

Abstract
Hyperglycemia has toxic effects on almost all cells in the body. Ophthalmic complications of hyperglycemia are most profound in cornea and retina. Seventy percent of diabetics suffer from corneal complications, collectively called diabetic keratopathy, which includes include recurrent erosions, delayed wound healing, ulcers, and edema. Confocal microscopy has permitted in vivo imaging of corneal nerves, which are also affected in diabetic subjects. Gene therapies upregulating MNNG HOS transforming gene (cMet) and/or downregulating MMP10 and cathepsin S are potential future therapies for diabetic keratopathy.
Diabetic retinopathy (DR) is the most common cause of blindness in people over the age of 50. There is accumulating evidence that DR is an inflammatory disease. The initial events in animal models of DR are increased vascular permeability and leukostasis. This binding of leukocytes to the endothelium results from an increase in intracellular adhesion molecule-1 (ICAM-1) on the retinal capillary endothelium (EC) and expression of CD11/CD18 on the surface of the activated leukocyte. We have observed polymorphonuclear leukocytes (PMNs) at sites of EC vascular dysfunction in diabetic retinas as well as choroid. Anti-inflammatory drugs like etanercept, aspirin, or meloxicam reduce leukostasis and EC death. Future therapies may include repopulation of the acellular capillaries after EC and pericyte death with vascular progenitors made from the patient's own blood cells.
Keywords: diabetes, retinopathy, choroidopathy, cornea, inflammation
Introduction
Hyperglycemia has toxic effects on almost all cells in the body. Ophthalmic complications of hyperglycemia are most profound in cornea and retina. The cornea experiences 4-fold higher glucose in diabetic tear film than in control tears. Seventy percent of diabetics suffer from corneal complications collectively called diabetic keratopathy. Retina accounts for the majority of visual loss in diabetics, and diabetic retinopathy is the most common cause of blindness in people over the age of 50. In addition, the diabetic choroid has vascular–related changes similar to those in the diabetic retina.
Diabetic Cornea
The diabetic cornea suffers from cellular dysfunction and dysfunctional repair mechanisms, which include recurrent erosions, delayed wound healing, ulcers, and edema. In addition and undoubtedly related to epithelial dysfunction, alterations in epithelial basement membrane occur. Also, neuropathy occurs in diabetic cornea, resulting in loss of corneal sensation and innervation,1 which may be related to corneal epithelial defects. There is a suggestion that neuropathy is causing corneal epithelial defects.2 The prominence and early onset of neuropathy in diabetic cornea have recently been documented and quantified in human subjects by Midena and associates and in Adrey Zhivov's lab using confocal imaging.1,3,4 This offers the possibility of noninvasive detection of early changes in the diabetic eye that may be predictive of diabetic retinopathy.
Corneal nerve alterations also occur in the limbal area, where there are corneal epithelial stem cells at the cornea–scleral junction that are the source of new cells for the central corneal epithelium. However, there is no definitive marker for these progenitor cells, which delays our understanding of these cells and the stimuli for their differentiation and migration.
Several techniques have been developed to study corneal wound healing. All of the cells of cornea can be grown in culture, and their ability to migrate and proliferate can then be evaluated in vitro. However, these cells can be difficult to propagate in large numbers as primary cultures, and a single cell type does not represent the response of the whole diabetic cornea. There are animal models that have many changes similar to those in human diabetic corneas. For example, the Goto-Kaizaki type 2 diabetic rat's cornea has poor epithelial wound healing,5 and neuropathy occurs in the corneas of rats with streptozocin (STZ)-induced diabetes.6 Another model is rat or human corneas kept ex vivo; wound healing is studied in these preparations. Central wounds are made in the corneas, and the cornea is placed in serum-free medium at the air–liquid interface.7 Delayed wound healing and epithelial marker abnormalities appear in human diabetic corneas in culture; but they lack innervation, so an important participant in corneal response is missing.8 Using this wound healing assay ex vivo, it was determined that cathepsin S and MMP10 overexpression inhibits wound healing and downregulates phospho-protein kinase B (p-Akt, a serine threonine-specific protein kinase important in glucose metabolism) in normal corneas.9 c-Met (MNNG HOS transforming gene), a proto-oncogene encoding hepatocyte growth factor receptor, is a key element in wound healing as demonstrated when cMet is upregulated and the rate of corneal wound healing increases.10 The effect is more significant with simultaneous inhibition of MMP10 and cathepsin S by small hairpin RNA (shRNA). The positive effect is due to restoration of hepatocyte growth factor (HGF) signaling, marker patterns, and wound healing. This same gene therapy at the limbus only normalizes wound healing at the limbus.
Early Changes in Diabetic Retina
Diabetic retinopathy (DR) is a microvasculopathy in that the microvasculature leaks serum, increased vascular permeability, and capillaries are lost early in the disease. Hyperglycemia and mitochondrial and extracellular reactive oxygen species (ROS) are toxic to endothelial cells (ECs), pericytes, and neurons, resulting in their death early in DR. There is accumulating evidence that low-grade inflammation underlies the vascular complications of DR.11–13 Inflammation is a nonspecific response of the body to tissue injury in which leukocytes are recruited to the inflamed tissue. Diabetic retinopathy is categorized best as a chronic low-level inflammation in which there are elevated systemic cytokines like TNF-α and IL-1β and elevated numbers of circulating activated leukocytes.14–16
Nishiwaki and associates observed “leukostasis,” sticking and retention of leukocytes labeled with acridine orange, in real time within the microvasculature of diabetic rats.17 Leukostasis was observed as early as 2 weeks after induction of diabetes by STZ. This is approximately the same time frame as for leakage of the inner blood–retinal barrier (BRB) in rats, which has been observed as early as 1 week after induction of diabetes.18,19 These two events appear to be the earliest changes that occur in the STZ diabetic rodent retina. The extent of leukostasis was the same at 1 month as it was at 11 months in diabetic mice, suggesting that leukostasis is chronic, like other diabetic pathologic processes.20
There are three possible mechanisms for leukostasis.21 One mechanism could be decreased retinal blood flow and perfusion pressure. There is little agreement on blood flow or perfusion pressure in diabetic retina, perhaps because different instruments were used and patients of different cohorts were at different stages of DR.22 The number of static leukocytes increases in STZ-induced diabetic rats compared to nondiabetics, whereas the leukocyte velocity is not different from that in controls,23,24 so blood flow does not seem to contribute to leukostasis.
A second mechanism could be the narrowing of capillary lumens during diabetes. There is an increase in the potent vasoconstrictor endothelin in diabetic retina, and increased expression of endothelin receptors (ET-R) has been reported on diabetic retinal pericytes.25 ET-1 (endothelin-1) is also elevated in the plasma of diabetic patients.26 Furthermore, ET-R agonists reduced leukostasis in diabetic rat retina. The degree of constriction in the human diabetic (nonspecific esterase) retina and its role in leukostasis are still undetermined.
The final mechanism is increased leukocyte–endothelial adhesion in retinal blood vessels. Leukocytes from diabetic patients have increased levels of CD11a, CD11b, and CD18.19 Increased numbers of activated polymorphonuclear leukocytes (PMNs), which express CD11b and CD18 on their surface, circulate in diabetics compared to controls.16 Diabetic PMNs are larger and more rigid than normal PMNs and must be compressed to fit through normal retinal capillaries.27,28 We have observed PMNs at sites of endothelial cell dysfunction (loss of adenosine diphosphatase [ADPase] activity29) in human diabetic retina (Fukushima I and Lutty G, unpublished data, 1997) (Fig. 1) and in the retinas of spontaneously diabetic monkeys.30 In diabetic monkeys, increased number of PMNs was related to dyslipidemia and hypertension. Increased leukostasis also occurs in the Tokushima Fatty rats (OLTEF), which also has dyslipidemia as well.31 The oxidative burst of a PMN can injure or kill ECs.28
Figure 1.

Adenosine diphosphatase (ADPase) activity and nonspecific esterase activity (red, indicating PMNs) in a diabetic retina (left). Bright-field (A) and dark-field illumination (B) and a section (C) after embedding in glycol methacrylate are shown for the same capillary segment (triple arrow). ADPase activity, an indicator of endothelial cell function,29 is lost at the site of PMN binding. When PMNs in retina were counted (D), there were significantly more PMNs in diabetic retina than in control subjects. Alkaline phosphatase activity (APase, blue) and nonspecific esterase (NSE) activity (red) in a normal choroid (E) and in a diabetic choroid (F). PMNs in the diabetic choroid are present at sites of choriocapillaris dysfunction, loss of APase activity (F).45 PMNs in diabetic and nondiabetic choroid were counted, and all areas of diabetic choroid had more PMNs than did normal choroid (G).43
In addition to increased activated leukocytes circulating in diabetics, the levels of leukocyte adhesion molecules are elevated in diabetics. We observed elevated ICAM-1, which is associated with firm adherence of activated leukocytes, in human diabetic retina compared to controls.32 A similar elevation in ICAM-1 was observed in ECs of the diabetic rat retina.33 Leukostasis in diabetic rats was inhibited by intravenous administration of an antibody against ICAM-1.34 Leukostasis can also be inhibited by blocking the counterreceptor for ICAM-1 on the leukocytes, CD18.20,35 Increased leukostasis does not occur in retina when diabetes is induced in ICAM-1- or CD18-deficient mice.20
Diabetic Retinopathy
Endothelial cell death from hyperglycemia or leukocyte oxidative burst and subsequent increased vascular permeability appear to occur before pericyte dropout occurs. Once pericyte death occurs, all that remains are acellular capillaries that are basically collagenous tubes. There is no blood flow in the collagenous tubes,36 so the sensory retina adjacent becomes hypoxic. This hypoxic environment upregulates VEGF, which stimulates further elevated vascular permeability,37,38 increased expression of ICAM-1,39 and EC proliferation.40 Increased permeability of fluid and protein can result in diabetic macular edema (DME). Diabetic macular edema is the common cause of visual function loss in both nonproliferative and proliferative DR. Increased proliferation of ECs initially causes intraretinal microvascular abnormalities (IRMA), small abnormal vascular formations in areas lacking viable capillaries.41 Increased EC proliferation is probably also responsible for formation of microaneurysms in which ECs proliferate in the absence of pericytes, the cell type that normally keeps ECs quiescent. Finally, EC proliferation and migration from veins and venules can result in the formation of preretinal neovascularization, the hallmark of proliferative retinopathy.
Diabetic Choroidopathy
We have reported changes similar to DR in diabetic choroid. We observed dropout of choriocapillaris in diabetic choroid (Fig. 2), and there was a direct correlation between PMN number and area of choriocapillaris (CC) dropout (Fig. 1).42,43 There was a 4-fold greater area of acellular capillaries in diabetic choroid compared with aged control subjects. Although the CC constitutively expresses ICAM-1, the relative level was increased in CC and was present in all choroidal blood vessels of diabetics.32 We also observed upregulation of P-selectin in diabetic choroidal blood vessels. The severity of CC dropout was related to the thickness of Bruch's membrane deposits in diabetic choroid; the thickest deposits were present over areas of complete CC loss (Fig. 2D), suggesting that the deposits were related to loss of CC transport.42 Other features of diabetic choroidopathy were intrachoroidal and extrachoroidal neovascularization. The intrachoroidal neovascularization consisted of capillary networks deep in choroid near the lamina fusca, the border of choroid and sclera.44 Extrachoroidal neovascularization was observed within Bruch's membrane and between Bruch's membrane and RPE.45 Both forms of neovascularization were mostly at the equator or more peripheral in choroid. The extrachoroidal neovascularization was often autoinfarcted; that is, only collagenous tubes remained.
Figure 2.
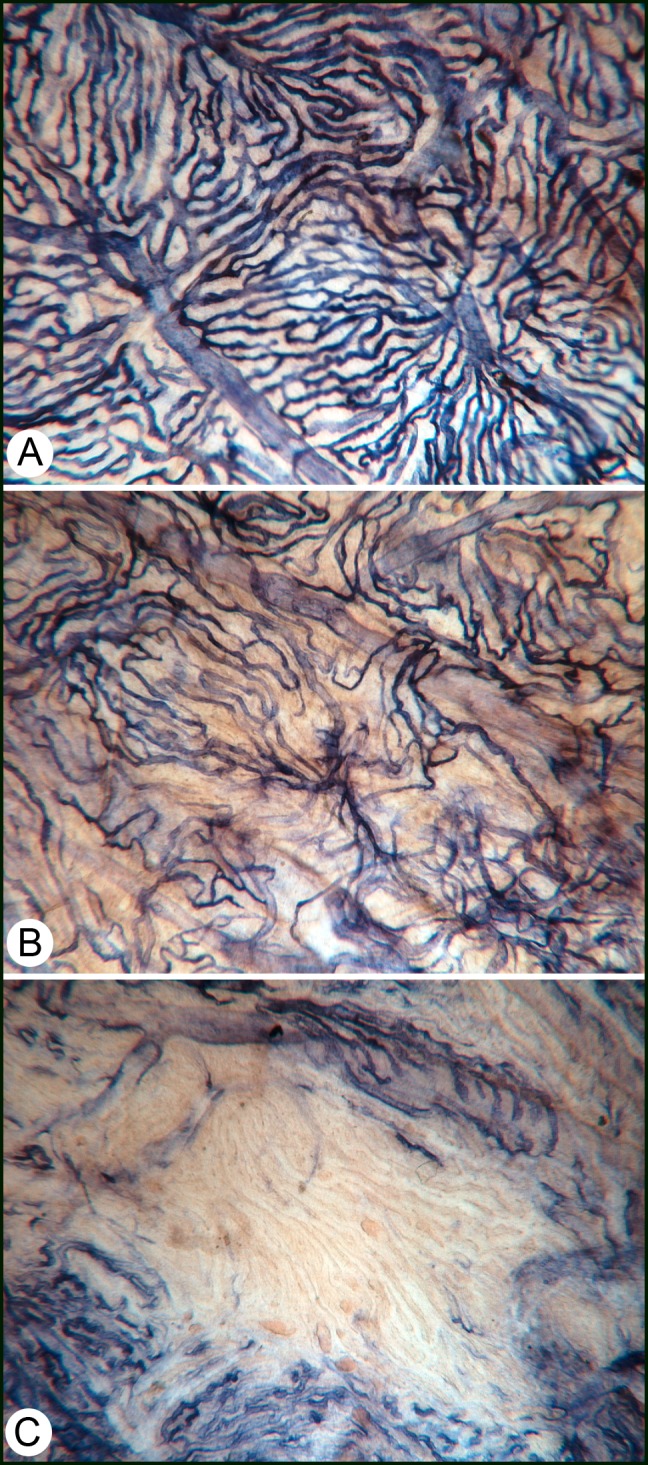
Alkaline phosphatase staining in a diabetic choroid. (A) Some areas of the choroid look normal and have organized lobules. However, on closer examination, the choriocapillaris (CC) has tortuous areas and some attenuated and constricted capillaries. (B) This area has a diffuse loss of CC, which was associated with deposits on Bruch's membrane.42 (C) An area of complete loss of CC, which was associated with the thickest deposits on Bruch's membrane in diabetic subjects.
Future Therapies for Diabetic Retinopathy and Choroidopathy
Experimental work by Antonia Joussen and Anthony Adamis demonstrated that death of ECs related to leukostasis can be prevented by blocking or genetically eliminating either ICAM-1 or CD18.11,20 Joussen has demonstrated that increased leukostasis and subsequent EC death can be prevented with anti-inflammatory agents. High-dose aspirin reduces expression of CD11a, CD11b, and CD18.20 High-dose aspirin, etanercept (soluble TNFR-Fc, tumor necrosis factor-1 linked to a human FC region), and high-dose meloxicam (a cyclooxygenase 2 inhibitor) reduced leukostasis and suppressed BRB breakdown in diabetic rats.20
Another therapeutic approach would be to repopulate the acellular capillaries with vascular progenitor cells. This approach was pioneered by Maria Grant, who demonstrated its feasibility.46,47 However, she and others found that diabetic endothelial progenitor cells (EPCs) were deficient in homing ability and, therefore, had limited engraftment capacity.48 The homing efficiency of diabetic EPCs can be increased by treatment of the cells with nitric oxide.49 We have generated vascular progenitors from CD34+ cord blood cells induced nonvirally into pluripotent stem cells (iPSCs).50 iPSCs were trained to be vascular progenitors on fibronectin substratum with high levels of VEGF.51 Of the four populations that resulted, CD31+/CD146+ cells were selected and expanded. When these cells are injected into vitreous of NOD/Scid mice that had experienced ischemia/reperfusion injury to retina resulting in acellular capillaries, the cells homed to the abluminal surface of the acellular capillaries, taking a pericyte position. When the cells were delivered intravenously, they were engrafted in a lumenal position, suggesting that they were assuming the role of endothelial cells (Fig. 3) (Park T, Bhutto I, Zimmerlin L, et al., manuscript submitted, 2013). The hypoxic adjacent retina makes both stromal-derived factor-1 (SDF-1) and VEGF, which would provide the stimulus for homing of vascular progenitors to the acellular capillaries. Our future work will focus on repopulation of acellular capillaries with our iPSC vascular progenitors in diabetic animals. The use of iPSCs made from CD34+ cells in blood brings us closer to autologous regenerative medicine for acellular capillaries.
Figure 3.

Engraftment of vascular progenitors (green) into capillaries of a NOD/Scid mouse retina that experienced I/R injury. The cells were CD31+/146+ iPSCs made from cord blood CD34+ cells. The capillaries are stained with anti-collagen 4 (red).
Conclusions
In conclusion, hyperglycemia has far-reaching effects on the eye. In cornea, it causes diabetic keratopathy, and in retina and choroid, directly or indirectly, it kills vascular and neuronal cells. The obvious therapy is maintenance of normoglycemia or compliance by the patient. Once the eye has been exposed to hyperglycemia long-term, basement membranes have accumulated toxic advanced glycosylation end products and cell death has occurred. Future therapies for diabetic keratopathy could involve gene therapy to upregulate c-Met and/or downregulate cathepsin S and MMP10. The cornea is a desirable site for gene therapy because it could be given topically and remain local in its effects.
The retinal and choroidal damage from hyperglycemia appears related to local inflammation. VEGF, IL-1β, and TNF-α are elevated in serum and in the local milieu (Fig. 4). TNF-α can activate nuclear factor–κβ (NK-κβ light-chain–enhancer of activated B cells), which upregulates genes involved in inflammation and ROS production. This causes upregulation of ICAM-1, which binds activated leukocytes in diabetes (Fig. 4). Leukocyte oxidative burst injures ECs so that vascular leakage occurs at these sites. The sites may be repaired by adjacent cells but may also be the site of repeated occlusive events. Since a PMN is so large and rigid in diabetics, it fills the lumen when firmly adherent. A PMN's life is short, so repeated occlusion and then PMN death mean a local ischemia/reperfusion (I/R) event each time a PMN binds to the site, creating a continuum of small I/R events in the capillary segment. If extracellular matrix is exposed, platelet fibrin thrombi will form, which can signal the end of flow in that vascular segment (Fig. 4). This also occurs in diabetic choriocapillaris. The acellular capillaries are sites where autologous vascular progenitors could be used to repopulate the ECs as well as pericyte niches.
Figure 4.
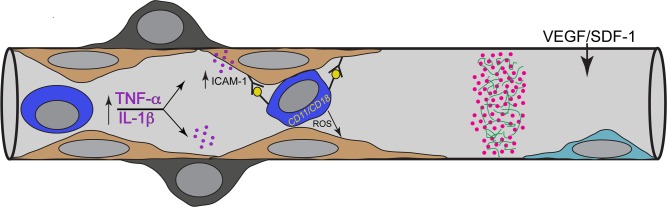
Schematic representation of possible events in which leukostasis results in acellular capillaries. From left to right, there are high levels of TNF-α and IL-1β circulating in diabetics, which are potent upregulators of ICAM-1 by endothelial cells. ICAM-1 firmly binds neutrophils (PMNs or dark blue cell), which are activated and express CD11/CD18 on their surface. Once bound, these PMNs can create an oxidative burst, which injures endothelial cells (ECs). From repeated occlusions, ECs are lost, a platelet/fibrin thrombus (pink dots in green fibers) forms on exposed basement membrane, and eventually pericytes are lost, yielding an acellular capillary. A potential therapy might be repopulation of the acellular capillaries with vascular progenitors (light blue cell), which home to the site because of SDF-1 and VEGF upregulated expression in adjacent hypoxic retina.
Acknowledgments
The author thanks Scott McLeod for creating the figures and Jingtai Cao, Ichiro Fukushima, Sam Kim, and Scott McLeod for their excellent work on diabetic retinopathy and choroidopathy. The author also thanks Alex Ljubimov for sharing his wisdom on diabetic keratopathy.
Supported by the Juvenile Diabetic Research Foundation, American Heart Association Established Investigator Award, National Institutes of Health Grants RO1 EY09357-18 (GL) and RO1 EY016151-06 and EY-01765 (Wilmer), and a grant from the Maryland Stem Cell Research Fund (TEDCO).
Disclosure: G.A. Lutty, None
References
- 1. Zhivov A, Winter K, Hovakimyan M, et al. Imaging and quantification of subbasal nerve plexus in healthy volunteers and diabetic patients with or without retinopathy. PLoS One. 2013; 8: e52157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. He J, Bazan HE. Mapping the nerve architecture of diabetic human corneas. Ophthalmology. 2012; 119: 956–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Midena E, Brugin E, Ghirlando A, Sommavilla M, Avogaro A. Corneal diabetic neuropathy: a confocal microscopy study. J Refract Surg. 2006; 22: S1047–S1052 [DOI] [PubMed] [Google Scholar]
- 4. Midena E, Cortez M, Miotto S, Gambato C, Cavarzeran F, Ghirlando A. Confocal microscopy of corneal sub-basal nerve plexus: a quantitative and qualitative analysis in healthy and pathologic eyes. J Refract Surg. 2009; 25: S125–S130 [DOI] [PubMed] [Google Scholar]
- 5. Chikama T, Wakuta M, Liu Y, Nishida T. Deviated mechanism of wound healing in diabetic corneas. Cornea. 2007; 26: S75–S81 [DOI] [PubMed] [Google Scholar]
- 6. Yin J, Huang J, Chen C, Gao N, Wang F, Yu FS. Corneal complications in streptozocin-induced type I diabetic rats. Invest Ophthalmol Vis Sci. 2011; 52: 6589–6596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Foreman DM, Pancholi S, Jarvis-Evans J, McLeod D, Boulton ME. A simple organ culture model for assessing the effects of growth factors on corneal re-epithelialization. Exp Eye Res. 1996; 62: 555–564 [DOI] [PubMed] [Google Scholar]
- 8. Kabosova A, Kramerov AA, Aoki AM, Murphy G, Zieske JD, Ljubimov AV. Human diabetic corneas preserve wound healing, basement membrane, integrin and MMP-10 differences from normal corneas in organ culture. Exp Eye Res. 2003; 77: 211–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Saghizadeh M, Kramerov AA, Yaghoobzadeh Y, et al. Adenovirus-driven overexpression of proteinases in organ-cultured normal human corneas leads to diabetic-like changes. Brain Res Bull. 2010; 81: 262–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saghizadeh M, Kramerov AA, Yu FS, Castro MG, Ljubimov AV. Normalization of wound healing and diabetic markers in organ cultured human diabetic corneas by adenoviral delivery of c-Met gene. Invest Ophthalmol Vis Sci. 2010; 51: 1970–1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Adamis AP. Is diabetic retinopathy an inflammatory disease? Br J Ophthalmol. 2002; 86: 363–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kern TS. Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp Diabetes Res. 2007; 2007: 95103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Noda K, Nakao S, Ishida S, Ishibashi T. Leukocyte adhesion molecules in diabetic retinopathy. J Ophthalmol. 2012; 2012: 279037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clausell N, Kalil P, Biolo A, Molossi S, Azevedo M. Increased expression of tumor necrosis factor-alpha in diabetic macrovasculopathy. Cardiovasc Pathol. 1999; 8: 145–151 [DOI] [PubMed] [Google Scholar]
- 15. Clausen P, Jacobsen P, Rossing K, Jensen JS, Parving HH, Feldt-Rasmussen B. Plasma concentrations of VCAM-1 and ICAM-1 are elevated in patients with Type 1 diabetes mellitus with microalbinuria and overt nephropathy. Diabet Med. 2000; 17: 644–649 [DOI] [PubMed] [Google Scholar]
- 16. Wierusz-Wysocki B, Wysocki H, Siekierka H, Wykretowicz A, Szcaepanik A, Klimas R. Evidence of polymorphonuclear neutrophils (PMN) activation in patients with insulin-dependent diabetes mellitus. J Leukoc Biol. 1987; 42: 519–523 [DOI] [PubMed] [Google Scholar]
- 17. Nishiwaki H, Ogura Y, Kimura H, Kiryu J, Honda Y. Quantitative evaluation of leukocyte dynamics in retinal microcirculation. Invest Ophthalmol Vis Sci. 1995; 36: 123–130 [PubMed] [Google Scholar]
- 18. Joussen AM, Murata T, Tsujikawa A, Kirchhof B, Bursell S-E, Adamis AP. Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol. 2001; 158: 147–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Joussen AM, Poulaki V, Mitsiades N, et al. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-a suppression. FASEB J. 2002; 16: 438–440 [DOI] [PubMed] [Google Scholar]
- 20. Joussen AM, Poulaki V, Le ML, et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004; 18: 1450–1452 [DOI] [PubMed] [Google Scholar]
- 21. Hughes JM. Leukostasis and inflammation: crucial steps in the development of diabetic retinopathy or epiphenomenon. In: Hughes JM. ed Endothelial Dysfunction in Experimental Models of Preclinical Diabetic Retinopathy. Amsterdam: Academic Medical Center-University of Amsterdam; 2010. [Google Scholar]
- 22. Grunwald JE, Bursell S-E. Hemodynamic changes as early markers of diabetic retinopathy. Curr Opin Endocrinol Diabetes. 1996; 3: 298–306 [Google Scholar]
- 23. Abiko T, Abiko A, Clermont AC, et al. Characterization of retinal leukostasis and hemodynamics in insulin resistance and diabetes: role of oxidants and protein kinase-C activation. Diabetes. 2003; 52: 829–837 [DOI] [PubMed] [Google Scholar]
- 24. Miyamoto K, Hiroshiba N, Tsujikawa A, Ogura Y. In vivo demonstration of increased leukocyte entrapment in retinal microcirculation of diabetic rats. Invest Ophthalmol Vis Sci. 1998; 39: 2190–2194 [PubMed] [Google Scholar]
- 25. Chakravarthy U, Hayes RG, Stitt AW, Douglas A. Endothelin expression in ocular tissues of diabetic and insulin-treated rats. Invest Ophthalmol Vis Sci. 1997; 38: 2144–2151 [PubMed] [Google Scholar]
- 26. Takagi C, King GL, Takagi H, Lin YW, Clermont AC, Bursell SE. Endothelin-1 action via endothelin receptors is a primary mechanism modulating retinal circulatory response to hyperoxia. Invest Ophthalmol Vis Sci. 1996; 37: 2099–2109 [PubMed] [Google Scholar]
- 27. Braun R, Fisher T, Meiselman H, Hatchell D. Decreased deformability of polynuclear leukocytes in diabetic cats. Microcirculation. 1996; 3: 271–278 [DOI] [PubMed] [Google Scholar]
- 28. Schmid-Schönbein GW. Granulocyte activation and capillary obstruction. Monogr Atheroscler. 1990; 15: 150–159 [PubMed] [Google Scholar]
- 29. Lutty GA, McLeod DS. Phosphatase enzyme histochemistry for studying vascular hierarchy, pathology, and endothelial cell dysfunction in retina and choroid. Vision Res. 2005; 45: 3504–3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim SY, Johnson MA, McLeod DS, Alexander T, Hansen BC, Lutty GA. Neutrophils are associated with capillary closure in spontaneously diabetic monkey retinas. Diabetes. 2005; 54: 1534–1542 [DOI] [PubMed] [Google Scholar]
- 31. Lu ZY, Bhutto IA, Amemiya T. Retinal changes in Otsuka long-evans Tokushima fatty rats (spontaneously diabetic rat)--possibility of a new experimental model for diabetic retinopathy. Jpn J Ophthalmol. 2003; 47: 28–35 [DOI] [PubMed] [Google Scholar]
- 32. McLeod DS, Lefer DJ, Merges C, Lutty GA. Enhanced expression of intracellular adhesion molecule-1 and P-selectin in the diabetic human retina and choroid. Am J Pathol. 1995; 147: 642–653 [PMC free article] [PubMed] [Google Scholar]
- 33. Nozaki M, Ogura Y, Hirabayashi Y, Saishin Y, Shimada S. Enhanced expression of adhesion molecules of the retinal vascular endothelium in spontaneous diabetic rats. Ophthalmic Res. 2002; 34: 158–164 [DOI] [PubMed] [Google Scholar]
- 34. Miyamoto K, Khosrof S, Bursell SE, et al. Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc Natl Acad Sci U S A. 1999; 96: 10836–10841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Barouch F, Miyamoto K, Allport J, et al. Integrin-mediated neutrophil adhesion and retinal leukostasis in diabetes. Invest Ophthalmol Vis Sci. 2000; 41: 1153–1158 [PubMed] [Google Scholar]
- 36. Kohner EM, Henkind P. Correlation of fluorescein angiogram and retinal digest in diabetic retinopathy. Am J Ophthalmol. 1970; 69: 403–414 [DOI] [PubMed] [Google Scholar]
- 37. Tolentino MJ, Husain D, Theodosiadis P, et al. Angiography of fluorestricted anti-vascular endothelial growth factor antibody and dextrans in experimental choroidal neovascularization. Arch Ophthalmol. 2000; 118: 78–84 [DOI] [PubMed] [Google Scholar]
- 38. Tolentino MJ, McLeod DS, Taomoto M, Otsuji T, Adamis AP, Lutty GA. Pathological features of vascular endothelial growth factor-induced retinopathy in non-human primates. Am J Ophthalmol. 2002; 133: 373–385 [DOI] [PubMed] [Google Scholar]
- 39. Lu M, Perez V, Ma N, et al. VEGF increases retinal ICAM-1 expression in vivo. Invest Ophthalmol Vis Sci. 1999; 40: 1808–1812 [PubMed] [Google Scholar]
- 40. D'Amore PA. Mechanisms of retinal and choroidal neovascularization. Invest Ophthalmol Vis Sci. 1994; 35: 3974–3979 [PubMed] [Google Scholar]
- 41. Lutty GA, McLeod DS. A new technique for visualization of the human retinal vasculature. Arch Ophthalmol. 1992; 110: 267–276 [DOI] [PubMed] [Google Scholar]
- 42. Cao J, McLeod S, Merges CA, Lutty GA. Choriocapillaris degeneration and related pathologic changes in human diabetic eyes. Arch Ophthalmol. 1998; 116: 589–597 [DOI] [PubMed] [Google Scholar]
- 43. Lutty GA, Cao J, McLeod DS. Relationship of polymorphonuclear leukocytes (PMNs) to capillary dropout in the human diabetic choroid. Am J Pathol. 1997; 151: 707–714 [PMC free article] [PubMed] [Google Scholar]
- 44. Fukushima I, McLeod DS, Lutty GA. Intrachoroidal microvascular abnormality, a previously unrecognized form of choroidal neovascularization. Am J Ophthalmol. 1997; 124: 473–487 [DOI] [PubMed] [Google Scholar]
- 45. McLeod DS, Lutty GA. High resolution histologic analysis of the human choroidal vasculature. Invest Ophthalmol Vis Sci. 1994; 35: 3799–3811 [PubMed] [Google Scholar]
- 46. Jarajapu YP, Caballero S, Verma A, et al. Blockade of NADPH oxidase restores vasoreparative function in diabetic CD34+ cells. Invest Ophthalmol Vis Sci. 2011; 52: 5093–5104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shaw LC, Neu MB, Grant MB. Cell-based therapies for diabetic retinopathy. Curr Diab Rep. 2011; 11: 265–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Caballero S, Sengupta N, Afzal A, et al. Ischemic vascular damage can be repaired by healthy, but not diabetic, endothelial progenitor cells. Diabetes. 2007; 56: 960–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Segal MS, Shah R, Afzal A, et al. Nitric oxide cytoskeletal-induced alterations reverse the endothelial progenitor cell migratory defect associated with diabetes. Diabetes. 2006; 55: 102–109 [PubMed] [Google Scholar]
- 50. Park TS, Huo JS, Peters A, et al. Growth factor-activated stem cell circuits and stromal signals cooperatively accelerate non-integrated iPSC reprogramming of human myeloid progenitors. PLoS One. 2012; 7: e42838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Park TS, Zimmerlin L, Zambidis ET. Efficient and simultaneous generation of hematopoietic and vascular progenitors from human induced pluripotent stem cells. Cytometry A. 2013; 83: 114–126 [DOI] [PMC free article] [PubMed] [Google Scholar]