected underway.
To minimize any loss due to bacterial consumption, water samples were kept at ambient water temperature and analyzed within 3 h of sampling. Methanol was extracted from seawater across a semipermeable silicon membrane thermostated at 50 °C into a supply of clean nitrogen flowing directly into the PTR-MS, as described in ref. 16. The first of the triplicate samples was used to condition the membrane; reported Cw values represent the average of the latter two samples. The system was calibrated every 2 wk using water standards prepared by serial dilution of reagent-grade methanol. Calibration constants were stable over the entire cruise, varying less than 10%. Estimated as three times the noise of the nitrogen blanks, the detection limit for seawater methanol concentration was ∼6 nM.
For the computation of Ka, latitudinally bin-averaged flux and Ca were linearly interpolated to the times of water collection. Given the transect format of the cruise, uncertainties due to horizontal gradients were random and should not contribute to any bias in Ka. Any proportional error in Ca should also be reflected in the flux and so not affect Ka. Judging from a recent survey (46), uncertainties in H for methanol should be within 10%.
Acknowledgments
M.Y. thanks B. Huebert for guidance; P. Mason, A. Staff, and S. Howell for instrumentation support; J. Stephens and F. Hopkins for equipment setup; and M. Johnson, T. Bell, D. Woolf, and J. Dixon for scientific input. We gratefully acknowledge the National Oceanic and Atmospheric Administration Air Resources Laboratory for the provision of the HYSPLIT transport and dispersion model and READY Web site (http://ready.arl.noaa.gov). This work was supported by US National Science Foundation Grant OISE-1064405 and UK Natural Environment Research Council National Capability funding to Plymouth Marine Laboratory and the National Oceanography Centre, Southampton, United Kingdom. This research is a contribution to the international Surface Ocean Lower Atmosphere Study (SOLAS) and Integrated Project of Ocean Research (IMBER) projects and represents contribution no. 228 of the Atlantic Meridional Transect program.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: Data will be made available in the British Oceanographic Data Centre database (http://www.bodc.ac.uk) within two years from the completion of AMT-22.
References
- 1.Warneke C, et al. Acetone, methanol, and other partially oxidized volatile organic emissions from dead plant matter by abiological processes: Significance for atmospheric HOx chemistry. Global Biogeochem Cycles. 1999;13:9–17. [Google Scholar]
- 2.Guenther A, et al. Natural emissions of non-methane volatile organic compounds; carbon monoxide, and oxides of nitrogen from North America. Atmos Environ. 2000;34:2205–2230. [Google Scholar]
- 3.Heikes B, et al. Atmospheric methanol budget and ocean implication. Global Biogeochem Cycles. 2002;16(4):80-1–80-13. [Google Scholar]
- 4.Jacob D, et al. Global budget of methanol: Constraints from atmospheric observations. J Geophys Res. 2005;110:D08303. [Google Scholar]
- 5.Millet D, et al. New constraints on terrestrial and oceanic sources of atmospheric methanol. Atmos Chem Phys. 2008;8:6887–6905. [Google Scholar]
- 6.Millet D, et al. Formaldehyde distribution over North America: Implications for satellite retrievals of formaldehyde columns and isoprene emission. J Geophys Res. 2006;111:D24S02. [Google Scholar]
- 7.Duncan B, et al. Global budget of CO, 1988–1997: Source estimates and validation with a global model. J Geophys Res. 2007;112:D22301. [Google Scholar]
- 8.Karl T, et al. Exchange processes of volatile organic compounds above a tropical rain forest: Implications for modeling tropospheric chemistry above dense vegetation. J Geophys Res. 2004;109:D18306. [Google Scholar]
- 9.Williams J, et al. Measurements of organic species in air and seawater from the tropical Atlantic. Geophys Res Lett. 2004;31:L23S06. [Google Scholar]
- 10.Carpenter L, et al. Uptake of methanol to the North Atlantic Ocean surface. Global Biogeochem Cycles. 2004;18:GB4027. [Google Scholar]
- 11.Murrell C, McDonald I. 2000. Methylotrophy. Encyclopedia of Microbiology, ed Lederberg J, III (Academic, New York), pp 245–255.
- 12.Sun J, et al. One carbon metabolism in SAR11 pelagic marine bacteria. PLoS ONE. 2011;6(8):e23973. doi: 10.1371/journal.pone.0023973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dixon J, Beale R, Nightingale P. Rapid biological oxidation of methanol in the tropical Atlantic: significance as a microbial carbon source. Biogeosciences. 2011;8:2707–2716. [Google Scholar]
- 14.Dixon J, Nightingale P. Fine-scale variability in methanol uptake and oxidation: From the microlayer to 1000 m. Biogeosciences. 2012;9:2961–2972. [Google Scholar]
- 15.Kameyama S, et al. High-resolution measurement of multiple volatile organic compounds dissolved in seawater using equilibrator inlet–proton transfer reaction-mass spectrometry (EI–PTR-MS) Mar Chem. 2010;122:59–73. doi: 10.1021/ac901630h. [DOI] [PubMed] [Google Scholar]
- 16.Beale R, Liss PS, Dixon JL, Nightingale PD. Quantification of oxygenated volatile organic compounds in seawater by membrane inlet-proton transfer reaction/mass spectrometry. Anal Chim Acta. 2011;706(1):128–134. doi: 10.1016/j.aca.2011.08.023. [DOI] [PubMed] [Google Scholar]
- 17.Beale R, et al. Methanol, acetaldehyde and acetone in the surface waters of the Atlantic Ocean. J Geophys Res. 2013 doi: 10.1002/jgrc.20322. [DOI] [Google Scholar]
- 18.Singh H, et al. Oxygenated volatile organic chemicals in the oceans: Inferences and implications based on atmospheric observations and air-sea exchange models. Geophys Res Lett. 2003;30:1862. [Google Scholar]
- 19.Liss P, Slater P. Flux of gases across the air–sea interface. Nature. 1974;247:181–184. [Google Scholar]
- 20.Wanninkhof R, Asher W, Ho D, Sweeney C, McGillis W. Advances in quantifying air–sea gas exchange and environmental forcing. Annu Rev Mar Sci. 2009;1:213–244. doi: 10.1146/annurev.marine.010908.163742. [DOI] [PubMed] [Google Scholar]
- 21.Snider J, Dawson G. Tropospheric light alcohols, carbonyls, and acetonitrile: Concentrations in the southwestern United States and Henry’s law data. J Geophys Res. 1985;90D:3797–3805. [Google Scholar]
- 22.Hicks B, Wesely M, Lindberg S, Bromberg S. 1986. Proceedings of the NAPAP Workshop on Dry Deposition (National Oceanic and Atmospheric Administration, Oak Ridge, TN), pp 25–27.
- 23.Duce R, et al. The atmospheric input of trace species to the world ocean. Global Biogeochem Cycles. 1991;5:193–259. [Google Scholar]
- 24.Fairall C, Bradley E, Hare J, Grachev A, Edson J. Bulk parameterization of air-sea fluxes: Updates and verification for the COARE algorithm. J Clim. 2003;16:571–591. [Google Scholar]
- 25.Mackay D, Yeun AT. Mass transfer coefficient correlations for volatilization of organic solutes from water. Environ Sci Technol. 1983;17(4):211–217. doi: 10.1021/es00110a006. [DOI] [PubMed] [Google Scholar]
- 26.Liss P. Processes of gas exchange across an air-water interface. Deep Sea Res. 1973;20:221–238. [Google Scholar]
- 27.Marandino C, De Bruyn W, Miller S, Prather M, Saltzman S. Oceanic uptake and the global atmospheric acetone budget. Geophys Res Lett. 2005;32:L15806. [Google Scholar]
- 28.Faloona I, et al. Sulfur dioxide in the tropical marine boundary layer: Dry deposition and heterogeneous oxidation observed during the Pacific Atmospheric Sulfur Experiment. J Atmos Chem. 2010;63:13–32. [Google Scholar]
- 29.Draxler R, Rolph G. 2013. HYSPLIT model. Available at http://ready.arl.noaa.gov/HYSPLIT.php. Accessed January 25, 2013.
- 30.Read KA, et al. Multiannual observations of acetone, methanol, and acetaldehyde in remote tropical Atlantic air: Implications for atmospheric OVOC budgets and oxidative capacity. Environ Sci Technol. 2012;46(20):11028–11039. doi: 10.1021/es302082p. [DOI] [PubMed] [Google Scholar]
- 31.McCarren J, et al. Microbial community transcriptomes reveal microbes and metabolic pathways associated with dissolved organic matter turnover in the sea. Proc Natl Acad Sci USA. 2010;107(38):16420–16427. doi: 10.1073/pnas.1010732107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fairall C, Bradley E, Rogers D, Edson J, Yong G. Bulk parameterization of air-sea fluxes for Tropical Ocean-Global Atmosphere Coupled-Ocean Atmosphere Response Experiment. J Geophys Res. 1996;101(C2):3747–3764. [Google Scholar]
- 33.Johnson M. A numerical scheme to calculate temperature and salinity dependent air‐water transfer velocities for any gas. Ocean Sci. 2010;6:913–932. [Google Scholar]
- 34.Liss P. Chemistry of the sea-surface microlayer. In: Riley JP, Skirrow G, editors. Chemical Oceanography. 2nd Ed. London: Academic; 1975. pp. 193–244. [Google Scholar]
- 35.Hardy J, Apts C. The sea-surface microlayer: Phytoneuston productivity and effects of atmospheric particulate matter. Mar Biol. 1984;82:293–300. [Google Scholar]
- 36.Wurl O, Wurl E, Miller L, Johnson K, Vagle S. Formation and global distribution of sea-surface microlayers. Biogeosciences. 2011;8:121–135. [Google Scholar]
- 37.Williams P, et al. Chemical and microbiological studies of sea-surface films in the Southern Gulf of California and off the West Coast of Baja California. Mar Chem. 1986;19:17–98. [Google Scholar]
- 38.Soloviev A, Lukas R. The Near-Surface Layer of the Ocean, Structure, Dynamics and Application. The Netherlands: Springer; 2006. pp. 143–216. [Google Scholar]
- 39.Monod A, Carlier P. Impact of clouds on the tropospheric ozone budget: Direct effect of multiphase photochemistry of soluble organic compounds. Atmos Environ. 1999;33:4431–4446. [Google Scholar]
- 40.Mopper K, Zhou X. Hydroxyl radical photoproduction in the sea and its potential impact on marine processes. Science. 1990;250(4981):661–664. doi: 10.1126/science.250.4981.661. [DOI] [PubMed] [Google Scholar]
- 41.Yang M, Beale R, Smyth T, Blomquist B. Measurements of OVOC fluxes by eddy covariance using a proton-transfer-reaction mass spectrometer—method development at a coastal site. Atmos Chem Phys. 2013;13:6165–6184. [Google Scholar]
- 42.Edson J, Hinton A, Prada K, Hare J, Fairall C. Direct covariance flux estimates from mobile platforms at sea. J Atmos Ocean Technol. 1998;15:547–562. [Google Scholar]
- 43.Kaimal J, Wyngaard J, Izumi Y, Coté O. Spectral characteristics of surface layer turbulence. Q J R Meteorol Soc. 1972;98:563–589. [Google Scholar]
- 44.Bariteau L, et al. Determination of oceanic ozone deposition by ship-borne eddy covariance flux measurements. Atmos. Meas. Tech. 2010;3:441–455. [Google Scholar]
- 45.Blomquist B, Huebert B, Fairall C, Faloona I. Determining the sea-air flux of dimethylsulfide by eddy correlation using mass spectrometry. Atmos. Meas. Tech. 2010;3:1–20. [Google Scholar]
- 46.Warneck P. A note on the temperature dependence of Henry’s Law coefficients formethanol and ethanol. Atmos Environ. 2006;40:7146–7151. [Google Scholar]