

Introduction
Reviews of information about AMD, cataract, and glaucoma make it apparent that while each eye tissue has its own characteristic metabolism, structure, and function, there are common perturbations to homeostasis that are associated with age-related dysfunction. The commonalities appeared to be biochemical stresses and their sequelae. Recognition of shared etiologic factors for age-related debilities allows rationalization of comparable risk factor-disease incidence relationships—such as nutritional risk factors for AMD and cataract (as well as cardiovascular disease and diabetes)—and informs about potential new therapeutic avenues, such as stress reducers (i.e., antioxidants) and/or proteolysis enhancers. It also maximizes the return on the investment in research effort and costs. For example, drugs or nutrients that protect against AMD may also prove effective against cataract, glaucoma, or/and other age-related neurodegenerative debilities.
This article summarizes cell biologic and biochemical changes in aging and age-related diseases of the eye. Clearly, this is a larger challenge with a richer literature than can be properly treated in a short review such as this. In this short review, we focus on age-related stresses and current and anticipated means to diminish the stress. Recognizing that almost all age-related diseases such as Alzheimer and Parkinson diseases, cataract, AMD, glaucoma, diabetes, and the premature aging diseases such as progeria, have in common the accumulation of damaged proteins, we select three aspects of age-related biochemical changes that are common to most eye tissues: oxidative stresses; problems associated with and/or due to damaged proteins that accumulate in the retina, lens, and cornea; and intracellular degradative capacities that usually keep levels of damaged proteins in check in early life or when tissues are not stressed, but that may fail upon stress or aging (Figs. 1, 2). We offer apologies to investigators whose work we do not cite or can acknowledge only via reviews.1
Figure 1.
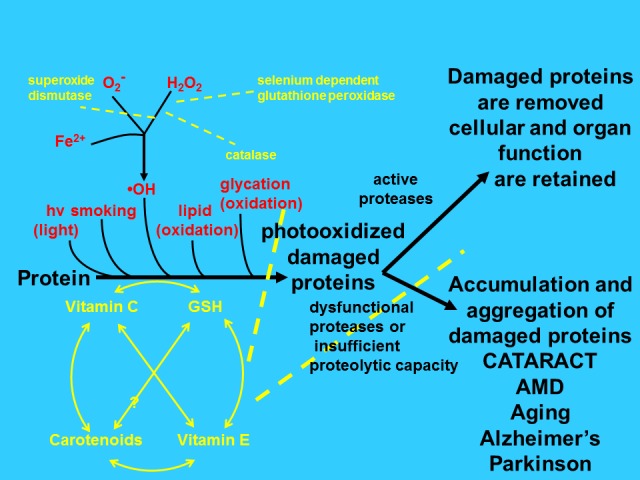
Scheme of proposed relationship between chronic stress, protective capacities, proteolytic editing machinery and age-related disease. When young, proteins are intact and cell and tissue functions are retained. Upon exposure to various stresses (red) including consuming higher glycemic index diets the proteins are damaged and become dysfunctional and cytotoxic. Adequate levels of antioxidants and antioxidant enzymes (yellow) ameliorate these stresses (dashed lines) and preserve proteolytic function. If sufficient proteolytic capacity exists, damaged proteins can be removed and function retained (upper right). If, however, the proteolytic machines (ubiquitin proteolytic pathway and autophagic lysosomal pathways) are rendered insufficient, proteins will aggregate and cytotoxicity will ensue, resulting in compromised tissue function, including cataracts, AMD, and glaucoma (lower right).
Figure 2.
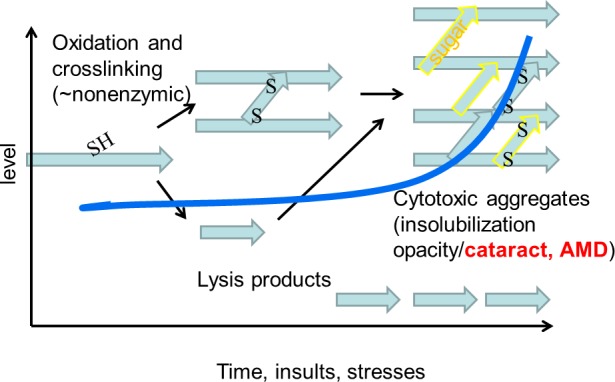
Upon aging, proteins are damaged. This damage includes various modifications such as oxidations, reaction with other moieties such as sugar derivatives, cross linking (yellow), and lysis. These processes accelerate upon aging (follow blue line) and parallel rates of development of age-related diseases. The aggregates result in cataracts in the lens and in AMD-related deposits in aging retinal RPE.
The most rapidly growing segment of many societies is the elderly. The prevalence of cataract, AMD, and glaucoma accelerates with age. Among those who are aged 75 years or older, prevalence rates of cataract, AMD, and glaucoma are approximately 60%, 15%, and 20% of the population, respectively. These estimates almost double for people aged just 10 years older. Like most tissues in general, most eye tissues suffer from the accumulation of damaged proteins. Such accumulation appears to involve post-synthetic modifications to proteins and limits on the proteolytic capacities that are normally available to degrade and remove the altered or obsolete proteins before they transform into cytotoxic aggregates. Collectively, we call the sum of synthesis, post-synthetic modification, editing and removal of proteins “proteopoise.” Compromises to proteopoise are also thought to be etiologic for many age-related neuropathies and premature aging syndromes.1–7 Herein, we work our way from the anterior of the eye, or cornea, through to the lens and on to the posterior segment or retina, recalling common themes of age-related changes and protein quality control.
Age-Related Changes in the Cornea, Lens, and Retina
The cornea is a multilayered tissue containing three distinct cellular layers, epithelium, stroma, and endothelium, and two membrane structures: Bowman's layer, separating the epithelium and stroma; and Descemet's membrane, separating the stroma from the endothelium. The major functions of the cornea are to protect the rest of the eye from environmental insults and to refract light.
Structural and biochemical changes have been noted in all layers of the cornea upon aging. The corneal epithelium becomes more permeable with age,8 possibly due to alterations in the distribution of α6 and β4 integrins, transmembrane receptors that mediate the attachment between a cell and its surroundings.9 Age-related alterations in the human (diurnal) cornea appear to involve cumulative, prolonged ultraviolet radiation exposure as well as stresses that are associated with aging per se. This leads to the generation of reactive oxygen species that, in turn, cause oxidative stress. Accordingly, it is not surprising that protein oxidation is a frequent insult to the cornea.10 This involves advanced glycation–end products (AGEs) that form due to a nonenzymatic reaction between proteins and aldehydes and ketones, most of which are derived from sugars. Levels of AGEs increase upon aging in corneal collagen, lens, and probably all eye tissues11 and may be further increased by diabetes or due to consuming high-glycemic index diets.6,12–14 AGE-modified collagen may contribute to the increase in collagen fibrils and decreased corneal flexibility observed upon aging.15,16 Age-related thickening of Bowman's layer and Descemet's membrane also involves post-synthetic modification.17,18
Additional evidence of age-related oxidative damage derives from analyses of genetic material. Genomic and mitochondrial DNA are damaged with age and corneas from older donors show increased 8-OHdG, a marker of DNA oxidation, a consequence—at least in part—of the age-related compromise in DNA damage repair capacity.19,20
Ascorbate and glutathione are important nonenzymatic antioxidants in the cornea. Ascorbate levels are significantly higher in the cornea than in the serum or aqueous humor.21,22 Surprisingly, there is a trend toward increased ascorbate, glutathione peroxidase, and the antioxidant cytoglobin in aged human corneas.21 However, there was a decrease in both mRNA and protein expression of superoxide dismutase-1 and γ-glutamyl–transpeptidase activity.19,23 Similarly, in aged rabbit corneas, the levels of glutathione peroxidase, superoxide dismutase, and catalase were significantly decreased.22 Because these antioxidant enzymes provide critical protection against oxidative stress, age-related losses in their activity would confer enhanced susceptibility to stress.1,24–26
With mounting stress, it is not surprising that damaged proteins and other potentially harmful moieties also accumulate. Mutations also cause accumulation of abnormal proteins, many of which have been etiologically associated with disease. An example is optineurin. Although incompletely characterized at present, optineurin appears to have roles in apoptosis, inflammation, vasoconstriction, morphogenesis, membrane, and vesicle trafficking, as well as in transcription activation. Mutations in optineurin have been related to risk for glaucoma, and are also found in inclusions in patients with amyotrophic lateral sclerosis (ALS).27,28 Such accumulation of altered proteins are thought to be exacerbated by insufficient proteolytic or other degradative capacity and etiologically related to many premature age-related diseases including Parkinson, Alzheimer, and ALS.2,4,5,29,30
Corneal endothelial cells show increased expression of p21cip1 and p16INK4a31,32 and senescence-associated beta galactosidase33 during aging, consistent with diminished proliferative capacity and cell density with age.34 Since p21cip1 is a substrate of the ubiquitin proteasome proteolytic pathway, it is possible that intracellular proteolytic capacities are compromised upon aging (see Age-Related Changes In Proteolytic Capacities During Aging In Cornea, Lens And Retina section).
Light coming from the cornea must pass through the fiber cells of the lens nucleus en route to the retina. Of all the tissues in the eye, it is probably easiest to recognize deficits in proteopoise in the lens. Fiber cells are functionally analogous to a fiber optic. When young, they are filled with a clear solution of native proteins. The lens is also equipped with very high levels of glutathione, ascorbate, and antioxidant enzymes.1 However, upon aging and stress, these levels decline and the antioxidant enzymes are rendered less active. Consequently, proteins are gradually modified, often by oxidation, deamidations, racemizations, and they lyse or aggregate and precipitate.1–3,6,7,24–26 In recent years, there has been increased interest in the damage that is caused by elevated levels of dietary sugars, or AGEs because elevations in intake of carbohydrates has recently been related to enhanced risk for cataract, AMD, cardiovascular disease, and diabetes.12,35 The rates of accumulation of many of these post-synthetic alterations appear to accelerate upon aging (Figs. 1, 2).
The retina is composed of a myriad of cell types. They can be very roughly divided into neural retina, RPE, and the choroidal vessels that feed the rear of the retina. The choroidal vessels at the rear of the eye supply nutrients and oxygen to the outer layer of the retina, and actively transport waste away from the retina. Clearly, they are essential for maintaining retinal health, but upon aging, they are partially lost in humans.36,37 In contrast with thinning in humans, in mice there is evidence of increased choroidal thickness upon aging.38 Retinal pigmented epithelial cells and photoreceptors are also lost, particularly in AMD.
Multiple studies have identified oxidative stress as an etiologic factor in AMD.39–41 The retina a fertile environment for oxidative stress. This is due to the presence of two blood supplies, the highly oxygenated environment, along with the presence of high levels of photosensitizers and readily oxidizable lipid, protein and carbohydrate substrates. This is exacerbated by a huge proteolytic burden, particularly in the RPE, due to the requirement to degrade the tips of photoreceptor outer segments that are shed nightly. Oxidative stress is indicated by the contents of basal laminar deposits and drusen that herald the onset of AMD and by the marked increase in risk for AMD in smokers. Additionally, Handa and colleagues found evidence of AGE modification of choroidal proteins in an aged donor, who exhibited no age related eye disease,42,43 and this was expanded by observations of elevated levels of AGEs and harbingers of AMD in older animals that consumed higher glycemic index diets.44 Importantly, we find a systemic burden indicated by the higher levels of AGEs throughout the eye and many bodily tissues of mice that consumed higher GI diets.12 Emphasizing that this is a diet GI-AMD risk relationship, there is increased risk for each category of AMD in people who consume the highest GI diets.45 Since people who consume higher GI diets are at increased risk of AMD, as well as cardiovascular disease and diabetes, it is likely that the accumulation of AGEs and disease are mechanistically linked and that treatments for one malady may bear benefits for other etiologically linked debilities.46 Importantly, findings from the Age-Related Eye Diseases Study 2 indicate that intake of elevated levels of vitamins C and E as well as zinc and lutein confer some protection against progress of intermediate to advanced AMD.47 Clearly, it would be of greatest interest to find the means to avoid onset of AMD.
Genetic variations in complement factor H confer major risk for AMD, but the mechanism is unknown. The hypothesis that oxidative stress or its sequelae are involved in risk for AMD was corroborated by Weismann's recent observation that mutant complement factor H cannot detoxify lipid oxidation products as effectively as the normal protein.48 This is the first mechanistic link between robust epidemiologic associations regarding risk for AMD and the multiple studies that have associated genetic mutations, particularly genes that regulate immune and inflammatory responses, with risk for AMD.49,50 The relationship between inflammation and risk for AMD is also supported by observations of increased numbers of macrophages in the choroid of aged mice,38 as well as increased levels of prostaglandin, PGE2, and its receptor PGE2-EP2 in the choroid of aged rats.51 The age-altered cytokine profiles and macrophage responses to retinal laser insult52 inform about new targets for therapy.
Age-Related Changes in Proteolytic Capacities During Aging in the Cornea, Lens, and Retina
In addition to post-synthetic modifications, damaged or obsolete proteins may accumulate because they are not recognized as obsolete, or because of insufficient proteolytic capacity.2,3,12,30 Such proteins may aggregate, as is observed in cataract, or be otherwise cytotoxic. Recognition of damaged proteins in the cytoplasm is usually accomplished by ubiquitination. In the ubiquitin pathway, ubiquitin is attached to substrates by the sequential activities of E1 (ubiquitin-activating enzymes); E2s (ubiquitin-conjugating enzymes); and E3s (ubiquitin ligases). The exquisite selectivity of ubiquitin conjugation is achieved by the combinatorial activities of dozens of E2s together with hundreds of E3s.53 Ubiquitinated proteins are delivered to the proteasome where the ubiquitin is removed and recycled and the substrate is degraded to peptides that are eventually reduced to amino acids by aminopeptidases.54 Selectivity is also achieved by the many deubiquitinating enzymes that, by regulating the extent of ubiquitination, control access of the ubiquitinated substrate to the proteasome. An important observation regarding links between proteolytic capacities and oxidative status derives from observation that all the conjugating enzymes of the UPS are sulfhydryl enzymes and that the GSH/GSSG ratio controls their activities.55
A parallel—if less selective—pathway involves autophagy. In this pathway, parts of the cytoplasm are engulfed by a membrane and delivered to the lysosome for degradation. Multiple recent papers document that the UPS and autophagic pathways work in concert.12,56 Calpains also complement the UPS and autophagosomal pathways and recent discoveries indicate that these pathways may be linked (Liu and Taylor, unpublished observations, 2013).
What are the proteolytic capacities in cornea, lens, and retina and what is their fate and functional status during aging and upon stress? In the cornea, the larger literature to date deals with extracellular proteases and collagenolysis.57 Evidence regarding additional proteolytic capacities is being developed, in part, via high-throughput analytical methods.58
Since protein aggregation and precipitation is clearly related to cataract, much effort has been devoted to documenting proteolytic capacities in the lens and their fate upon oxidative stress and aging. While lenses contain all of the ubiquitin, lysosomal, and calpain machineries in the young nucleated cells, by virtue of the lens fibers degrading their organelles, any opportunity for renewal and much of the autophagic lysosomal proteolytic capacity is lost during differentiation and aging. There are also some species differences, such as in the complement of calpains in lenses from different species. Ubiquitin-conjugating activity decreases in old rat lens as compared with younger rat lens. The conjugating activity is also decreased from the outer cortex (newer fiber cells) to the lens nucleus (oldest fiber cells).59–61 In the human lens, the three peptidase activities of the proteasome did not change with aging, but they are found at decreased levels in cataractous lenses, consistent with a requirement for these activities to rid lens cells of damaged proteins.62 As noted above, even when the cells have the full complement of ubiquitination enzymes, increased oxidative stress can diminish their efficacy (Fig. 1). Similarly, oxidative modifications to substrates may alter access to these systems. Thus, it has been demonstrated that while mild oxidative stress or deamidation makes some substrates better proteasomal substrates, glycation has the reverse effect.12,63 Aminopeptidase activities also decline with age and certainly in older developmental zones of the same lens.54
It is probable that the lens neutral proteinase first described by Van Heyningen and later studied by Wagner, and which was found at reduced activity in the older, central regions of the lens, is the proteasome.64 An age-related decline in functional proteasome was also reported in brains, mainly due to associated decreased assembly of 26S proteasome.65 Interestingly, overexpression of the proteasome subunit Rpn11 can prevent the decline of proteasome activity and prevent accumulation of neurodegeneration-related polyQ. Rpn6 also appears to play roles in 20S and 26S proteasome assembly66 and ectopic expression of Rpn6 is sufficient to increase proteasomal assembly and activity.67 Thus, modulation of proteasome subunits may be a viable approach to enhancing proteasomal activity lost during aging.
In the neural retina, the chymotrypsin-like activity of the proteasome decreases with aging, but this decrease was not observed in RPE.68,69 Our unpublished results indicate that the chymotrypsin-like activity decreases both in retina and RPE upon aging.70
Surprisingly, there is no report on age-related changes in calpain and autophagy in the retina or lens. However, experimental impairment of the ubiquitin proteasome pathway or autophagy causes cataract,71,72 indicating that the ubiquitin proteasome system (UPS) and autophagy play roles in maintaining lens transparency. Both pathways are engaged in the removal of AGEs.12
Furthermore, UPS and calpain activity are linked by virtue of the relationship between UPS-dependent regulation of connexins that regulate calcium influx. Importantly, calpain activity and its cleavage products increase in cataract and cause ER stress that, in turn, increases calcium release and calpain activation in lens epithelial cells.73 The finding of increased levels of calpain-cleaved spectrin in patients with neuronal diseases could be a biomarker of such diseases.74 Calpain activity also increases in the brain with some age-related diseases. In partial corroboration of these findings, systemic inhibition of calpain appears to prevent Alzheimer related lesions in a mouse model of the disease.75,76
Chaperones clearly interact with the cellular proteolytic machines. There appears to be an age-dependent decline in chaperone-mediated autophagy, at least in liver. This seems to be due to a decrease in the lysosomal receptor Lamp-2A.77 Importantly, restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function,78 including the removal of modified proteins. Evidence for salutary effects of chaperones is accumulating in eye tissues.79,80
Unmet Needs and Opportunities
Taken together, it is clear that a vast array of antioxidant and proteolytic capacities maintain homeostasis, when they are functional and unimpaired. However, upon aging or stress, these capacities are inactivated and become insufficient. Thus, there ensues a vicious cycle of stress, accumulation of damaged proteins, and diminished proteolytic capacity that leads to accelerated accumulation of cytotoxic materials, and disease. We know that adequate nutrition is crucial for maintenance of eye function. Accordingly, massive educational campaigns should be instituted that relate risk for blindness to nutrition, smoking, obesity and exercise. These are bound to bear positive results. We could eliminate much age-related disease if salutary behavior changes or treatment could be implemented earlier. To encourage this, we need to determine biomarkers that will allow us to anticipate incipient disease. Current technologies make this an achievable objective. Education about healthy lifestyles and eating habits is probably the least costly and most effective investment with regard to achieving prolonged eye function. It has recently been demonstrated that electron recycling agents can delay mitochondrial disease. Since the age-related debilities noted here all have origins in oxidative stress, it is likely that such agents can also delay formation of the offending moieties and/or preserve function of the degradative machines (UPS, autophagic/lysosomal) that remove oxidized products. Recent findings that degradation of neurodegeneration-related proteins can be enhanced should be translated to therapy for other age-related protein precipitation diseases including cataract, AMD, and possibly glaucoma. New and more potent antioxidants can be discovered or constructed for delivery to eyes to enhance protection against oxidative stress. Elucidating components of age-related vision loss that are not due to retina or lens damage will provide needed understanding and open up new alternatives for vision restoration for people with neurologic damage.
Given the promise for success and the low cost of this research relative to the gains in sight maintenance from its successful completion, it is clear that additional funding would be a wise investment.
Acknowledgments
Supported by USDA 1950-510000-060-01A, Johnson and Johnson Focused Giving, and a gift from Alcon via John Lang. The work was also supported by EY RO1 13250 and EY RO1 21212. The authors alone are responsible for the content and writing of the paper.
Disclosure: E.A. Whitcomb, None; F. Shang, None; A. Taylor, None
References
- 1. Taylor A. Nutritional and Environmental Influences on the Eye. Boca Raton, FL: CRC Press; 1999. [Google Scholar]
- 2. Shang F, Taylor A. Role of the ubiquitin-proteasome in protein quality control and signaling: implication in the pathogenesis of eye diseases. Prog Mol Biol Transl Sci. 2012; 109: 347–396 [DOI] [PubMed] [Google Scholar]
- 3. Shang F, Taylor A. Roles for the ubiquitin-proteasome pathway in protein quality control and signaling in the retina: implications in the pathogenesis of age-related macular degeneration. Mol Aspects Med. 2012; 33: 446–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006; 313: 1604–1610 [DOI] [PubMed] [Google Scholar]
- 5. Cohen E, Dillin A. The insulin paradox: aging, proteotoxicity and neurodegeneration. Nat Rev Neurosci. 2008; 9: 759–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tessier F, Obrenovich M, Monnier VM. Structure and mechanism of formation of human lens fluorophore LM-1. Relationship to vesperlysine A and the advanced Maillard reaction in aging, diabetes, and cataractogenesis. J Biol Chem. 1999; 274: 20796–20804 [DOI] [PubMed] [Google Scholar]
- 7. Monnier VM, Sell DR, Genuth S. Glycation products as markers and predictors of the progression of diabetic complications. Ann N Y Acad Sci. 2005; 1043: 567–581 [DOI] [PubMed] [Google Scholar]
- 8. Chang SW, Hu FR. Changes in corneal autofluorescence and corneal epithelial barrier function with aging. Cornea. 1993; 12: 493–499 [DOI] [PubMed] [Google Scholar]
- 9. Trinkaus-Randall V, Tong M, Thomas P, Cornell-Bell A. Confocal imaging of the alpha 6 and beta 4 integrin subunits in the human cornea with aging. Invest Ophthalmol Vis Sci. 1993; 34: 3103–3109 [PubMed] [Google Scholar]
- 10. Shoham A, Hadziahmetovic M, Dunaief JL, Mydlarski MB, Schipper HM. Oxidative stress in diseases of the human cornea. Free Radic Biol Med. 2008; 45: 1047–1055 [DOI] [PubMed] [Google Scholar]
- 11. Shamsi FA, Partal A, Sady C, Glomb MA, Nagaraj RH. Immunological evidence for methylglyoxal-derived modifications in vivo. Determination of antigenic epitopes. J Biol Chem. 1998; 273: 6928–6936 [DOI] [PubMed] [Google Scholar]
- 12. Uchiki T, Weikel KA, Jiao W, et al. Glycation-altered proteolysis as a pathobiologic mechanism that links dietary glycemic index, aging, and age-related disease (in non diabetics). Aging Cell. 2012; 11: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Obrenovich ME, Monnier VM. Glycation stimulates amyloid formation. Sci Aging Knowledge Environ. 2004; 2004: pe3 [DOI] [PubMed] [Google Scholar]
- 14. Zong H, Ward M, Stitt AW., AGEs RAGE. and diabetic retinopathy. Curr Diab Rep. 2011; 11: 244–252 [DOI] [PubMed] [Google Scholar]
- 15. Daxer A, Misof K, Grabner B, Ettl A, Fratzl P. Collagen fibrils in the human corneal stroma: structure and aging. Invest Ophthalmol Vis Sci. 1998; 39: 644–648 [PubMed] [Google Scholar]
- 16. Malik NS, Moss SJ, Ahmed N, Furth AJ, Wall RS, Meek KM. Ageing of the human corneal stroma: structural and biochemical changes. Biochim Biophys Acta. 1992; 20: 222–228 [DOI] [PubMed] [Google Scholar]
- 17. Alvarado J, Murphy C, Juster R. Age-related changes in the basement membrane of the human corneal epithelium. Invest Ophthalmol Vis Sci. 1983; 24: 1015–1028 [PubMed] [Google Scholar]
- 18. Johnson DH, Bourne WM, Campbell RJ. The ultrastructure of Descemet's membrane. I. Changes with age in normal corneas. Arch Ophthalmol. 1982; 100: 1942–1947 [DOI] [PubMed] [Google Scholar]
- 19. Joyce NC, Harris DL, Zhu CC. Age-related gene response of human corneal endothelium to oxidative stress and DNA damage. Invest Ophthalmol Vis Sci. 2011; 52: 1641–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Joyce NC, Zhu CC, Harris DL. Relationship among oxidative stress, DNA damage, and proliferative capacity in human corneal endothelium. Invest Ophthalmol Vis Sci. 2009; 50: 2116–2122 [DOI] [PubMed] [Google Scholar]
- 21. Brubaker RF, Bourne WM, Bachman LA, McLaren JW. Ascorbic acid content of human corneal epithelium. Invest Ophthalmol Vis Sci. 2000; 41: 1681–1683 [PubMed] [Google Scholar]
- 22. Cejkova J, Vejrazka M, Platenik J, Stipek S. Age-related changes in superoxide dismutase, glutathione peroxidase, catalase and xanthine oxidoreductase/xanthine oxidase activities in the rabbit cornea. Exp Gerontol. 2004; 39: 1537–1543 [DOI] [PubMed] [Google Scholar]
- 23. Redmond P, Burnham JM, Langford MP, Misra RP, Redens TB, Texada DE. Age-related decrease in human corneal gamma-glutamyltranspeptidase activity. Cornea. 2013; 32: e121–e126 [DOI] [PubMed] [Google Scholar]
- 24. Lou MF. Redox regulation in the lens. Prog Retin Eye Res. 2003; 22: 657–682 [DOI] [PubMed] [Google Scholar]
- 25. Yan H, Harding JJ, Xing K, Lou MF. Revival of glutathione reductase in human cataractous and clear lens extracts by thioredoxin and thioredoxin reductase, in conjunction with alpha-crystallin or thioltransferase. Curr Eye Res. 2007; 32: 455–463 [DOI] [PubMed] [Google Scholar]
- 26. Xing KY, Lou MF. Effect of age on the thioltransferase (glutaredoxin) and thioredoxin systems in the human lens. Invest Ophthalmol Vis Sci. 2010; 51: 6598–6604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Minegishi Y, Iejima D, Kobayashi H, et al. Enhanced optineurin E50K-TBK1 interaction evokes protein insolubility and initiates familial primary open-angle glaucoma. Hum Mol Genet. 2013; 21: 21 [DOI] [PubMed] [Google Scholar]
- 28. Sirohi K, Chalasani ML, Sudhakar C, Kumari A, Radha V, Swarup G. M98K-OPTN induces transferrin receptor degradation and RAB12-mediated autophagic death in retinal ganglion cells. Autophagy. 2013; 9: 510–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morimoto RI, Cuervo AM. Protein homeostasis and aging: taking care of proteins from the cradle to the grave. JGerontol A Biol Sci Med Sci. 2009; 64: 167–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001; 292; 1552–1555 [DOI] [PubMed] [Google Scholar]
- 31. Joyce NC. Cell cycle status in human corneal endothelium. Exp Eye Res. 2005; 81: 629–638 [DOI] [PubMed] [Google Scholar]
- 32. Wang Y, Zang X, Chen P. High expression of p16INK4a and low expression of Bmi1 are associated with endothelial cellular senescence in the human cornea. Mol Vis. 2012; 18: 803–815 [PMC free article] [PubMed] [Google Scholar]
- 33. Song Y, Lin FY, Yin F, et al. Phosphonosulfonates are potent, selective inhibitors of dehydrosqualene synthase and staphyloxanthin biosynthesis in Staphylococcus aureus. J Med Chem. 2009; 52: 976–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roszkowska AM, Colosi P, D'Angelo P, Ferreri G. Age-related modifications of the corneal endothelium in adults. Int Ophthalmol. 2004; 25: 163–166 [DOI] [PubMed] [Google Scholar]
- 35. Chiu C-J, Liu S, Willett WC, et al. Informing food choices and health outcomes by use of the dietary glycemic index. Nutr Revs. In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ramrattan RS, van der Schaft TL, Mooy CM, de Bruijn WC, Mulder PG, de Jong PT. Morphometric analysis of Bruch's membrane, the choriocapillaris, and the choroid in aging. Invest Ophthalmol Vis Sci. 1994; 35: 2857–2864 [PubMed] [Google Scholar]
- 37. Spaide RF. Age-related choroidal atrophy. Am J Ophthalmol. 2009; 147: 801–810 [DOI] [PubMed] [Google Scholar]
- 38. Xu P, Duong DM, Seyfried NT, et al. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009; 137: 133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mettu PS, Wielgus AR, Ong SS, Cousins SW. Retinal pigment epithelium response to oxidant injury in the pathogenesis of early age-related macular degeneration. Mol Aspects Med. 2012; 33: 376–398 [DOI] [PubMed] [Google Scholar]
- 40. Jarrett SG, Boulton ME. Consequences of oxidative stress in age-related macular degeneration. Mol Aspects Med. 2012; 33: 399–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Handa JT. How does the macula protect itself from oxidative stress? Mol Aspects Med. 2012; 33: 418–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Handa JT, Verzijl N, Matsunaga H, et al. Increase in the advanced glycation end product pentosidine in Bruch's membrane with age. Invest Ophthalmol Vis Sci. 1999; 40: 775–779 [PubMed] [Google Scholar]
- 43. Farboud B, Aotaki-Keen A, Miyata T, Hjelmeland LM, Handa JT. Development of a polyclonal antibody with broad epitope specificity for advanced glycation endproducts and localization of these epitopes in Bruch's membrane of the aging eye. Mol Vis. 1999; 5: 11 [PubMed] [Google Scholar]
- 44. Weikel KA, Fitzgerald P, Shang F, et al. Natural history of age-related retinal lesions that precede AMD in mice fed high or low glycemic index diets. Invest Ophthalmol Vis Sci. 2012; 53: 622–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chiu CJ, Milton RC, Gensler G, Taylor A. Association between dietary glycemic index and age-related macular degeneration in nondiabetic participants in the Age-Related Eye Disease Study. Am J Clin Nutr. 2007; 86: 180–188 [DOI] [PubMed] [Google Scholar]
- 46. Weikel KA, Chiu CJ, Taylor A. Nutritional modulation of age-related macular degeneration. Mol Aspects Med. 2012; 33: 318–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Group TA-REDSAR Lutein + zeaxanthin and omega-3 fatty acids for age-related macular degeneration: the Age-Related Eye Disease Study 2 (AREDS2) randomized clinical trial. JAMA. 2013; 309: 2005–2015 [DOI] [PubMed] [Google Scholar]
- 48. Weismann D, Hartvigsen K, Lauer N, et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature. 2011; 478: 76–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gorin MB. Genetic insights into age-related macular degeneration: controversies addressing risk, causality, and therapeutics. Mol Aspects Med. 2012; 33: 467–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Han M, Giese G, Schmitz-Valckenberg S, et al. Age-related structural abnormalities in the human retina-choroid complex revealed by two-photon excited autofluorescence imaging. J Biomed Opt. 2007; 12: 024012 [DOI] [PubMed] [Google Scholar]
- 51. Steinle JJ, Sharma S, Smith CP, McFayden-Ketchum LS. Normal aging involves modulation of specific inflammatory markers in the rat retina and choroid. J Gerontol A Biol Sci Med Sci. 2009; 64: 325–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kelly J, Ali Khan A, Yin J, Ferguson TA, Apte RS. Senescence regulates macrophage activation and angiogenic fate at sites of tissue injury in mice. J Clin Invest. 2007; 117: 3421–3426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002: 82: 374–428 [DOI] [PubMed] [Google Scholar]
- 54. Taylor A. Aminopeptidases. 1st ed. Austin, TX: Landes Bioscience; 1996. [Google Scholar]
- 55. Jahngen-Hodge J, Obin MS, Gong X, et al. Regulation of ubiquitin-conjugating enzymes by glutathione following oxidative stress. J Biol Chem. 1997; 272: 28218–28226 [DOI] [PubMed] [Google Scholar]
- 56. Riley BE, Kaiser SE, Shaler TA, et al. Ubiquitin accumulation in autophagy-deficient mice is dependent on the Nrf2-mediated stress response pathway: a potential role for protein aggregation in autophagic substrate selection. J Cell Biol. 2010; 191: 537–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Graff J, Kahn M, Samiei A, et al. A dietary regimen of caloric restriction or pharmacological activation of SIRT1 to delay the onset of neurodegeneration. J Neurosci. 2013; 33: 8951–8960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Karring H, Poulsen ET, Runager K, et al. Serine protease HtrA1 accumulates in corneal transforming growth factor beta induced protein (TGFBIp) amyloid deposits. Mol Vis. 2013; 19: 861–876 [PMC free article] [PubMed] [Google Scholar]
- 59. Shang F, Gong X, Taylor A. Activity of ubiquitin-dependent pathway in response to oxidative stress. Ubiquitin-activating enzyme is transiently up-regulated. J Biol Chem. 1997; 272: 23086–23093 [DOI] [PubMed] [Google Scholar]
- 60. Shang F, Gong X, Palmer HJ, Nowell TR, Jr, , Taylor A. Age-related decline in ubiquitin conjugation in response to oxidative stress in the lens. Exp Eye Res. 1997; 64: 21–30 [DOI] [PubMed] [Google Scholar]
- 61. Pereira P, Shang F, Hobbs M, Girao H, Taylor A. Lens fibers have a fully functional ubiquitin-proteasome pathway. Exp Eye Res. 2003; 76: 623–631 [DOI] [PubMed] [Google Scholar]
- 62. Zetterberg M, Petersen A, Sjostrand J, Karlsson J. Proteasome activity in human lens nuclei and correlation with age, gender and severity of cataract. Curr Eye Res. 2003; 27: 45–53 [DOI] [PubMed] [Google Scholar]
- 63. Dudek EJ, Lampi KJ, Lampi JA, et al. Ubiquitin proteasome pathway-mediated degradation of proteins: effects due to site-specific substrate deamidation. Invest Ophthalmol Vis Sci. 2010; 51: 4164–4173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wagner BJ, Margolis JW, Abramovitz AS, Fu SCJ. Differential inhibition of two proteolytic activities in bovine lens neutral-proteinase preparations. Biochem J. 1985; 228: 517–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tonoki A, Kuranaga E, Tomioka T, et al. Genetic evidence linking age-dependent attenuation of the 26S proteasome with the aging process. Mol Cell Biol. 2009; 29: 1095–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pathare GR, Nagy I, Bohn S, et al. The proteasomal subunit Rpn6 is a molecular clamp holding the core and regulatory subcomplexes together. Proc Natl Acad Sci U S A. 2012; 109: 149–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Vilchez D, Morantte I, Liu Z, et al. RPN-6 determines C. elegans longevity under proteotoxic stress conditions. Nature. 2012; 489: 263–268 [DOI] [PubMed] [Google Scholar]
- 68. Louie JL, Kapphahn RJ, Ferrington DA. Proteasome function and protein oxidation in the aged retina. Exp Eye Res. 2002; 75; 271–284 [PubMed] [Google Scholar]
- 69. Kapphahn RJ, Giwa BM, Berg KM, et al. Retinal proteins modified by 4-hydroxynonenal: identification of molecular targets. Exp Eye Res. 2006; 83: 165–175 [DOI] [PubMed] [Google Scholar]
- 70. Fernandes AF, Zhou J, Zhang X, et al. Oxidative inactivation of the proteasome in retinal pigment epithelial cells. A potential link between oxidative stress and up-regulation of interleukin-8. J Biol Chem. 2008; 283: 20745–20753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Caceres A, Shang F, Wawrousek E, et al. Perturbing the ubiquitin pathway reveals how mitosis is hijacked to denucleate and regulate cell proliferation and differentiation in vivo. PLoS One. 2010; 5: e13331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Morishita H, Eguchi S, Kimura H, et al. Deletion of autophagy-related 5 (Atg5) and Pik3c3 genes in the lens causes cataract independent of programmed organelle degradation. J Biol Chem. 2013; 288: 11436–11447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Elanchezhian R, Palsamy P, Madson CJ, Lynch DW, Shinohara T. Age-related cataracts: homocysteine coupled endoplasmic reticulum stress and suppression of Nrf2-dependent antioxidant protection. Chem Biol Interact. 2012; 200: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yan XX, Jeromin A. Spectrin Breakdown Products (SBDPs) as Potential Biomarkers for Neurodegenerative Diseases. Curr Transl Geriatr Exp Gerontol Rep. 2012; 1: 85–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Medeiros R, Kitazawa M, Chabrier MA, et al. Calpain inhibitor A-705253 mitigates Alzheimer's disease-like pathology and cognitive decline in aged 3xTgAD mice. Am J Pathol. 2012; 181: 616–625 [DOI] [PubMed] [Google Scholar]
- 76. Getz GS. Calpain inhibition as a potential treatment of Alzheimer's disease. Am J Pathol. 2012; 181: 388–391 [DOI] [PubMed] [Google Scholar]
- 77. Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. J Biol Chem. 2000; 275: 31505–31513 [DOI] [PubMed] [Google Scholar]
- 78. Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med. 2008: 14; 959–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bian Q, Fernandes AF, Taylor A, Wu M, Pereira P, Shang F. Expression of K6W-ubiquitin in lens epithelial cells leads to upregulation of a broad spectrum of molecular chaperones. Mol Vis. 2008; 14: 403–412 [PMC free article] [PubMed] [Google Scholar]
- 80. Marques C, Guo W, Pereira P, et al. The triage of damaged proteins: degradation by the ubiquitin-proteasome pathway or repair by molecular chaperones. FASEB J. 2006; 20: 741–743 [DOI] [PMC free article] [PubMed] [Google Scholar]