

Significance
Pseudoxanthoma elasticum (PXE) is a heritable disease characterized by mineralization of the skin, eyes, and arteries, for which no effective treatment exists. PXE is caused by inactivating mutations in the gene encoding the transporter ABCC6. PXE is a metabolic disease caused by the absence of an unknown factor in the circulation. Our study indicates that the factor that normally prevents PXE is pyrophosphate, which is provided to the circulation in the form of nucleoside triphosphates via an ABCC6-dependent mechanism. Our findings provide leads for the treatment of this intractable disease.
Keywords: ATP secretion, ectopic calcification, MRP6, ENPP1
Abstract
Pseudoxanthoma elasticum (PXE) is an autosomal recessive disease characterized by progressive ectopic mineralization of the skin, eyes, and arteries, for which no effective treatment exists. PXE is caused by inactivating mutations in the gene encoding ATP-binding cassette sub-family C member 6 (ABCC6), an ATP-dependent efflux transporter present mainly in the liver. Abcc6−/− mice have been instrumental in demonstrating that PXE is a metabolic disease caused by the absence of an unknown factor in the circulation, the presence of which depends on ABCC6 in the liver. Why absence of this factor results in PXE has remained a mystery. Here we report that medium from HEK293 cells overexpressing either human or rat ABCC6 potently inhibits mineralization in vitro, whereas medium from HEK293 control cells does not. Untargeted metabolomics revealed that cells expressing ABCC6 excrete large amounts of nucleoside triphosphates, even though ABCC6 itself does not transport nucleoside triphosphates. Extracellularly, ectonucleotidases hydrolyze the excreted nucleoside triphosphates to nucleoside monophosphates and inorganic pyrophosphate (PPi), a strong inhibitor of mineralization that plays a pivotal role in several mineralization disorders similar to PXE. The in vivo relevance of our data are demonstrated in Abcc6−/− mice, which had plasma PPi levels <40% of those found in WT mice. This study provides insight into how ABCC6 affects PXE. Our data indicate that the factor that normally prevents PXE is PPi, which is provided to the circulation in the form of nucleoside triphosphates via an as-yet unidentified but ABCC6-dependent mechanism.
Pseudoxanthoma elasticum (PXE) is an autosomal recessive disease characterized by late-onset and progressive ectopic mineralization of skin, eyes, and arteries (1). This prototypical connective tissue disease affects ∼1 in 50,000 persons worldwide, for whom there is no effective therapy (2). Along with relatively benign (but stigmatizing) skin lesions, PXE causes progressive loss of vision and cardiovascular complications (2). The severity of these symptoms is highly variable among patients, even within families (3).
In 2000, multiple research groups reported that PXE is caused by inactivating mutations in the ATP-binding cassette sub-family C member 6 (ABCC6) gene (4–7). ABCC6, also known as multidrug resistance-associated protein 6 (MRP6), is a member of the C branch of the superfamily of ATP-binding cassette (ABC)-transporters, which use the energy provided by the hydrolysis of ATP to transport substrates across a membrane (8). ABCC6 expression is high in the liver, lower in the kidney, and very low or absent in other tissues, including affected skin, eyes, and arteries (5, 9). In the liver, ABCC6 is present in the sinusoidal (basolateral) membrane of hepatocytes, facing the central circulation (10–12).
Abcc6−/− mice faithfully recapitulate most of the symptoms of PXE and have been indispensable for showing that PXE is a metabolic disease (13–16). Abcc6−/− muzzle skin that mineralizes in Abcc6−/− mice does not mineralize when grafted onto WT mice, and muzzle skin of WT mice mineralizes only when grafted onto Abcc6−/− mice (17). Moreover, surgically joining the systemic circulation of Abcc6−/− mice with that of WT mice halts mineralization in Abcc6−/− mice (18). Thus, PXE is caused not by a lack of functional ABCC6 in the affected tissues, but rather by the absence of a factor that is normally provided to the circulation by an ABCC6-dependent mechanism. The nature of this factor has remained elusive.
In vitro vesicular uptake experiments have confirmed that ABCC6, like its close homolog ABCC1, is an efflux transporter, transporting a handful of glutathione-conjugates and the cyclic peptide BQ-123 (10, 19, 20). The short list of identified ABCC6 substrates does not include any compound obviously relevant for PXE, and the only substrate shared by human ABCC6 (hABCC6) and rat ABCC6 (rABCC6) is not an endogenous compound, but rather the synthetic cyclic peptide BQ-123. Based on the clinical similarities of PXE to other diseases, vitamin K conjugates (21) and adenosine (22) have been proposed as substrates, but none of these are actually transported (23, 24).
Thus, despite all efforts, the mechanism by which ABCC6 prevents ectopic mineralization remains a mystery, and no attractive new hypotheses about the nature of the physiological ABCC6 substrate have been proposed. In the present study, we used untargeted metabolomics to screen for ABCC6-dependent factors that inhibit mineralization, to shed more light on the mechanism by which ABCC6 prevents pathological mineralization.
Results
ABCC6-Conditioned Medium Inhibits Mineralization in Vitro.
PXE is a metabolic disease caused by the absence of a factor, the presence of which depends on ABCC6 in the liver (17). Jiang and coworkers (14, 18) previously showed that in an in vitro system using vascular smooth muscle cells, serum from Abcc6+/+ mice inhibits mineralization to a greater extent than serum from Abcc6−/− mice. We took a similar approach to test whether a mineralization-inhibiting factor is also present in medium from HEK293 cells overexpressing human ABCC6 (HEK293/hABCC6) or rat ABCC6 (HEK293/rABCC6). Both HEK293/hABCC6- and HEK293/rABCC6-conditioned medium potently inhibited mineralization of ATDC5 cells, whereas medium from control cells (HEK293/GFP) did not (Fig. 1). These results indicate that the factor that prevents mineralization in vivo is also extruded from cultured HEK293 cells containing ABCC6 in vitro.
Fig. 1.

Effect of human and rat ABCC6-conditioned medium on in vitro mineralization of ATDC5 cells. ATDC5 cells were seeded in a 24-well plate (2.8 × 104 cells/well) and cultured in propagation medium for 4 d and in differentiation medium for 7 d, before the addition of mineralization medium supplemented with 50% conditioned medium from HEK293/GFP (control), HEK293/rABCC6, or HEK293/hABCC6 cells (three different batches). After 2 d, calcium phosphate deposits were visualized using alizarin red staining (A), followed by solubilization in cetylpyridinium and quantitation at 550 nm (B). Data are mean ± SD (n = 6).
ABCC6-Conditioned Medium Contains High Levels of Nucleosides and Nucleotides.
To identify the ABCC6-dependent factor responsible for inhibition of mineralization, we generated global metabolite profiles of plasma samples as well as culture media using untargeted LC-MS metabolomics. In these profiles, we searched for ABCC6-related differences in metabolite levels, but we did not detect any compounds in plasma that were consistently more abundant in Abcc6+/+ mice (n = 9) than in littermate Abcc6−/− mice (n = 10). However, the differences between conditioned medium from ABCC6-overexpressing cells and control cells were overwhelming; the most profound differences were induced by the presence of rABCC6.
Using database searches (Human Metabolome Database and METLIN metabolome database), MS/MS fragmentation spectra, and reference standards, we identified many of these ABCC6-dependent compounds as nucleosides, nucleoside monophosphates, and nucleotide sugars (Table 1, control). The increased extracellular levels of these compounds were not related to cell lysis, given that the levels of oxidized glutathione were similar across all sample groups (Table 1, control). Besides inosine and guanosine, we detected no other ribonucleosides in the medium samples. Although the selected mass range (m/z 250–1,000) did not cover all ribonucleosides, we did not detect any other ribonucleosides in experiments with wider mass ranges either. The negative ionization applied for MS detection results in a higher sensitivity toward anionic nucleotides than toward basic ribonucleosides, which could explain why they were not detected. Even though nucleosides, nucleotides, and nucleotide sugars account for the major differences, we also found differences (both increases and decreases) in the levels of compounds that we could not identify. All raw data are available at the MetaboLights repository, and the processed data are accessible on XCMS Online (25, 26).
Table 1.
Differences in conditioned medium (24 h) of ABCC6-overexpressing and control cells in the absence (control) or presence of the ectonucleotidase inhibitors ARL67156 and AMP-PCP, as detected by untargeted metabolomics
| Compound | Control | ARL67156 + AMP-PCP | ||||
| HEK293 controls, peak area, AU | hABCC6, fold change/peak area, AU | rABCC6, fold change/peak area, AU | HEK293 controls, peak area, AU | hABCC6, fold change/peak area, AU | rABCC6, fold change/peak area, AU | |
| Nucleosides | ||||||
| Inosine | 9.48E+03 | 8.6*** | 79*** | 1.34E+06 | 0.9ns,† | 0.8ns,† |
| Guanosine | 8.65E+03 | 3.6ns | 23*** | 3.62E+04 | 0.7ns | 1.0ns |
| Nucleoside monophosphates | ||||||
| AMP | 8.60E+04 | 7.5*** | 99*** | 1.01E+08 | 0.9ns,† | 1.0ns,† |
| GMP | 5.52E+03 | 37*** | 401*** | 2.38E+05 | 8*** | 31*** |
| CMP‡ | 7.15E+01 | 158*** | 1,292*** | 4.29E+03 | 5.8*** | 29.2*** |
| UMP | 1.52E+04 | 10*** | 64*** | 7.61E+04 | 6.5*** | 26*** |
| TMP | 4.23E+03 | 1.1ns | 13.6*** | 9.57E+03 | 9.8*** | 22.8*** |
| dAMP | 6.78E+03 | 0.2ns | 5.0* | 4.07E+04 | 1.8*** | 5.9*** |
| Nucleoside diphosphates | ||||||
| ADP | 6.88E+04 | 0.9ns | 1.9ns | 1.88E+06 | 12*** | 26*** |
| GDP | 1.94E+02 | 2.1ns | 6.3ns | 1.45E+04 | 33*** | 162*** |
| CDP‡ | ND | ND | 1.22E+02ns,¶ | 3.76E+03 | 15.9*** | 44.4*** |
| UDP | ND | 1.32E+02ns,¶ | 1.38E+03ns,¶ | 4.60E+03 | 17*** | 99*** |
| TDP | ND | ND | ND | 1.54E+02 | 642*** | 1590*** |
| Nucleoside triphosphates | ||||||
| ATP | 5.31E+04 | 1.0ns | 1.0ns | 5.76E+04 | 15*** | 127*** |
| GTP | 4.79E+02 | 0.3ns | 1.2ns | 3.03E+03 | 4.6*** | 73*** |
| CTP | ND | ND | ND | ND | 2.41E+03***,¶ | 2.65E+04***,¶ |
| UTP | 1.52E+02 | NDns | NDns | ND | 8.63E+03***,¶ | 6.70E+04***,¶ |
| Nucleotide sugars | ||||||
| ADP-ribose§ | 7.41E+03 | 0.1ns | 2.4ns | 9.34E+03 | 2.7*** | 45*** |
| GDP-hexose‡,§ | ND | ND | 9.71E+04***,¶ | 4.98E+04 | 4.8*** | 20*** |
| GDP-deoxyhexose‡,§ | ND | ND | 1.40E+05***,¶ | 6.21E+04 | 2.7*** | 11*** |
| UDP-glucuronic acid | 5.25E+03 | NDns | 1.7ns | 2.70E+05 | 1.5*** | 2.5*** |
| UDP-glucose | 6.85E+01 | 6.6ns | 168*** | 1.07E+05 | 1.6** | 3.9*** |
| UDP-N-acetylhexosamine§ | 1.17E+05 | 3.3*** | 19.4*** | 7.47E+06 | 1.2ns | 1.4* |
| CMP-N-acetylneuramic acid | 4.71E+04 | 1.7ns | 3.5** | 6.84E+04 | 1.7ns | 2.7** |
| Miscellaneous | ||||||
| Oxidized glutathione | 1.77E+06 | 1.4ns | 1.2ns | 1.81E+06 | 1.4ns | 1.3ns |
These differences are not due to cell lysis, as demonstrated by the similar levels of oxidized glutathione in all samples. Peak areas are shown as mean values (n = 6). ND, not detected.
Breakdown of AMP-PCP generated high levels of AMP and inosine and masked potential differences.
Putative identification, no reference standard tested.
In those cases where a metabolite was detected only for ABCC6-containing cells, the peak area (in italic) is shown instead of fold-change.
The exact identity of the sugar moiety was not determined.
P < 0.05, **P < 0.01, ***P < 0.001 (all in bold); ns, P not significant, two-sided Student t test, ABCC6 (n = 3) vs. HEK293 + HEK293/GFP (n = 6).
ABCC6 Increases Extracellular Pyrophosphate Levels.
None of the compounds found to be increased in medium of ABCC6-containing cells could be linked directly to mineralization. A differentially present compound with an exceptionally high abundance and fold change in medium of ABCC6-containing cells was adenosine monophosphate (AMP). Although AMP itself is not known to affect mineralization, adenosine triphosphate (ATP) does have a role in the prevention of ectopic mineralization. ATP can be hydrolyzed to AMP and the mineralization inhibitor inorganic pyrophosphate (PPi) by ectonucleotide pyrophosphatase-phosphodiesterase (ENPP)-type ectonucleotidases (27). PPi was not detected by our untargeted metabolomics method, however, owing to the chosen mass range. Thus, we tested whether PPi levels were affected by the presence of ABCC6 using a specific enzymatic assay. The PPi levels in HEK293/hABCC6 and HEK293/rABCC6 medium increased rapidly over time, reaching micromolar levels that were already 19- and 85-fold higher than those in control media, respectively, after only 24 h (Fig. 2A). Lomashvili et al. (28) previously reported that PPi levels as low as 5 µM strongly inhibit mineralization of aortic vascular smooth muscle cells in vitro; thus, the high PPi levels in our ABCC6-conditioned medium are sufficient to inhibit mineralization.
Fig. 2.

Effect of ABCC6 expression and ectonucleotidase inhibition on nucleotide and pyrophosphate levels in culture medium. Control cells (HEK293, ○-dashed line and HEK293/GFP, □-dashed line) and ABCC6-overexpressing cells (HEK293/hABCC6, ▲-solid line and HEK293/rABCC6, ▼-solid line) were seeded in a six-well plate (7.5 × 105 cells/well) and grown to confluence overnight. Medium was replaced with control medium (A–C) or medium containing 1 mM ARL67156 plus 0.5 mM AMP-PCP (D–F), and samples were collected for LC-MS metabolomics and PPi (A and D) quantification. Metabolite levels were determined semiquantitatively by accurate mass extraction from the LC-MS data. Data are mean ± SD (n = 3). AU, arbitrary units.
Pyrophosphate Is Formed from Nucleoside Triphosphates by Ectonucleotidases.
To provide further evidence that nucleoside triphosphates are secreted by ABCC6-containing cells, we incubated cells in the presence of the ectonucleotidase inhibitors ARL67156 and β,γ-methyleneadenosine triphosphate (AMP-PCP). Ectonucleotidase inhibition strongly diminished the ABCC6-dependent increase in extracellular PPi (Fig. 2 A and D). Inversely, this inhibition resulted in an ABCC6-dependent increase in ATP (Fig. 2 B and E). Similar results were obtained for the other nucleoside triphosphates (Fig. S1), although ATP was the most abundant.
The identified differences detected in the 24-h medium samples with ectonucleotidase inhibitors are summarized in Table 1. In the presence of inhibitors, we also detected increased nucleoside diphosphate levels in ABCC6-conditioned medium. These nucleoside diphosphates presumably are derived from the high levels of nucleoside triphosphates. Although the absolute levels of nucleotide sugars are increased on ectonucleotidase inhibition, the differences between ABCC6-overexpressing cells and control cells are generally reduced (Table 1 and Fig. 2 C and F). The differences detected in the absence of inhibitors are probably an indirect effect caused by the high amounts of nucleotides in medium of ABCC6-containing cells, which may competitively inhibit the breakdown of nucleotide sugars by ectonucleotidases. In conclusion, our data indicate that nucleoside triphosphates are the predominant class of compounds secreted by ABCC6-containing cells.
PXE-Associated ABCC6V1298F Does Not Increase Extracellular Pyrophosphate Levels.
To confirm that only expression of functional ABCC6 raises extracellular PPi levels, we generated Flp-In T-REx 293 cells containing inducible rABCC6, hABCC6, or hABCC6V1298F constructs. hABCC6V1298F is properly routed to the plasma membrane, but its transport activity is <10% of that of WT hABCC6 owing to the V1298F mutation in one of its ATP-binding domains (12, 20). Importantly, this mutation occurs in patients with PXE (12, 20). On induction by doxycyclin, all three cell lines expressed ABCC6 (Fig. S2). Flp-In T-REx 293 cells are designed to express inserted constructs at a similar level, and this was the case in our experiments as well (Fig. S2). Extracellular PPi levels increased on induction of rABCC6 (Fig. 3A) and hABCC6 (Fig. 3B), but not on induction of hABCC6V1298F (Fig. 3B), demonstrating that ATP hydrolysis is required for nucleotide release. The comparable PPi levels found in the medium of cells expressing rABCC6 or hABCC6 indicate that these orthologs have no intrinsic differences in catalytic activity. Thus, the higher activity of the HEK293/rABCC6 cells compared with HEK293/hABCC6 cells is most likely caused by a difference in expression levels.
Fig. 3.

Effect of induced rABCC6, hABCC6, or hABCC6V1289F expression on PPi levels in culture medium. Control cells (Flp-In T-REx 293) and Flp-In T-REx 293 containing inducible rABCC6 (A), hABCC6, or hABCC6V1298F (B) constructs were seeded in 24-well plates (2 × 105 cells/well) and grown to confluence overnight. ABCC6 expression was induced by adding 1 µg/mL of doxycyclin (+dox). Each day, the medium was collected for PPi determination and replaced with fresh medium. Data are mean ± SD (n = 3). LLQ, lower limit of quantitation of the assay.
Nucleoside Triphosphates Are Not Transported by rABCC6 in Vesicular Transport Assays.
Because ABCC6 functions as an efflux pump (10, 19, 20), the most straightforward explanation for the increased secretion of nucleotides by ABCC6-containing cells is that these compounds are ABCC6 substrates. We tested whether nucleoside triphosphates are transported into rABCC6-containing, inside-out membrane vesicles in an ATP-dependent fashion. A major complicating factor in the use of ATP as a substrate in such assays is that ATP is hydrolyzed by the nucleotide-binding domains of rABCC6 to generate the energy required for transport. Moreover, we found that for human ABCC1, GTP and UTP can be hydrolyzed as well (Fig. S3).
Nevertheless, uptake of physiological concentrations of ATP (4 mM), GTP (500 µM), and UTP (20 µM) by rABCC6-containing vesicles was not greater than uptake by control vesicles in the presence of ATP (Fig. S4). The addition of glutathione, a cofactor required for other ABCC subfamily transporters, did not change these negative results (Fig. S4).
Importantly, the rABCC6-containing inside-out membrane vesicles were functionally active, being derived from the same cell line used in the metabolite profiling experiments and transporting the known rABCC6 substrate BQ-123 (10) (Fig. S5). Based on these experiments, we conclude that rABCC6 does not transport nucleoside triphosphates, but induces nucleotide release indirectly via an as-yet unknown mechanism.
In Vivo Relevance: Abcc6−/− Mice Have Reduced PPi Plasma Levels.
Results obtained in vitro do not necessarily apply in vivo. Notably, we did not detect reduced levels of nucleotides or nucleosides in plasma samples from Abcc6−/− mice. To assess whether the increased efflux of nucleoside triphosphates from ABCC6-containing cells has in vivo relevance, we evaluated the effect of the Abcc6 genotype on plasma PPi levels in vivo. Platelets release large amounts of the mineralization inhibitor PPi on activation (29), which might obscure detection of differences between Abcc6+/+ and Abcc6−/− mice. Thus, we measured PPi in platelet-free plasma. Similar levels of platelet factor 4 (PF4) across the sample groups ruled out differences in platelet activation before platelet removal (Fig. S6). In line with our in vitro data, the plasma concentration of PPi was significantly lower in Abcc6−/− mice compared with Abcc6+/+ mice, in both males (2.4-fold) and females (5.6-fold) (Fig. 4). The PPi levels of Abcc6+/− mice were not significantly different from those in Abcc6+/+ mice. These data clearly support a function of ABCC6 in the regulation of PPi levels in vivo.
Fig. 4.
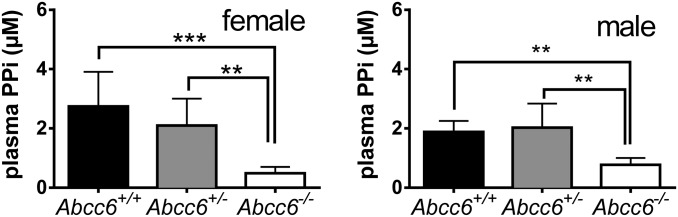
Effect of the Abcc6 genotype on PPi levels in mouse plasma. Platelet-free plasma was collected and analyzed with a specific PPi assay based on the PPi-dependent conversion of glucose-1-phosphate to glucuronic acid-6-phosphate. Data are mean ± SD. **P < 0.01; ***P < 0.001, one-way ANOVA (n = 7, 8, and 6 for females; n = 7, 6, and 5 for males).
Discussion
PXE is caused by inactivating mutations in the gene encoding the efflux transporter ABCC6. Thus far, it has been unclear why decreased activity of this hepatic efflux transporter results in the ectopic mineralization of skin, eyes, and blood vessels, and which antimineralization factor is missing in the circulation of PXE patients. We now provide evidence that ABCC6 indirectly raises plasma PPi, a known antimineralization factor (28). The loss of ABCC6 in Abcc6−/− mice significantly reduced plasma PPi levels, and transfection of HEK293 cells with rat or human ABCC6 constructs resulted in the accumulation of substantial levels of PPi in the culture medium.
How ABCC6 accomplishes this PPi accumulation remains unclear. We found that the presence of ABCC6 in HEK293 cells results in the excretion of ATP and other nucleoside triphosphates, which are rapidly converted into PPi and nucleoside monophosphates by an ENPP-type ectonucleotidase. ABCC6 was unable to transport nucleoside triphosphates in our vesicular transport experiments, however.
Because ABCC6, like its close homolog ABCC1, functions as a transporter, we favor a model in which ABCC6 secretes an as-yet unidentified factor X that induces nucleoside triphosphate release in HEK293 cells and peripheral tissues. Thus far, all of our experiments aimed at identifying X have failed. Our metabolomic analyses of plasma and culture medium did not reveal X. Even experiments in which [3H]adenosine-labeled HEK293/GFP cells were exposed to HEK293/rABCC6-conditioned medium did not result in the induction of [3H]ATP release (Fig. S7). Under these conditions, [3H]adenosine is rapidly converted into [3H]ATP and labeled HEK293/rABCC6 cells secrete the label (Fig. S7).
There are obvious reasons why the experiments have not yielded evidence of factor X. Factor X could be unstable, directly bound to cells, or slowly acting. It is important to stress that our data do not exclude the possibility that ABCC6 mediates nucleotide release by another, indirect mechanism. In general, nucleotide release can be mediated through vesicular release or ion channels, but an endogenous compound that can induce this release is unknown (30). Although highly controversial, it should be noted that other members of the ABC family, including ABCB1 (P-glycoprotein) and ABCC7 (CFTR), also have been associated with ATP release (31, 32); however, we did not observe increased nucleotide release from HEK293 cells overexpressing ABCB1, ABCC1, ABCC3, ABCC4, or ABCC5 in our laboratory.
Regardless of the mechanism, our demonstration that ABCC6 induces the release of nucleoside triphosphates, resulting in PPi production by an ENPP-type ectonucleotidase, firmly links PXE to a group of calcification disorders related to low PPI levels. Mutations in ectonucleotide pyrophosphatase-phosphodiesterase 1 (ENPP1) cause generalized arterial calcification of infancy (GACI), a disease with clinical symptoms overlapping those of PXE (33, 34). The similarity between GACI and PXE is underscored by the recent observation that both GACI and PXE can be caused by mutations in ENPP1 as well as in ABCC6 (35). Another mineralization disorder, arterial calcification due to deficiency of CD73 (ACDC), is caused by the absence of functional ecto-5′-nucleotidase (NT5E, or CD73), an enzyme that converts extracellular AMP to adenosine and phosphate (22, 36). The absence of NT5E results in ectopic mineralization owing to reduced extracellular formation of adenosine, which leads to increased expression of tissue-nonspecific alkaline phosphatase (TNAP), a PPi-degrading enzyme (36). Based on the similarities in ACDC, GACI, and PXE, others have hypothesized that ABCC6 prevents PXE through a mechanism overlapping the ENPP1-NT5E pathway (37, 38). Our findings provide direct experimental support for this hypothesis and position ABCC6 immediately upstream of the ENPP1-NT5E pathway.
In addition to TNAP, ENPP1, and NT5E, the PPi channel ANK also regulates extracellular PPi levels (39). Differences in these PPi-regulating factors may counteract or aggravate the loss of ABCC6 and could explain the high variability in PXE penetrance. It would be interesting to investigate whether PXE penetrance is associated with decreased extracellular PPi levels and specific genetic variants of genes that control these levels.
Several modifier genes, including GGCX, SPP1, and MGP, have been associated with the clinical manifestations of PXE (2), but none of these genes has been directly associated with PPi. Likewise, oral magnesium supplementation, which has been used to treat Abcc6−/− mice (40), does not affect serum PPi levels (41). Most likely, these factors modify the PXE phenotype via mechanisms not involving PPi.
Alterations in the abundance of other antimineralization factors have been described in PXE patients and Abcc6−/− mice (14). For example, serum levels of the mineralization inhibitor fetuin A are slightly reduced in PXE patients and Abcc6−/− mice (14, 42). Production of fetuin A in the liver was similar in WT and Abcc6−/− mice, however (14). The reduced serum fetuin A levels in Abcc6−/− mice might be related to binding of circulating fetuin A to the mineralizing lesions, as observed in other mineralization disorders (43). Similarly, matrix Gla protein (MGP) was found to be reduced and undercarboxylated in PXE patients and Abcc6−/− mice (44, 45). Given that carboxylated MGP binds to hydroxyapatite crystals, the reduced MGP levels could be secondary to mineralization rather than causative (46).
Our metabolomic screen did not detect consistent differences between plasma samples from Abcc6+/+ and Abcc6−/− mice, whereas many differences were found in conditioned medium. Most likely, the nucleosides and nucleotides that we detected in vitro were quickly dispersed, metabolized, or reused in vivo, resulting in levels below the limit of detection of our metabolomic screen. Nevertheless, we detected decreased plasma PPi levels in ABCC6-deficient mice with a specific enzymatic assay, demonstrating that our in vitro findings apply in vivo.
Our data indicate that PPi is the factor that normally prevents PXE. If reduced PPi levels cause mineralization, then PPi supplementation could prevent it. Indeed, daily i.p. administration of PPi was found to inhibit vascular mineralization in rats and mice with experimentally induced renal failure; however, the short plasma half-life and lack of a suitable dosage form make PPi an unattractive candidate for supplementation therapy in humans (47, 48). Treatment with bisphosphonates, a class of nonhydrolyzable PPi analogs, has been reasonably successful in GACI patients (49, 50), but data are lacking for PXE patients (35, 51). Alternatively, administration of the hypothetical factor X could be a potential cure for PXE. We are continuing our experiments to determine how ABCC6 induces nucleotide release.
Materials and Methods
ABCC6-Overexpressing Cells.
cDNAs encoding hABCC6 [kindly provided by András Váradi, Institute of Enzymology, Budapest (20)], rABCC6 [kindly provided by Bruno Stieger, University of Zurich, Zurich (10)], and GFP were subcloned into pDonr223. The V1298F mutation was introduced into hABCC6 with the Quick Change II Site-Directed Mutagenesis Kit (Agilent), using the following primers: 5′-GAAGGTGGGCATCTTTGGCAGGACCGG-3′ (forward) and 5′-CCGGTCCTGCCAAAGATGCCCACCTTC-3′ (reverse). cDNAs encoding hABCC6, rABCC6, and GFP were subcloned into the pQCXIP expression vector and transfected into HEK293 cells using calcium phosphate precipitation. The presence of rABCC6 [K14 antibody, provided by Bruno Stieger (10)] and hABCC6 [M6II-7 antibody (11)] was confirmed in clones resistant to puromycin (2 µg/mL) by immunoblot analysis.
cDNAs encoding rABCC6, hABCC6, and hABCC6V1298F were subcloned into the pcDNA5-FRT-TO expression vector that was made Gateway-compatible by introduction of the ccdB cassette (attR1-CmR-ccdB-attR2). Flp-In T-REx 293 cells were transfected with a 1:9 mixture of cDNA in pcDNA5-FRT-TO and pOG44 (Invitrogen) using lipofectamine (Invitrogen), according to the manufacturer’s instructions. The presence of rABCC6 and hABCC6 was confirmed after induction with 1 µg/mL of doxyxyclin in clones resistant to hygromycin (100 µg/mL) by immunoblot analysis as described above.
Abcc6−/− Mice.
Abcc6−/− mice were generated as described previously (16), and back-crossed into a C57BL/6 background (52). WT Abcc6+/+ mice, heterozygous Abcc6+/− mice, and Abcc6−/− mice were housed in constant-temperature rooms under a 12-h light/12-h dark cycle and received food and water ad libitum. All mouse handling and experimental procedures were conducted in accordance with institutional guidelines for animal care and use.
In Vitro Mineralization Assay.
The in vitro mineralization assay using ATDC5 cells was based on Huitema et al. (53) and is described in detail in SI Materials and Methods.
Cell Culture and Ectonucleotidase Inhibition.
HEK293 and Flp-In T-REx 293 cells were cultured in DMEM (Invitrogen) supplemented with 10% FCS and 100 U/mL pen/strep at 37 °C and 5% CO2 under humidifying conditions. HEK293 cells were seeded in six-well plates (Costar; Corning) in triplicate at a density of 7.5 × 105 cells per well and allowed to grow to confluence overnight. At this point, the medium was replaced with 2 mL of fresh medium with or without 1 mM ARL67156 (Tocris) plus 0.5 mM AMP-PCP (Sigma-Aldrich). Samples (200 µL) were collected after 0, 1, 2, 4, 8, and 24 h and frozen at −20 °C until further processing. Flp-In T-REx 293 cells were seeded in 24-well plates (2 × 105 cells/well) and grown to confluence overnight. Expression was induced by adding 1 µg/mL of doxycyclin. Medium (0.5 mL) was collected for PPi determination, and replaced with fresh medium daily for 4 d.
Plasma Collection.
Whole blood was collected in syringes containing 7.5 IU of heparin by cardiac puncture and kept on ice until separation of plasma and erythrocytes by centrifugation (1,000 × g at 4 °C for 20 min). The plasma layer was collected, depleted of platelets by filtration (2,200 × g at 4 °C for 20 min) through a Centrisart I 300,000-kDa mass cutoff filter (Sartorius), and stored at −20 °C until further processing.
Untargeted Metabolomics.
Untargeted metabolomics was performed using ion-pairing liquid chromatography coupled to an LTQ-Orbitrap Discovery (Thermo Fisher Scientific) operated in the negative ionization mode, as detailed in SI Materials and Methods.
Pyrophosphate Assay.
Pyrophosphate was determined enzymatically as described by O’Neill et al. (47), modified for a 96-well format. First, a blank was prepared for each sample by incubating 20 µL of sample with 0.175 U pyrophosphorylase (from baker’s yeast; Sigma-Aldrich) for 1 h at 37 °C. To these blanks and 20 µL of the untreated sample were added 100 µL of prewarmed assay mix [20 µM NADP, 8 µM UDP glucose, 0.5 U/mL UDP pyrophoshorylase, 3.7 U/mL phosphoglucomutase, 0.1 U/mL glucose-6-phosphate dehydrogenase (all from Sigma-Aldrich), and 0.003 µCi/mL [14C]UDP glucose (ARC Radiochemicals) in 70 mM Tris⋅HCl (pH 7.6), 90 mM KCl, 5 mM MgCl2, and 100 µM AMP (Sigma-Aldrich)]. After incubation for 30 min at 37 °C, the 96-well plate was cooled on ice, and 200 µL of 3% activated charcoal (Sigma-Aldrich) in water was added. The plate was left on ice for 15 min and shaken every 5 min. Finally, a 250-µL sample was filtered over a Millipore HTS Hi-Flow FB 96-well filter plate, and 200 µL of the filtrate was analyzed by liquid scintillation counting using Ultima Gold liquid scintillation mixture (PerkinElmer). The PPi-related radioactivity was calculated by subtracting the blanks from the untreated samples. A calibration curve ranging from 0 to 20 µM PPi was constructed in DMEM or PPi-free human plasma on each 96-well plate.
Vesicular Uptake Assays.
Membrane vesicles were prepared from HEK293/GFP and HEK293/rABCC6 cells as described preciously (54). Vesicular uptake was assessed using a rapid filtration technique (55), as described in detail in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Leonie Huitema, Bruno Stieger and András Váradi for providing reagents and helpful discussions, and Alfred Schinkel for a critical reading of the manuscript. This work was funded in part by PXE international.
Footnotes
The authors declare no conflict of interest.
Data deposition: Untargeted metabolomics data are available through MetaboLights, www.ebi.ac.uk/metabolights (accession no. MTBLS61) and XCMS Online, https://xcmsonline.scripps.edu/jobs.php?action=listpublicshares (ID 1009444–1009446).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1319582110/-/DCSupplemental.
References
- 1.Neldner KH. Pseudoxanthoma elasticum. Clin Dermatol. 1988;6(1):1–159. doi: 10.1016/0738-081x(88)90003-x. [DOI] [PubMed] [Google Scholar]
- 2.Uitto J, Váradi A, Bercovitch L, Terry PF, Terry SF. Pseudoxanthoma elasticum: Progress in research toward treatment. Summary of the 2012 PXE international research meeting. J Invest Dermatol. 2013;133(6):1444–1449. doi: 10.1038/jid.2013.20. [DOI] [PubMed] [Google Scholar]
- 3.Uitto J, Li Q, Jiang Q. Pseudoxanthoma elasticum: Molecular genetics and putative pathomechanisms. J Invest Dermatol. 2010;130(3):661–670. doi: 10.1038/jid.2009.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Le Saux O, et al. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat Genet. 2000;25(2):223–227. doi: 10.1038/76102. [DOI] [PubMed] [Google Scholar]
- 5.Bergen AA, et al. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat Genet. 2000;25(2):228–231. doi: 10.1038/76109. [DOI] [PubMed] [Google Scholar]
- 6.Ringpfeil F, Lebwohl MG, Christiano AM, Uitto J. Pseudoxanthoma elasticum: Mutations in the MRP6 gene encoding a transmembrane ATP-binding cassette (ABC) transporter. Proc Natl Acad Sci USA. 2000;97(11):6001–6006. doi: 10.1073/pnas.100041297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Struk B, et al. Mutations of the gene encoding the transmembrane transporter protein ABC-C6 cause pseudoxanthoma elasticum. J Mol Med (Berl) 2000;78(5):282–286. doi: 10.1007/s001090000114. [DOI] [PubMed] [Google Scholar]
- 8.Borst P, Evers R, Kool M, Wijnholds J. A family of drug transporters: Tthe multidrug resistance-associated proteins. J Natl Cancer Inst. 2000;92(16):1295–1302. doi: 10.1093/jnci/92.16.1295. [DOI] [PubMed] [Google Scholar]
- 9.Kool M, van der Linden M, de Haas M, Baas F, Borst P. Expression of human MRP6, a homologue of the multidrug resistance protein gene MRP1, in tissues and cancer cells. Cancer Res. 1999;59(1):175–182. [PubMed] [Google Scholar]
- 10.Madon J, Hagenbuch B, Landmann L, Meier PJ, Stieger B. Transport function and hepatocellular localization of mrp6 in rat liver. Mol Pharmacol. 2000;57(3):634–641. doi: 10.1124/mol.57.3.634. [DOI] [PubMed] [Google Scholar]
- 11.Scheffer GL, et al. MRP6 (ABCC6) detection in normal human tissues and tumors. Lab Invest. 2002;82(4):515–518. doi: 10.1038/labinvest.3780444. [DOI] [PubMed] [Google Scholar]
- 12.Le Saux O, et al. Expression and in vivo rescue of human ABCC6 disease-causing mutants in mouse liver. PLoS ONE. 2011;6(9):e24738. doi: 10.1371/journal.pone.0024738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gorgels TG, et al. Abcc6 deficiency in the mouse leads to calcification of collagen fibers in Bruch’s membrane. Exp Eye Res. 2012;104:59–64. doi: 10.1016/j.exer.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 14.Jiang Q, Li Q, Uitto J. Aberrant mineralization of connective tissues in a mouse model of pseudoxanthoma elasticum: Systemic and local regulatory factors. J Invest Dermatol. 2007;127(6):1392–1402. doi: 10.1038/sj.jid.5700729. [DOI] [PubMed] [Google Scholar]
- 15.Klement JF, et al. Targeted ablation of the abcc6 gene results in ectopic mineralization of connective tissues. Mol Cell Biol. 2005;25(18):8299–8310. doi: 10.1128/MCB.25.18.8299-8310.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gorgels TG, et al. Disruption of Abcc6 in the mouse: Novel insight in the pathogenesis of pseudoxanthoma elasticum. Hum Mol Genet. 2005;14(13):1763–1773. doi: 10.1093/hmg/ddi183. [DOI] [PubMed] [Google Scholar]
- 17.Jiang Q, Endo M, Dibra F, Wang K, Uitto J. Pseudoxanthoma elasticum is a metabolic disease. J Invest Dermatol. 2009;129(2):348–354. doi: 10.1038/jid.2008.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang Q, et al. Parabiotic heterogenetic pairing of Abcc6−/−/Rag1−/− mice and their wild-type counterparts halts ectopic mineralization in a murine model of pseudoxanthoma elasticum. Am J Pathol. 2010;176(4):1855–1862. doi: 10.2353/ajpath.2010.090983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Belinsky MG, Chen ZS, Shchaveleva I, Zeng H, Kruh GD. Characterization of the drug resistance and transport properties of multidrug resistance protein 6 (MRP6, ABCC6) Cancer Res. 2002;62(21):6172–6177. [PubMed] [Google Scholar]
- 20.Iliás A, et al. Loss of ATP-dependent transport activity in pseudoxanthoma elasticum-associated mutants of human ABCC6 (MRP6) J Biol Chem. 2002;277(19):16860–16867. doi: 10.1074/jbc.M110918200. [DOI] [PubMed] [Google Scholar]
- 21.Borst P, van de Wetering K, Schlingemann R. Does the absence of ABCC6 (multidrug resistance protein 6) in patients with pseudoxanthoma elasticum prevent the liver from providing sufficient vitamin K to the periphery? Cell Cycle. 2008;7(11):1575–1579. doi: 10.4161/cc.7.11.6005. [DOI] [PubMed] [Google Scholar]
- 22.Markello TC, et al. Vascular pathology of medial arterial calcifications in NT5E deficiency: Implications for the role of adenosine in pseudoxanthoma elasticum. Mol Genet Metab. 2011;103(1):44–50. doi: 10.1016/j.ymgme.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fülöp K, et al. ABCC6 does not transport vitamin K3-glutathione conjugate from the liver: Relevance to pathomechanisms of pseudoxanthoma elasticum. Biochem Biophys Res Commun. 2011;415(3):468–471. doi: 10.1016/j.bbrc.2011.10.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szabó Z, Váradi A, Li Q, Uitto J. ABCC6 does not transport adenosine: Relevance to pathomechanism of pseudoxanthoma elasticum. Mol Genet Metab. 2011;104(3):421. doi: 10.1016/j.ymgme.2011.07.013. author reply 422. [DOI] [PubMed] [Google Scholar]
- 25.Tautenhahn R, Patti GJ, Rinehart D, Siuzdak G. XCMS Online: A Web-based platform to process untargeted metabolomic data. Anal Chem. 2012;84(11):5035–5039. doi: 10.1021/ac300698c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haug K, et al. MetaboLights—an open-access general-purpose repository for metabolomics studies and associated meta-data. Nucleic Acids Res. 2013;41(Database issue):D781–D786. doi: 10.1093/nar/gks1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stefan C, Jansen S, Bollen M. NPP-type ectophosphodiesterases: Unity in diversity. Trends Biochem Sci. 2005;30(10):542–550. doi: 10.1016/j.tibs.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 28.Lomashvili KA, Cobbs S, Hennigar RA, Hardcastle KI, O’Neill WC. Phosphate-induced vascular calcification: Role of pyrophosphate and osteopontin. J Am Soc Nephrol. 2004;15(6):1392–1401. doi: 10.1097/01.asn.0000128955.83129.9c. [DOI] [PubMed] [Google Scholar]
- 29.Silcox DC, Jacobelli S, McCarty DJ. Identification of inorganic pyrophosphate in human platelets and its release on stimulation with thrombin. J Clin Invest. 1973;52(7):1595–1600. doi: 10.1172/JCI107336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lazarowski ER. Vesicular and conductive mechanisms of nucleotide release. Purinergic Signal. 2012;8(3):359–373. doi: 10.1007/s11302-012-9304-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roman RM, et al. Hepatocellular ATP-binding cassette protein expression enhances ATP release and autocrine regulation of cell volume. J Biol Chem. 1997;272(35):21970–21976. doi: 10.1074/jbc.272.35.21970. [DOI] [PubMed] [Google Scholar]
- 32.Reisin IL, et al. The cystic fibrosis transmembrane conductance regulator is a dual ATP and chloride channel. J Biol Chem. 1994;269(32):20584–20591. [PubMed] [Google Scholar]
- 33.Rutsch F, et al. Mutations in ENPP1 are associated with “idiopathic” infantile arterial calcification. Nat Genet. 2003;34(4):379–381. doi: 10.1038/ng1221. [DOI] [PubMed] [Google Scholar]
- 34.Le Boulanger G, et al. An unusual severe vascular case of pseudoxanthoma elasticum presenting as generalized arterial calcification of infancy. Am J Med Genet A. 2010;152A(1):118–123. doi: 10.1002/ajmg.a.33162. [DOI] [PubMed] [Google Scholar]
- 35.Nitschke Y, et al. Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am J Hum Genet. 2012;90(1):25–39. doi: 10.1016/j.ajhg.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.St Hilaire C, et al. NT5E mutations and arterial calcifications. N Engl J Med. 2011;364(5):432–442. doi: 10.1056/NEJMoa0912923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rutsch F, Nitschke Y, Terkeltaub R. Genetics in arterial calcification: Pieces of a puzzle and cogs in a wheel. Circ Res. 2011;109(5):578–592. doi: 10.1161/CIRCRESAHA.111.247965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nitschke Y, Rutsch F. Genetics in arterial calcification: Lessons learned from rare diseases. Trends Cardiovasc Med. 2012;22(6):145–149. doi: 10.1016/j.tcm.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 39.Harmey D, et al. Concerted regulation of inorganic pyrophosphate and osteopontin by akp2, enpp1, and ank: An integrated model of the pathogenesis of mineralization disorders. Am J Pathol. 2004;164(4):1199–1209. doi: 10.1016/S0002-9440(10)63208-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kupetsky-Rincon EA, Li Q, Uitto J. Magnesium reduces carotid intima-media thickness in a mouse model of pseudoxanthoma elasticum: A novel treatment biomarker. Clin Transl Sci. 2012;5(3):259–264. doi: 10.1111/j.1752-8062.2011.00390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Granda JL, Falvo KA, Bullough PG. Pyrophosphate levels and magnesium oxide therapy in osteogenesis imperfecta. Clin Orthop Relat Res. 1977;(126):228–231. [PubMed] [Google Scholar]
- 42.Hendig D, et al. Role of serum fetuin-A, a major inhibitor of systemic calcification, in pseudoxanthoma elasticum. Clin Chem. 2006;52(2):227–234. doi: 10.1373/clinchem.2005.059253. [DOI] [PubMed] [Google Scholar]
- 43.Price PA, Williamson MK, Nguyen TM, Than TN. Serum levels of the fetuin-mineral complex correlate with artery calcification in the rat. J Biol Chem. 2004;279(3):1594–1600. doi: 10.1074/jbc.M305199200. [DOI] [PubMed] [Google Scholar]
- 44.Boraldi F, et al. Matrix gla protein and alkaline phosphatase are differently modulated in human dermal fibroblasts from PXE patients and controls. J Invest Dermatol. 2013;133(4):946–954. doi: 10.1038/jid.2012.460. [DOI] [PubMed] [Google Scholar]
- 45.Li Q, Jiang Q, Schurgers LJ, Uitto J. Pseudoxanthoma elasticum: Reduced gamma-glutamyl carboxylation of matrix gla protein in a mouse model (Abcc6−/−) Biochem Biophys Res Commun. 2007;364(2):208–213. doi: 10.1016/j.bbrc.2007.09.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O’Young J, et al. Matrix Gla protein inhibits ectopic calcification by a direct interaction with hydroxyapatite crystals. J Am Chem Soc. 2011;133(45):18406–18412. doi: 10.1021/ja207628k. [DOI] [PubMed] [Google Scholar]
- 47.O’Neill WC, Lomashvili KA, Malluche HH, Faugere MC, Riser BL. Treatment with pyrophosphate inhibits uremic vascular calcification. Kidney Int. 2011;79(5):512–517. doi: 10.1038/ki.2010.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Riser BL, et al. Daily peritoneal administration of sodium pyrophosphate in a dialysis solution prevents the development of vascular calcification in a mouse model of uraemia. Nephrol Dial Transplant. 2011;26(10):3349–3357. doi: 10.1093/ndt/gfr039. [DOI] [PubMed] [Google Scholar]
- 49.Glatz AC, Pawel BR, Hsu DT, Weinberg P, Chrisant MR. Idiopathic infantile arterial calcification: Two case reports, a review of the literature and a role for cardiac transplantation. Pediatr Transplant. 2006;10(2):225–233. doi: 10.1111/j.1399-3046.2005.00414.x. [DOI] [PubMed] [Google Scholar]
- 50.Rutsch F, et al. GACI Study Group Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy. Circ Cardiovasc Genet. 2008;1(2):133–140. doi: 10.1161/CIRCGENETICS.108.797704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Q, Schumacher W, Jablonski D, Siegel D, Uitto J. Cutaneous features of pseudoxanthoma elasticum in a patient with generalized arterial calcification of infancy due to a homozygous missense mutation in the ENPP1 gene. Br J Dermatol. 2012;166(5):1107–1111. doi: 10.1111/j.1365-2133.2012.10811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gorgels TG, et al. Vitamin K supplementation increases vitamin K tissue levels but fails to counteract ectopic calcification in a mouse model for pseudoxanthoma elasticum. J Mol Med (Berl) 2011;89(11):1125–1135. doi: 10.1007/s00109-011-0782-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huitema LF, et al. The nitric oxide donor sodium nitroprusside inhibits mineralization in ATDC5 cells. Calcif Tissue Int. 2006;78(3):171–177. doi: 10.1007/s00223-005-1233-y. [DOI] [PubMed] [Google Scholar]
- 54.Reid G, et al. Characterization of the transport of nucleoside analog drugs by the human multidrug resistance proteins MRP4 and MRP5. Mol Pharmacol. 2003;63(5):1094–1103. doi: 10.1124/mol.63.5.1094. [DOI] [PubMed] [Google Scholar]
- 55.van de Wetering K, et al. Multidrug resistance proteins 2 and 3 provide alternative routes for hepatic excretion of morphine-glucuronides. Mol Pharmacol. 2007;72(2):387–394. doi: 10.1124/mol.107.035592. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.