

Significance
Solar water splitting into H2 and O2 with visible light has been achieved by a molecular assembly. The dye sensitized photoelectrosynthesis cell configuration combined with core–shell structures with a thin layer of TiO2 on transparent, nanostructured transparent conducting oxides (TCO), with the outer TiO2 shell formed by atomic layer deposition. In this configuration, excitation and injection occur rapidly and efficiently with the injected electrons collected by the nanostructured TCO on the nanosecond timescale where they are collected by the planar conductive electrode and transmitted to the cathode for H2 production. This allows multiple oxidative equivalents to accumulate at a remote catalyst where water oxidation catalysis occurs.
Abstract
Artificial photosynthesis and the production of solar fuels could be a key element in a future renewable energy economy providing a solution to the energy storage problem in solar energy conversion. We describe a hybrid strategy for solar water splitting based on a dye sensitized photoelectrosynthesis cell. It uses a derivatized, core–shell nanostructured photoanode with the core a high surface area conductive metal oxide film––indium tin oxide or antimony tin oxide––coated with a thin outer shell of TiO2 formed by atomic layer deposition. A “chromophore–catalyst assembly” 1, [(PO3H2)2bpy)2Ru(4-Mebpy-4-bimpy)Rub(tpy)(OH2)]4+, which combines both light absorber and water oxidation catalyst in a single molecule, was attached to the TiO2 shell. Visible photolysis of the resulting core–shell assembly structure with a Pt cathode resulted in water splitting into hydrogen and oxygen with an absorbed photon conversion efficiency of 4.4% at peak photocurrent.
Photosynthesis uses the energy of the sun with water as the reducing agent to drive the reduction of carbon dioxide to carbohydrates with oxygen as a coproduct through a remarkably complex process. At photosystem II, a subsystem imbedded in the thylakoid membrane where O2 is produced, light absorption, energy migration, electron transfer, proton transfer, and catalysis are all used in multiple stepwise chemical reactions which are carefully orchestrated at the molecular level (1, 2).
Photosynthesis solves the problem of energy storage by biomass production but with low solar efficiencies, typically <1%. In artificial photosynthesis with solar fuels production, the goal is similar but the targets are either hydrogen production from water splitting, Eq. 1, or reduction of carbon dioxide to a carbon-based fuel, Eq. 2 (3, 4). Different strategies for solar fuels have evolved (5, 6). In one, direct bandgap excitation of semiconductors creates electron–hole pairs which are then used to drive separate half-reactions for water oxidation (2H2O → O2 + 4H+ + 4e−) and water/proton reduction (2H+ + 2e− → H2) (7–9).
Here, we report a hybrid strategy for solar water splitting, the dye sensitized photoelectrosynthesis cell (DSPEC). It combines the electron transport properties of semiconductor nanocrystalline thin films with molecular-level reactions (10). In this approach, a chromophore–catalyst molecular assembly acts as both light absorber and catalyst. It is bound to the surface of a “core–shell,” nanostructured, transparent conducting oxide film. The core structure consists of a nanoparticle film of either tin-doped indium oxide (nanoITO), or antimony-doped tin oxide (nanoATO), deposited on a fluoride-doped tin oxide (FTO) glass substrate. The shell consists of a conformal TiO2 nanolayer applied by atomic layer deposition (ALD). The resulting “photoanode,” where water oxidation occurs, is connected to a Pt cathode for proton reduction to complete the water splitting cell.
A diagram for the photoanode in the DSPEC device is shown in Fig. 1. It illustrates the interface and the structure of chromophore–catalyst assembly 1, [(PO3H2)2bpy)2Rua(4-Mebpy-4′-bimpy)Rub(tpy)(OH2)]4+ ((PO3H2)2bpy is 4,4′-bisphosphonato-2,2′-bipyridine; 4-Mebpy-4′-bimpy is 4-(methylbipyridin-4′-yl)-N-benzimid-N′-pyridine; tpy is 2,2′:6′,2″-terpyridine)). It also illustrates the dynamic events that occur at the derivatized interface following light absorption by the surface-bound chromophore.
Fig. 1.

Illustrating interface binding, assembly structure, and a kinetic scheme following MLCT excitation of the chromophore in the assembly [((PO3H2)2bpy)2Rua(4-Mebpy-4′-bimpy)Rub(tpy)(OH2)]4+ (1) on core–shell nanoITO/TiO2. Excitation of the chromophore is followed by electron injection, electron migration through the core–shell structure, and intraassembly electron transfer oxidation of the catalyst.
Synthesis, characterization, and water oxidation catalysis by assembly 1 on nanoITO, abbreviated as 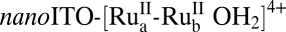 , have been investigated and the results are described in a parallel publication (11). The mechanism, established earlier for single-site molecular catalysts (12, 13), is shown in Scheme 1. Oxidative activation by stepwise 3e−/2H+ oxidation of the catalyst gives the intermediate
, have been investigated and the results are described in a parallel publication (11). The mechanism, established earlier for single-site molecular catalysts (12, 13), is shown in Scheme 1. Oxidative activation by stepwise 3e−/2H+ oxidation of the catalyst gives the intermediate 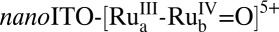 which is reactive toward water oxidation. It undergoes rate-limiting O-atom transfer to a water molecule in the surrounding solvent to give the peroxide intermediate,
which is reactive toward water oxidation. It undergoes rate-limiting O-atom transfer to a water molecule in the surrounding solvent to give the peroxide intermediate, 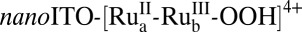 . This intermediate undergoes further oxidation and loss of O2 and reenters the catalytic cycle. In Scheme 1, “RDS” denotes the rate-determining step.
. This intermediate undergoes further oxidation and loss of O2 and reenters the catalytic cycle. In Scheme 1, “RDS” denotes the rate-determining step.
Scheme 1.
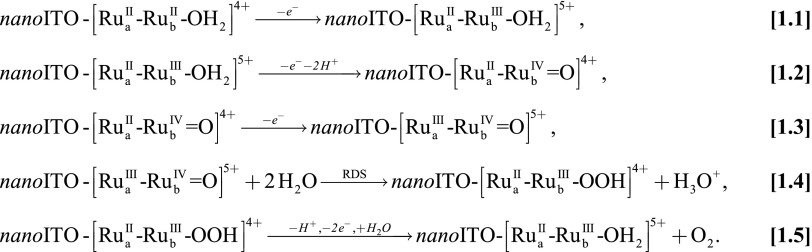
Mechanism of electrocatalytic water oxidation by 1 in 0.1 M HClO4 (11).
As illustrated in Fig. 1, on oxide semiconductor surfaces, metal-to-ligand charge transfer (MLCT) excitation of the chromophore in 1 results in excited state formation and electron injection into the conduction band of the semiconductor, Eq. 3. The absorption spectrum of the surface-bound assembly is shown in Fig. S1. It is dominated in the visible region by an absorption at λmax = 450 nm with ε ∼ 19,000 M-1⋅cm−1 (pH dependent) arising from overlapping MLCT absorptions at the chromophore and, to a lesser degree, at the catalyst.
Nanosecond transient absorption measurements on 1 and the model chromophore, [Ru(4,4′-(PO3H2bpy)(bpy)2]2+ (RuP) in the core–shell nanoITO/TiO2 structure depicted in Fig. 2, in 0.1 M HClO4 with excitation at 425 nm and monitoring at 450 nm, show that injection, Eq. 3, occurs within 20 ns after the laser pulse. Recovery of the bleach at 450 nm following the laser flash by back electron transfer, Eq. 4, occurs with complex kinetics (Table 1) as observed previously for related surface-bound complexes on TiO2 (14, 15), but on a noticeably longer average timescale for 1 compared with the model RuP, consistent with hole transfer to the catalyst.
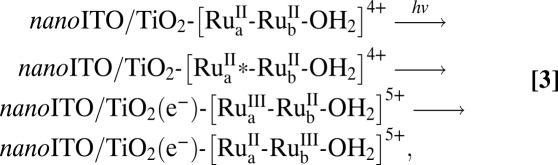 |
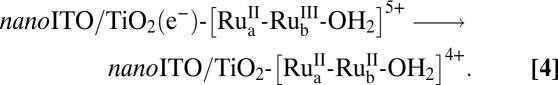 |
The reaction sequence in Eqs. 3 and 4, the mechanism of water oxidation by the assembly in Scheme 1, and the microscopic events illustrated in Fig. 1 reveal the underlying challenges to efficient DSPEC water splitting: 1) Three single-photon, single-electron excitation events are required for a single oxidative activation cycle to reach the key precursor intermediate, 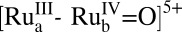 . 2) Under ambient sunlight in the nanostructured films, the rate of solar insolation is 1–2 s−1, which limits the rate of water oxidation to 0.25–0.5 s−1 per O2 given the stoichiometry of the reaction (16, 17). With back electron transfer occurring on a timescale of μs to ms, and three injection–oxidation cycles required to activate the catalyst, its reactive form is present in trace amounts under ambient solar illumination and efficiencies for water splitting are negligible. Photolysis of assembly 1 on mesoscopic thin films of TiO2 results in negligible photocurrents above background.
. 2) Under ambient sunlight in the nanostructured films, the rate of solar insolation is 1–2 s−1, which limits the rate of water oxidation to 0.25–0.5 s−1 per O2 given the stoichiometry of the reaction (16, 17). With back electron transfer occurring on a timescale of μs to ms, and three injection–oxidation cycles required to activate the catalyst, its reactive form is present in trace amounts under ambient solar illumination and efficiencies for water splitting are negligible. Photolysis of assembly 1 on mesoscopic thin films of TiO2 results in negligible photocurrents above background.
Fig. 2.

Kinetics of absorbance changes probed at 450 nm following 3.2-mJ, 425-nm pulsed laser excitation for RuP- and 1-derivatized FTO|nanoITO/TiO2 electrodes immersed in argon-degassed, aqueous pH 4.6 solutions with 0.5 M LiClO4 and 20 mM acetate/acetic acid buffer at 22 ± 1 °C. The first 10 µs displayed here were collected in a separate experiment from the longer timescale (i.e., out to 400 µs) data for improved data quality. Both data sets are presented together here (dark gray, RuP; black, 1) with the fits shown in red and blue, respectively.
Table 1.
Best-fit parameters to the triexponential decay function shown below obtained from transient absorption kinetic measurements with 450-nm monitoring at 22 ± 1 °C with 3.2-mJ, 425-nm excitation for RuP- and 1-derivatized FTO|nanoITO/TiO2 electrodes immersed in aqueous pH 4.6 solutions with 0.5 M LiClO4 and 20 mM acetate/acetic acid buffer
| Complex | A1 | τ1 | A2 | τ2 | A3 | τ3 | <τ> |
| 10 µs | |||||||
| RuP | 0.13 | 75 ns | 0.15 | 1.4 µs | 0.71 | 36 µs | 36 µs |
| 1 | 0.46 | 30 ns | 0.17 | 546 ns | 0.38 | 88 µs | 88 µs |
| 400 µs | |||||||
| RuP | 0.28 | 4 µs | 0.36 | 35 µs | 0.36 | 248 µs | 219 µs |
| 1 | 0.43 | 0.5 µs | 0.16 | 46 µs | 0.41 | 812 µs | 795 µs |
The first 10 µs and first 400 µs were fit separately to independent triexponential functions: ΔA = A1·exp(−t/τ1) + A2·exp(−t/τ2) + A3·exp(−t/τ3) with <τ> = (A1·(τ1)2 + A2·(τ2)2 + A3·(τ3)2)/(A1τ1 + A2τ2 + A3τ3).
There are several competing processes at the photoanode that dictate cell performance toward water splitting following injection. One is back electron transfer. Another is the rate of transport of injected electrons to the underlying FTO collector electrode. For a micrometers-thick TiO2 mesoscopic film typically used in such applications, transport of the injected electrons occurs on the ms timescale (15, 18). Achieving high DSPEC efficiencies requires either decreasing the rate of back electron transfer to the assembly or the electron transit time through the nanostructured film to the FTO electrode.
We report here a core–shell approach based on the latter. It overcomes unfavorable interfacial electron transfer kinetics by greatly decreasing the dimension of the TiO2 layer while preserving light absorption. In this approach, a thin layer of TiO2 is formed by ALD on the outside of an optically transparent nanoITO or nanoATO (generically nanoTCO) substrate. The latter are transparent conducting oxide (TCO) materials whose preparations and properties are described elsewhere (19).
Fig. 3 shows a transmission electron micrograph (TEM) of a core–shell structure consisting of a 3.6-nm layer of TiO2 on a 3.2-μm-thick nanoITO film with individual ITO nanoparticles 40–50 nm in diameter. In this and experiments described below, the core–shell nanoITO/TiO2 structure was prepared by 60 ALD cycles at 0.06 nm per cycle to give the outer 3.6-nm shell. In these thin TiO2 shells, rapid electron injection and slow electron transport continue to occur. However, at these dimensions, the transit time through the semiconductor to the conductive nanoTCO core is greatly decreased with injected electrons reaching the nanoITO core. Electrons injected into the outer TiO2 layer are transported rapidly to the FTO collector electrode for transmission through the external circuit to the cathode for H2 production. Stepwise excitation–injection cycles build up the multiple oxidative equivalents at the catalyst at the photoanode for oxidation of water to O2.
Fig. 3.

TEM depicting the core–shell nanostructure made by 60 ALD cycles of TiO2 deposited onto a nanoITO film on FTO glass.
The photo-driven equivalent of Scheme 1 is shown in Scheme 2. Under the conditions of the photolysis experiments–pH 4.6, 20 mM acetate/acetic acid buffer, 0.5 M LiClO4–oxidation of 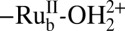 to
to 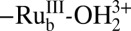 , with pKa,1 = 4.4, is followed by loss of a proton to give
, with pKa,1 = 4.4, is followed by loss of a proton to give 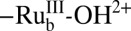 (11).
(11).
Scheme 2.

DSPEC water splitting with 1 on FTO|(core–shell)nanoITO/TiO2 in pH 4.6, 20 mM acetate/acetic acid buffer, 0.5 M LiClO4.
H2 Evolution and Photocurrent Measurements.
Fig. 4 shows the results of short-term, current-density–time measurements at the photoanode of a photoelectrochemical cell (PEC) in pH 4.6, 0.5 M LiClO4, 20 mM acetate/acetic acid buffer with 445-nm photolysis. The cell consisted of a 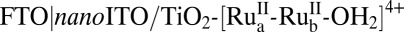 core–shell photoanode and Pt wire as cathode (SI Text). As found in previous studies (20, 21), the photocurrent was maximized with an applied bias of 0.20 V vs. normal hydrogen electrode (NHE) to match the potential for hydrogen evolution at the cathode. Integrated light intensities under the conditions of the experiment ranged from <10% to >90% of ambient sunlight at 100 mW/cm2. Negligible photocurrents were observed under the same conditions for the assembly on TiO2.
core–shell photoanode and Pt wire as cathode (SI Text). As found in previous studies (20, 21), the photocurrent was maximized with an applied bias of 0.20 V vs. normal hydrogen electrode (NHE) to match the potential for hydrogen evolution at the cathode. Integrated light intensities under the conditions of the experiment ranged from <10% to >90% of ambient sunlight at 100 mW/cm2. Negligible photocurrents were observed under the same conditions for the assembly on TiO2.
Fig. 4.

Current-density–time curves for photoelectrochemical experiments performed at 22 ± 1 °C using assembly 1 on a 3.6-nm TiO2 shell formed by ALD on nanoITO-coated FTO electrodes immersed in pH 4.6, 20 mM acetate/acetic acid buffer (0.5 M in LiClO4) as a function of 445-nm light intensity: 91 mW (red), 58 mW (black), 7.8 mW (blue). Experiments were performed in the presence of air with 0.2 V vs. NHE applied voltage bias. Counter electrode, Pt wire; reference electrode, Ag/AgCl (3 M NaCl; 0.207 V vs. NHE).
Fig. S2 shows the current–time behavior of the photoanode over a photolysis period of 30 min with 445-nm (FWHM ∼20 nm) irradiation. In this experiment, evolution of H2 at the cathode was measured as described in SI Text. Over the course of the experiment, the current decreased from 175 to 20 µA. Based on the light absorbed by the assembly at the excitation wavelength, the absorbed photon conversion efficiency was 4.4% calculated at peak photocurrent. The estimated Faradaic efficiency for H2 production was ∼86%. The decrease in photocurrent with time is currently under investigation and appears to arise from an instability toward ligand substitution in the Ru(III) form of the chromophore in the assembly, TiO2-[RuaIII-]3+. Note the change in absorption spectrum before and after photolysis in Fig. S3.
O2 Measurements.
A modified version of the rotating ring-disk electrode (RRDE) technique was used to measure O2. Murray and coworkers have applied RRDE to the investigation of oxidation electrocatalysis by iridium dioxide nanoparticles (22, 23). The RRDE technique was used to establish rates and stoichiometries of water oxidation by 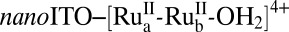 ; note Scheme 1 (11). In the RRDE experiments, a nanoparticle film of nanoITO was deposited on the glassy carbon disk of the RRDE electrode assembly by drop casting. In this experiment, oxygen generated at the disk during oxidative scans is detected at the ring by reduction of oxygen to water.
; note Scheme 1 (11). In the RRDE experiments, a nanoparticle film of nanoITO was deposited on the glassy carbon disk of the RRDE electrode assembly by drop casting. In this experiment, oxygen generated at the disk during oxidative scans is detected at the ring by reduction of oxygen to water.
For the photo-RRDE experiment, we replaced the commercially available glassy carbon disk electrode with a modified titanium disk. Electrode preparation is reported in Materials and Methods and is illustrated in Fig. 5. After depositing a nanoTCO (ITO or ATO) layer onto the Ti disk, a 3.6-nm TiO2 overlayer was deposited by ALD resulting in a nanoTCO(core)–TiO2(shell) nanostructure. ALD produced a uniform shell of TiO2 on the nanoTCO core; note the TEM image in Fig. 3. Fig. S4 shows cyclic voltammograms for 1-, RuP-, and underivatized core–shell nanoITO/TiO2 electrodes at pH 4.6 with added 20 mM acetate/acetic acid buffer in 0.5 M LiClO4 as electrolyte. RuP is the monomeric model [Ru(4,4′-(PO3H2bpy)(bpy)2]2+.
Fig. 5.

Illustrating the photo-RRDE experiment with a Ti disk and nanoTCO/TiO2 core–shell structure derivatized with 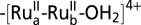 (1).
(1).
The results of a photo-RRDE experiment on a core–shell FTO|nanoATO/TiO2-1 electrode are shown in Fig. 6. As noted above, the current at the disk arises from the 4e− oxidation of water (2 H2O → O2 + 4 H++ 4 e−) and the current in the Pt ring from reduction of O2 to H2O after it is formed, O2 + 4 H++ 4 e− → 2 H2O. When the RRDE assembly is rotating (500 rpm), the appearance of current at the ring demonstrates O2 production at the disk (Fig. 6). Although not yet quantitative, these experiments do reliably and reproducibly demonstrate the production of O2 at the photoanode in tandem with H2 production at the cathode (24).
Fig. 6.

Current density response of the photo-RRDE disk and Pt ring electrodes at pH 4.6, 20 mM acetate/acetic acid buffer (0.5 M LiClO4) with assembly 1 on the surface of a 3.6-nm TiO2 shell deposited by ALD onto a nanoATO-coated Ti disk electrode. The rotation rate of the RRDE electrode assembly was 500 rpm. The applied voltage at the disk was held at 1 V while the potential at the platinum ring electrode was held at −0.35 V for O2 detection (V vs. Ag/AgCl 4 M KCl reference, 0.199 V vs. NHE); note ref. 24. Illumination was provided by the continuous output of a 473-nm DPSS laser (3.3 mW, LaserGlow model LRS-0473-PFM-00100-03). Light was cycled on or off through electronic TTL triggering of the laser.
Discussion
The observations made here, and the use of the core–shell nanoTCO/TiO2 configuration, provide the last piece of a puzzle about how to exploit chromophore–catalyst assemblies in DSPEC applications. The use of molecular assemblies is appealing because systematic synthetic changes and a modular approach can be used to obtain desirable properties by rapid iteration (25). The final output of the DSPEC is dictated by local interfacial dynamics with a core–shell advantage arising from the rapid transit of injected electrons through the thin TiO2 shell to the nanoTCO core for transfer to the cathode.
Initial per photon absorbed quantum efficiencies are 4–5% but with considerable latitude for improvement based on changes in the assembly and solution conditions. The local microscopic dynamics that dictate performance––injection, back electron transfer, competitive light absorption by the catalyst as Ru(II), rate of water oxidation––are all subject to synthetic modification. At low light levels, the cell dynamics are dictated by light absorption (Fig. 4), with evidence for rate-limiting water oxidation as the incident intensity is increased. Long-term instability of the assembly is being addressed by modifying reaction conditions, the nature of the chromophore, and stabilizing surface binding. The catalyst is stable toward water oxidation for extended periods under the conditions of the experiments as shown in the earlier electrochemical study (11).
Previous reports have appeared describing photocurrents and water oxidation by various combinations of chromophores and catalysts on TiO2 (26–31), most notably by Mallouk and coworkers (32), but typically with small photocurrents, low efficiencies, and limited stabilities. The core–shell molecular assembly approach described here offers a general platform for using chromophore–catalyst assemblies and it should open the door to a host of DSPEC applications.
Conclusions
We report here a nanoTCO-core–ALD-shell–chromophore–catalyst assembly for photoelectrochemical water splitting. This could provide a general platform for a variety of applications in solar fuels, green chemistry, and sensing.
Materials and Methods
Fabrication of nanoTCO|FTO Substrates.
A 3-g sample of nanoITO (Lihochem, Inc.) or nanoATO (Sigma-Aldrich Co.) powder was added to a mixture of acetic acid (3 g) and ethanol (10 mL) giving a 5-M solution/suspension (22 wt %). After brief manual shaking, this mixture was sonicated for 20 min. The colloidal suspension was further sonicated with a Branson ultrasonic horn outfitted with a flat microtip (70% power, 50% duty cycle; 5 min). FTO glass substrates, 4 cm × 2.2 cm, were prepared and cleaned by sonication in ethanol (EtOH) for 20 min followed by acetone for 20 min. Kapton tape was applied to one edge to maintain a defined area (1 cm × 2.5 cm). The nanoTCO colloidal suspension was coated on FTO glass substrates by a spin-coater (600 rpm, 10-s hold). nanoITO slides were annealed under air and then under 5% H2 with a method described previously (19). nanoATO slides were annealed just under air for 1 h at 500 °C. Annealed films were measured to be 3.2 ± 0.5 μm thick by surface profilometry. Once cooled to room temperature, nanoTCO electrodes were derivatized by overnight (∼16 h) immersion into 0.1 M HNO3 solutions containing ∼10−4–10−3 M Ru complex (RuP or 1).
Preparation of Ti|nanoTCO/TiO2 Disk Electrodes.
Titanium rod (commercial grade 2, McMaster-Carr Inc., model 89145K13) was machined into 4-mm-thick, 5-mm-diameter cylinders. After cleaning by sonication in EtOH for 20 min followed by acetone for 20 min, the nanoTCO solution–suspension described above was drop cast onto the flat surface of Ti. The solvent mixture was allowed to evaporate and the modified electrode was annealed under the same conditions as described above. After annealing, the electrodes were subjected to 60 ALD cycles TiO2 to create the core–shell nanostructures; Fig. S5. The resulting core–shell|Ti electrodes were further derivatized by overnight (∼16 h) immersion into 0.1 M HNO3 solutions containing ∼10−4–10−3 M Ru complex (RuP or 1). After dye loading, the modified electrodes were assembled as the disk of the Pt ring-disk electrode assembly (Pine Instruments Inc., model AFE6RIPT).
RRDE.
RRDE experiments were performed with a Pine Instruments bipotentiostat (model AFCBP1) and rotator (model AFMSRCE). The working electrode, namely the disk, was the previously described Ti|nanoTCO/TiO2 metal cylinder. The reference electrode, a standard Ag/AgCl electrode filled with 4 M KCl (Pine Instruments, model RREF0021, 0.199 V vs. NHE), was inserted into a double-junction reference port consisting of an electrolyte-filled, Vycor-tipped glass tube immersed in the primary electrolyte bath. The double junction was used to avoid chloride contamination of the primary electrolyte. The counter electrode, a platinum wire coil, was placed into an isolated glass tube with fine glass frit separator (Pine Instruments, model AFCTR5). The center of the exposed face of the working disk electrode was illuminated via the light output of a diode-pumped solid-state (DPSS) laser (Laserglow Inc., model LRS-0473-PFM-00100–05, 3.3 mW, 473 nm, 2-mm beam diameter). The laser was cycled on–off by electronic transistor–transistor logic (TTL) triggering from a pulse generator (Stanford Research Systems Inc., model DG535). Laser output power was determined using a Coherent FieldMax II-TOP power meter and Coherent LM-2 VIS power head.
All RRDE experiments were performed at 22 ± 1 °C by immersing the RRDE electrode assembly in aqueous solutions at pH 4.6 (20 mM acetate–acetic acid buffer, 0.5 M LiClO4) deaerated by bubbling with nitrogen gas for 20 min before experiments with the headspace continuously purged with nitrogen gas during experiments. The rotation rate of the RRDE electrode assembly was 500 rpm. Linear sweep voltammetry experiments in oxygen-saturated solutions were performed by using the platinum ring as the working electrode.
Nanosecond Transient Absorption Spectroscopy.
Transient absorption measurements used a commercially available laser flash photolysis apparatus (Edinburgh Instruments, Inc., model LP920) with laser excitation (425 nm, 3.2 mJ, 8-mm diameter, 5–7-ns FWHM) provided by a pulsed neodymium-doped yttrium aluminum garnet (Spectra-Physics, Inc., model Quanta-Ray LAB-170-10)/optical parametric oscillator (VersaScan-MB) laser combination. The repetition rate of the laser was matched to the rate at which the probe source was pulsed (i.e., intensified 50-fold compared with nonpulsed output), typically 1 Hz, although the laser flashlamps were fired at 10 Hz. Timing of the experiment was PC controlled via Edinburgh software (L900). The white light output of the LP920 probe source, a 450-W Xe lamp, was passed through a 40-nm long-pass color filter before passing through the sample. The LP920 was equipped with a multigrating detection monochromator outfitted with a Hamamatsu R928 photomultiplier tube (PMT) in a noncooled housing and a gated CCD (Princeton Instruments, PI-MAX3). The detector was software selectable with the PMT for monitoring transient absorption kinetics at a single wavelength (10-ns FWHM instrument response function, reliable data out to 400 μs, 300–900 nm) and the gated CCD for transient spectra covering the entire visible region (400–850 nm) at a given time after excitation with a typical gatewidth of 10 ns. For PMT measurements, spectral bandwidth was typically <5 nm with color filters placed after the sample but before the detection monochromator to eliminate laser scatter. Single-wavelength kinetic data were collected by averaging 10–100 sequences where one sequence refers to collection of laser-only data followed by pump–probe data. For timescales >10 μs, the probe-only data were also collected within the sequence because the strategy of using the linear portion before excitation to extrapolate the light intensity in the absence of the laser pulse was no longer valid due to a nonlinear temporal output of the pulsed probe source when viewed on longer timescales. Kinetic data were analyzed by using SigmaPlot (Systat, Inc.), Origin (OriginLab, Inc.), or L900 (Edinburgh, Inc.) software. Data were collected at room temperature (22 ± 1 °C).
Sample Preparation.
Transient absorption experiments were performed on samples prepared in the following manner. Derivatized FTO|nanoTCO/TiO2 electrodes (overall FTO slide size: 1.1 cm × 4.4 cm × 2.2 mm; the nanoTCO/TiO2 film covered a 1.1 cm × 1 cm area positioned at the lower end of the face of the slide) were inserted diagonally into a 1-cm path length cuvette that had been modified to incorporate a #15 O-ring joint at the top of the cuvette. Four mL of pH 4.6 (20 mM acetate buffer, 0.5 M LiClO4) solution were then added to the cuvette such that the derivatized portion of the FTO slide was completely immersed. The cuvette was sealed by attaching a separate Pyrex piece, via a mating #15 O-ring joint, modified with a sidearm and a Kontes vacuum-rated Teflon valve at the top. An argon (industrial grade, National Welders) degassing station consisting of a homebuilt glass manifold and inline oxygen scrubber, OxiClear model DGP250-R1, was used to bubble oxygen-free, argon gas through the solution via small Teflon tubing inserted through the sidearm for at least 45 min just before laser experiments to minimize effects due to the presence of oxygen. UV-visible absorption of samples was monitored before and after laser experiments to track assembly desorption–decomposition.
Supplementary Material
Acknowledgments
This research was supported primarily by the University of North Carolina Energy Frontier Research Center (UNC EFRC): Center for Solar Fuels, an Energy Frontier Research Center supported by the US Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences under Award DE-SC0001011 (supporting L.A., M.K.B., M.R.N., J.J.C., and R.A.B.). M.D.L. was supported by the Research Triangle Solar Fuels Institute. B.K. was supported by the US DOE, Office of Energy Efficiency and Renewable Energy, under Award 08NT0001925. W.S. was supported by the Center for Catalytic Hydrocarbon Functionalization, an EFRC funded by the US DOE, Office of Science, Office of Basic Energy Sciences, under Award DE-SC0001298 at the University of Virginia.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1319628110/-/DCSupplemental.
References
- 1.Nelson N, Yocum CF. Structure and function of photosystems I and II. Annu Rev Plant Biol. 2006;57(1):521–565. doi: 10.1146/annurev.arplant.57.032905.105350. [DOI] [PubMed] [Google Scholar]
- 2.Vinyard DJ, Ananyev GM, Dismukes GC. Photosystem II: The reaction center of oxygenic photosynthesis. Annu Rev Biochem. 2013;82(1):577–606. doi: 10.1146/annurev-biochem-070511-100425. [DOI] [PubMed] [Google Scholar]
- 3.Concepcion JJ, House RL, Papanikolas JM, Meyer TJ. Chemical approaches to artificial photosynthesis. Proc Natl Acad Sci USA. 2012;109(39):15560–15564. doi: 10.1073/pnas.1212254109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gust D, Moore TA, Moore AL. Solar fuels via artificial photosynthesis. Acc Chem Res. 2009;42(12):1890–1898. doi: 10.1021/ar900209b. [DOI] [PubMed] [Google Scholar]
- 5.Walter MG, et al. Solar water splitting cells. Chem Rev. 2010;110(11):6446–6473. doi: 10.1021/cr1002326. [DOI] [PubMed] [Google Scholar]
- 6.Bensaid S, Centi G, Garrone E, Perathoner S, Saracco G. Towards artificial leaves for solar hydrogen and fuels from carbon dioxide. ChemSusChem. 2012;5(3):500–521. doi: 10.1002/cssc.201100661. [DOI] [PubMed] [Google Scholar]
- 7.Cowan AJ, Durrant JR. Long-lived charge separated states in nanostructured semiconductor photoelectrodes for the production of solar fuels. Chem Soc Rev. 2013;42(6):2281–2293. doi: 10.1039/c2cs35305a. [DOI] [PubMed] [Google Scholar]
- 8.Smestad GP, Steinfeld A. Review: Photochemical and thermochemical production of solar fuels from H2O and CO2 using metal oxide catalysts. Ind Eng Chem Res. 2012;51(37):11828–11840. [Google Scholar]
- 9.Zou Z, Ye J, Sayama K, Arakawa H. Direct splitting of water under visible light irradiation with an oxide semiconductor photocatalyst. Nature. 2001;414(6864):625–627. doi: 10.1038/414625a. [DOI] [PubMed] [Google Scholar]
- 10.Alibabaei L, et al. Applications of metal oxide materials in dye sensitized photoelectrosynthesis cells for making solar fuels: Let the molecules do the work. J Mater Chem A. 2013;1:4133–4145. [Google Scholar]
- 11.Norris MR, Concepcion JJ, Fang Z, Templeton JL, Meyer TJ. Low overpotential water oxidation by a surface-bound Ruthenium-chromophore-Ruthenium-catalyst assembly. Angewandte Chemie International Edition. doi: 10.1002/anie.201305951. in press. [DOI] [PubMed] [Google Scholar]
- 12.Chen Z, Concepcion JJ, Jurss JW, Meyer TJ. Single-site, catalytic water oxidation on oxide surfaces. J Am Chem Soc. 2009;131(43):15580–15581. doi: 10.1021/ja906391w. [DOI] [PubMed] [Google Scholar]
- 13.Concepcion JJ, Tsai M-K, Muckerman JT, Meyer TJ. Mechanism of water oxidation by single-site ruthenium complex catalysts. J Am Chem Soc. 2010;132(5):1545–1557. doi: 10.1021/ja904906v. [DOI] [PubMed] [Google Scholar]
- 14.Ashford DL, et al. Photoinduced electron transfer in a chromophore-catalyst assembly anchored to TiO2. J Am Chem Soc. 2012;134(46):19189–19198. doi: 10.1021/ja3084362. [DOI] [PubMed] [Google Scholar]
- 15.Hagfeldt A, Boschloo G, Sun L, Kloo L, Pettersson H. Dye-sensitized solar cells. Chem Rev. 2010;110(11):6595–6663. doi: 10.1021/cr900356p. [DOI] [PubMed] [Google Scholar]
- 16.Gratzel M. The artificial leaf, bio-mimetic photocatalysis. CATTech. 1999;3:4–17. [Google Scholar]
- 17.O’Regan BC, Durrant JR. Kinetic and energetic paradigms for dye-sensitized solar cells: Moving from the ideal to the real. Acc Chem Res. 2009;42(11):1799–1808. doi: 10.1021/ar900145z. [DOI] [PubMed] [Google Scholar]
- 18.Mohammadpour R, Iraji Zad A, Hagfeldt A, Boschloo G. Comparison of trap-state distribution and carrier transport in nanotubular and nanoparticulate TiO(2) electrodes for dye-sensitized solar cells. ChemPhysChem. 2010;11(10):2140–2145. doi: 10.1002/cphc.201000125. [DOI] [PubMed] [Google Scholar]
- 19.Hoertz PG, Chen Z, Kent CA, Meyer TJ. Application of high surface area tin-doped indium oxide nanoparticle films as transparent conducting electrodes. Inorg Chem. 2010;49(18):8179–8181. doi: 10.1021/ic100719r. [DOI] [PubMed] [Google Scholar]
- 20.Song W, et al. Interfacial electron transfer dynamics for [Ru(bpy)2((4,4′-PO3H2)2bpy)]2+ sensitized TiO2 in a dye-sensitized photoelectrosynthesis cell: Factors influencing efficiency and dynamics. J Phys Chem C. 2011;115(14):7081–7091. [Google Scholar]
- 21.Fujishima A, Honda K. Electrochemical photolysis of water at a semiconductor electrode. Nature. 1972;238(5358):37–38. doi: 10.1038/238037a0. [DOI] [PubMed] [Google Scholar]
- 22.Nakagawa T, Bjorge NS, Murray RW. Electrogenerated IrO(x) nanoparticles as dissolved redox catalysts for water oxidation. J Am Chem Soc. 2009;131(43):15578–15579. doi: 10.1021/ja9063298. [DOI] [PubMed] [Google Scholar]
- 23.Nakagawa T, Beasley CA, Murray RW. Efficient electro-oxidation of water near its reversible potential by a mesoporous IrOx nanoparticle film. J Phys Chem C. 2009;113(30):12958–12961. [Google Scholar]
- 24.Concepcion JJ, Binstead RA, Alibabaei L, Meyer TJ. Application of the rotating ring-disc-electrode technique to water oxidation by surface-bound molecular catalysts. Inorg Chem. 2013;52(19):10744–10746. doi: 10.1021/ic402240t. [DOI] [PubMed] [Google Scholar]
- 25.Alstrum-Acevedo JH, Brennaman MK, Meyer TJ. Chemical approaches to artificial photosynthesis. 2. Inorg Chem. 2005;44(20):6802–6827. doi: 10.1021/ic050904r. [DOI] [PubMed] [Google Scholar]
- 26.Blakemore JD, et al. Half-sandwich iridium complexes for homogeneous water-oxidation catalysis. J Am Chem Soc. 2010;132(45):16017–16029. doi: 10.1021/ja104775j. [DOI] [PubMed] [Google Scholar]
- 27.Brimblecombe R, Koo A, Dismukes GC, Swiegers GF, Spiccia L. Solar driven water oxidation by a manganese molecular catalyst inspired by photosystem II. J Am Chem Soc. 2010;132(9):2892–2894. doi: 10.1021/ja910055a. [DOI] [PubMed] [Google Scholar]
- 28.Duan L, et al. A molecular ruthenium catalyst with water-oxidation activity comparable to that of photosystem II. Nat Chem. 2012;4(5):418–423. doi: 10.1038/nchem.1301. [DOI] [PubMed] [Google Scholar]
- 29.Duan L, Fischer A, Xu YH, Sun L. Isolated seven-coordinate Ru(IV) dimer complex with [HOHOH](-) bridging ligand as an intermediate for catalytic water oxidation. J Am Chem Soc. 2009;131(30):10397–10399. doi: 10.1021/ja9034686. [DOI] [PubMed] [Google Scholar]
- 30.Duan L, Xu YH, Tong LP, Sun L. Ce(IV)- and light-driven water oxidation by [Ru(terpy)(pic)3]2+ analogues: Catalytic and mechanistic studies. ChemSusChem. 2011;4(2):238–244. doi: 10.1002/cssc.201000313. [DOI] [PubMed] [Google Scholar]
- 31.Hull JF, et al. Highly active and robust Cp* iridium complexes for catalytic water oxidation. J Am Chem Soc. 2009;131(25):8730–8731. doi: 10.1021/ja901270f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao Y, et al. Improving the efficiency of water splitting in dye-sensitized solar cells by using a biomimetic electron transfer mediator. Proc Natl Acad Sci USA. 2012;109(39):15612–15616. doi: 10.1073/pnas.1118339109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.