

Summary
The prokaryotic CpG‐specific DNA methylase from Spiroplasma, SssI methylase, has been extensively used to methylate plasmid DNA in vitro to investigate the effects of methylation in vertebrate systems. Currently available methods to produce CpG‐methylated plasmid DNA have certain limitations and cannot generate large quantities of methylated DNA without cost or problems of purity. Here we describe an approach in which the SssI methylase gene has been introduced into the Escherichia coli bacterial genome under the control of an inducible promoter. Plasmid DNA propagated in this bacterium under conditions which induce the methylase gene result in significant (> 90%) CpG methylation. Methylated DNA produced by this approach behaves similarly to methylated DNA produced in vitro using the purified methylase. The approach is scalable allowing for the production of milligram quantities of methylated plasmid DNA.
Introduction
Bacteria have developed elaborate mechanisms to both recognize and destroy foreign DNA that may gain access to the organism. A series of DNA methylases and restriction endonucleases exist that can modify host DNA and cleave foreign DNA from a variety of sources including other bacterial species and mammalian cells. Similarly, mammalian species also have the ability to recognize microbial DNA and respond by activation of the immune system. This recognition of bacterial DNA by vertebrates is thought to be based on the frequency of a specific dinucleotide sequence, the CpG motif, and on the methylation status of this sequence (Krieg et al., 1998). The frequency of CpG dinucleotides are reduced three‐ to fourfold in vertebrate species compared with bacteria and the majority (∼70–80%) of vertebrate CpG motifs are methylated at position 5 of the cytosine residues (Bird, 1987). Unmethylated CpG motifs are recognized by the Toll‐like receptor 9 on vertebrate immune cells, including B cells, natural killer cells, macrophages and monocytes, leading to activation of the innate immune response and heightened immune surveillance (Cornelie et al., 2004a) (reviewed in Krieg, 2002). CpG methylation of mammalian DNA has further roles in transcriptional regulation as methylation influences the affinity of certain DNA‐binding proteins including histones and transcription factors (Schubeler et al., 2000).
Given the importance of the CpG motif and its methylation status, the ability to produce CpG‐methylated plasmid DNA would have several potential uses, including the study of mammalian transcriptional regulation, the propagation of certain cloned DNA repeat sequences that have increased stability when methylated (Nichol and Pearson, 2002), and the application to mammalian gene therapy approaches where methylated DNA may prevent the activation of the innate immune response (Chen et al., 2001). Currently available methods to produce CpG‐methylated DNA have certain drawbacks which have hampered their use, particularly in gene therapy applications where large quantities of pure plasmid DNA are needed. In vitro methylation of plasmid DNA with purified SssI methylase is very efficient, yet is limited by yield, cost (approximately one dollar per microgram of methylated plasmid) and contaminating methylase protein which could interfere with subsequent biological use (Renbaum et al., 1990). In another approach, co‐culture of the plasmid of interest with a plasmid expressing the SssI methylase can produce large amounts of methylated plasmid, yet the plasmid of interest is co‐purified with the methylase expressing plasmid leading to DNA contamination (Butler and Gotschlich, 1991). Therefore new approaches that enable the production of large quantities of pure CpG‐methylated plasmid DNA are desirable. Here we report on the generation of a transposon cassette that can be introduced into bacterial strains to facilitate regulated expression of the SssI methylase. With this system large quantities of methylated plasmid DNA can be obtained using standard culture conditions and routine plasmid purification methods. Plasmid DNA produced by these methods showed near complete methylation of CpG motifs and when introduced into mammalian cells exhibited altered transcriptional activities similar to DNA methylated in vitro. Furthermore, methylation of a plasmid containing a DNA transposon, Sleeping Beauty (SB), enhanced its transposition frequency in mammalian cells. This approach to generating methylated plasmid DNA will be useful as a tool to study the role of DNA methylation in transcriptional regulation and as a means to produce large‐scale quantities of methylated DNA for non‐viral gene delivery.
Results
Construction and integration of a bacterial transposon containing the methylase gene driven by an arabinose‐regulated promoter
The Spiroplasma SssI methylase gene was placed under the control of the PBAD promoter. This regulated expression cassette was then placed within the context of a Tn10 transposable element to facilitate integration of the methylase gene into the bacterial genome. These cloning steps generated the plasmid, pBSL180‐SssI (Fig. 1), which was introduced into a standard Escherichia coli bacterium (Top10) by transformation. Immediately following transformation, transposition was facilitated by induction of the Tn10 transposase. A drug‐selectable marker [kanamycin (Kan)] within the transposable element was used to isolate bacteria that had incorporated the SssI methylase into the bacterial chromosome. Several Kan‐resistant bacterial colonies were investigated for their ability to methylate bacterial chromosomal DNA. The addition of arabinose to the culture medium was able to partially block restriction endonuclease digestion of the bacterial chromosomal DNA by the methylation‐sensitive enzyme HpaII (Fig. 2A).
Figure 1.
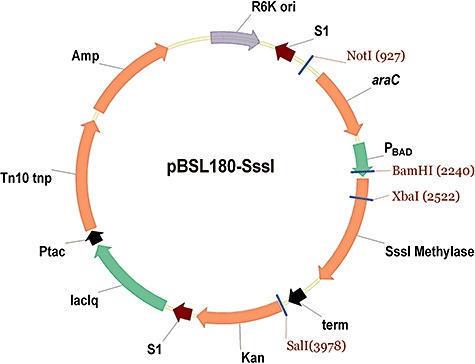
Schematic diagram of plasmid pBSL180‐SssI. To generate bacteria that express the SssI methylase under the control of an arabinose‐inducible promoter (PBAD), the following Tn10‐based transposon plasmid was generated. Adjacent to the SssI methylase is the drug resistance gene for kanamycin (Kan). The whole cassette; the araC, SssI methylase and Kan genes are surrounded by the Tn10 transposon elements (S1). The Tn10 transposase is under control of the lactose‐inducible promoter (lacIq). The plasmid contains an RK6 origin of replication making it incompetent for propagation in most E. coli strains, except those that are pir+ (e.g. strain EC100D, Epicentre).
Figure 2.
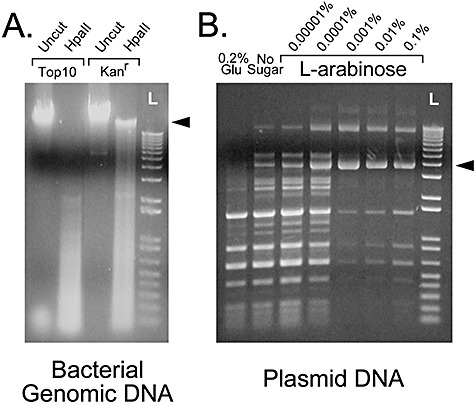
Arabinose‐regulated activity of the integrated SssI methylase gene in E. coli. A. Transformants were grown in LB medium containing kanamycin and arabinose 0.2% and bacterial genomic DNA was isolated. The extent of CpG methylation was determined by cutting with the restriction endonuclease HpaII and evaluating the extent of DNA cleavage. Comparisons were made with uncut genomic DNA of the same samples. Control bacterial DNA came from non‐transformed Top10 cells. The arrowhead points to the location of high‐molecular‐weight genomic DNA fragments. B. Transformants were electroporated with the plasmid pGreenLantern and grown in LB containing kanamycin and ampicillin with either 0.2% glucose, no sugar or arabinose at varying percentages (w/v). After overnight culture, mini‐prep DNA was cut with HpaII to analyse the methylation status of each plasmid. The arrowhead points to the location of uncut supercoiled plasmid DNA. L is a DNA ladder lane.
CpG methylation of plasmid DNA by bacteria harbouring the arabinose‐regulated methylase gene
The arabinose operon is negatively regulated by catabolite repression and therefore can be efficiently turned off if the bacteria are grown in the presence of glucose (0.2%). In the absence of any inducible sugar, the arabinose promoter will have some inherent activity and this activity can be further enhanced by adding increasing amounts of the inducer. Bacteria harbouring an integrated SssI methylase gene were transiently transfected with a plasmid and propagated in various conditions including 0.2% glucose, no sugar and varying amounts of arabinose. The degree of CpG methylation of the plasmid DNA was assessed by restriction endonuclease digestion with the enzyme HpaII. As shown in Fig. 2B, the degree of CpG methylation varied with the culture conditions. Surprisingly, the extent of CpG methylation did not directly correlate with increasing arabinose levels in the culture medium. An intermediate concentration of arabinose (0.001–0.01%) leads to optimal CpG methylation, whereas lower or higher concentrations (up to 2%) of the sugar lead to less DNA methylation. Based on estimates of band intensity from several different experiments, we estimate that > 90% of CpGs can be methylated under these optimal conditions (0.001% arabinose).
Although not shown here, the plasmid pBSL180‐SssI was integrated into a different strain of E. coli (DH10b‐trfA) and similar plasmid methylation experiments were performed. Identical findings were observed with the greatest extent of CpG plasmid methylation occurring at an arabinose concentration of 0.001%. Furthermore, the size of the plasmid does not seem to affect the extent of the methylation. The causes for incomplete methylation are unclear, yet it appears that a small percentage of plasmids are not methylated, either because they were newly generated, or because small populations of bacteria have lost the SssI methylase gene during propagation, perhaps due to toxicity. If higher degrees of plasmid DNA methylation are desired, overnight incubation of the in vivo methylated plasmids with significantly lower amounts (∼1 unit per 100 µg of plasmid) of purified SssI methylase can achieve near complete methylation. Despite the lack of complete methylation, this approach is still useful for the production of methylated DNA for various applications as the majority of the DNA is methylated.
Large‐scale production of methylated plasmid DNA
To ensure the results obtained in Fig. 2 could be repeated and were scaleable to larger quantities of plasmid propagation, we performed repeat bacterial transformations using two different plasmids. These bacteria were grown in various conditions including 0.2% glucose, no sugar and the optimal concentration of arabinose (0.001%). Plasmid DNA was obtained from overnight cultures of small quantities of bacteria (2 ml, mini‐prep), as well as larger quantities (500 ml, maxi‐prep) and the extent of CpG methylation was determined by restriction enzyme digestion (Fig. 3A and B). As expected, under inducing conditions both the small‐ and large‐scale plasmid propagations resulted in significant CpG methylation. To further demonstrate that the lack of restriction endonuclease activity of the HpaII enzyme was not due to alterations in the nucleotide sequence, the same DNA was cut with the methylation‐insensitive enzyme MspI, which gave the expected restriction pattern (Fig. 3B).
Figure 3.
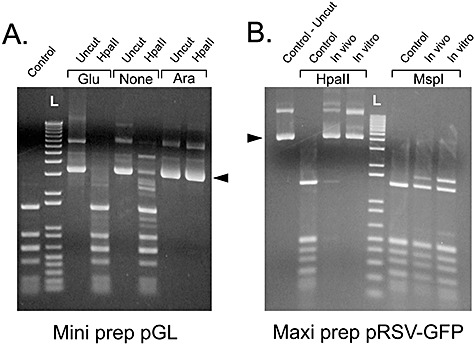
Small‐ and large‐scale production of methylated plasmid DNA. A. New transformants were generated to confirm the methylation effect. pGreenLantern (pGL) was electroporated into methylase‐expressing bacteria grown under several conditions including 0.2% glucose (Glu), no sugar (None) or 0.001% arabinose (Ara). Mini‐prep DNA was digested with HpaII to show the degree of methylation and uncut controls are provided for each sample (arrowhead). The control DNA (first lane) consisted of plasmid propagated in non‐methylating bacteria and cut with HpaII. B. Large‐scale maxi‐prep of the plasmid pRSV‐GFP grown in methylase‐expressing bacteria (In vivo). In addition, plasmid DNA was propagated in methylase‐negative bacteria (control), and an aliquot of this was exogenously methylated using purified SssI methylase (In vitro). Each DNA was cut with either HpaII (methylation‐sensitive) or MspI (methylation‐insensitive). Arrowhead illustrates the location of uncut plasmid DNA. L is the DNA ladder.
The large‐scale production of CpG‐methylated plasmid has application to the field of non‐viral gene delivery. Systemic administration of unmethylated microbial DNA is recognized by vertebrates resulting in increased release of pro‐inflammatory cytokines and an environment that stimulates immune recognition and destruction of cells transfected with the plasmid. In contrast, methylated plasmid DNA was able to block or significantly reduce the immune response to encoded or co‐administered protein antigens (Klinman et al., 1999; Cornelie et al., 2004b). Furthermore, reduced hepatotoxicity was observed in mice after receiving systemic administration of lipoplexes containing methylated plasmid DNA compared with unmethylated DNA (Loisel et al., 2001). Whether methylated plasmids will yield more efficient gene delivery with less toxicity in animal models will need to be further investigated; however, the creation of this system to produce methylated plasmid DNA provides a tool to address this question.
Comparison of the transcriptional activities of in vivo and in vitro methylated plasmid DNA
In order to demonstrate that the CpG‐methylated plasmid DNA produced within bacteria (in vivo) was similar to methylated DNA produced by incubation with purified SssI methylase (in vitro), an experiment was designed to test the biological effect of CpG methylation. It is well established that CpG DNA methylation negatively regulates RNA transcription from mammalian promoters. Previously, the Rous Sarcoma Virus (RSV) promoter has been shown to be highly sensitive to negative regulation by CpG methylation (Hsieh, 1994). We generated a RSV promoter construct driving expression of the reporter gene encoding green fluorescent protein (GFP) and used the extent of GFP fluorescence as a measure of promoter activity. Unmethylated pRSV‐GFP plasmid, as well as in vivo and in vitro methylated plasmid, was transiently transfected into the human embryonic kidney cell line 293T. Two days after transfection, the cells were analysed for GFP expression by a fluorescence‐activated cells sorting (FACS) machine. As expected, methylation reduced both the percentage of cells that were GFP positive, as well as the mean fluorescence intensity (MFI) of the GFP‐positive cells. Figure 4 summarizes the results from at least three independent experiments using two different plasmid DNA concentrations. The results indicate that both in vivo and in vitro methylated plasmid DNA undergoes transcriptional repression, although there was a trend for the in vivo methylated DNA to be less repressed, potentially due to incomplete methylation.
Figure 4.
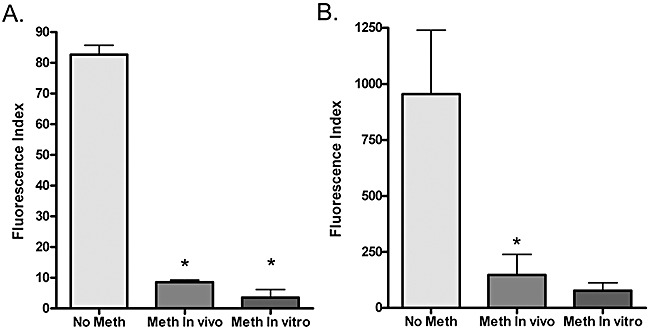
Graph of the effect of DNA methylation on RSV promoter activity. HEK293T cells were transiently transfected with the plasmid pRSV‐GFP that was either non‐methylated (No Meth), methylated in vivo (Meth In vivo) or methylated by incubation with purified SssI methylase (Meth In vitro). Two different DNA concentrations were transfected including low dose, 400 ng (A), or high dose 2 µg (B). The graph shows data from three or more independent experiments ± SD. The fluorescence index was determined my multiplying the %GFP‐positive cells times the mean fluorescence intensity of those cells. The asterisk (*) indicates statistical significance (P < 0.05) by Student's t‐test comparing in vivo with no methylation and in vitro with in vivo methylated.
Methylation of the SB DNA transposon enhances transposition
A previous publication had suggested that the SB transposon undergoes transposition more efficiently when the transposon is methylated (Yusa et al., 2004). To confirm these findings and to demonstrate that in vivo methylated transposon DNA can enhance transposition the following experiment was performed. The neomycin‐resistant SB transposon plasmid pMSZ‐CMV‐Neo contains a CpG‐depleted cytomegalovirus (CMV) promoter (Yew et al., 2002) and in theory should not be transcriptionally repressed by DNA methylation. Experiments looking at the activity of this promoter under methylated and unmethylated conditions show that methylation does diminish transcriptional activity to some degree (∼50–80% of normal). Despite this repression, the promoter activity was high enough to conduct an experiment in which the transposition activities of methylated and unmethylated transposons are compared. As shown in Fig. 5, in vivo methylated plasmid DNA did promote a higher number of neomycin‐resistant colonies compared with unmethylated controls which was statistically significant (P < 0.05). These data confirm that CpG‐methylated SB transposons are better substrates for transposition in vitro and could lead to enhanced transposition in animal models.
Figure 5.
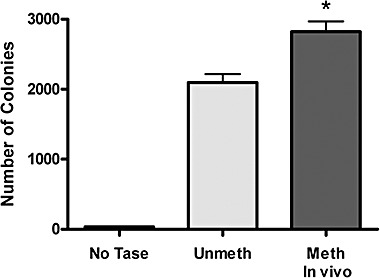
Transposition of methylated SB transposon. HeLa cells were transiently transfected with unmethylated (Unmeth) or in vivo methylated (Meth In vivo) neomycin‐resistant transposon DNA from the Sleeping Beauty transposon together with unmethylated plasmid expressing the transposase (pCMV‐HSB16). One set of cells received unmethylated transposon and no transposase (No Tase). Two days after transfection, cells are plated at low density and selected in drug for 2 weeks to generate colonies. The mean number of colonies from four or more independent experiments is graphed. A significant difference was observed showing increased transposition following methylation (*P < 0.003).
Experimental procedures
Plasmid construction
The plasmids pAIT2 (Renbaum et al., 1990), pBAD24 (Guzman et al., 1995) and pBSL180 (Alexeyev and Shokolenko, 1995) were obtained from the following sources, respectively: New England Biolabs, American Type Culture Collection and the National Institute of Genetics in Japan. To generate the Tn10 transposon plasmid pBSL180‐SssI, changes were first made to pBAD24 so that a portion of this plasmid could easily be cloned into pBSL180. Unique SalI and NotI sites were introduced into pBAD24 spanning the arabinose‐regulated expression cassette. The gene coding the Spiroplasma methylase (SssI) was amplified by PCR using the plasmid pAIT2 as template and the PCR product was cloned into the NcoI and SphI sites of the modified pBAD24. This plasmid was then cut with SalI and NotI to release the arabinose‐regulated SssI expression cassette and cloned into the SalI/NotI sites of pBSL180, generating pBSL180‐SssI.
To generate the SB transposon that contains a CpG‐depleted CMV promoter, the promoter/intron of the plasmid pGZB‐sCAT (Yew et al., 2002) was amplified by PCR and cloned in the place of the promoter and intron of the transposon pMSZ‐Neo (Liu et al., 2004).
Bacterial strains and culture
All bacterial strains were propagated in Luria–Bertani (LB) broth with the exception of DH10b‐trfA which were grown in low‐salt LB (5 g l−1 NaCL). Antibiotics used for selection included ampicillin (Amp) 100 µg ml−1, Kan 50 µg ml−1 and zeocin (Zeo) 25 µg ml−1. The plasmid pAIT2 and any other plasmid that contained a functionally active SssI methylase were propagated in bacterial strains that lack the methylation‐sensitive endonucleases McrA, McrBC and Mrr. pBAD24 was propagated in standard bacteria, yet once the SssI gene was cloned into this vector, subclones were maintained in Mrr‐ strains in the presence of 0.2% glucose. All pBSL180 plasmids contain the R6K origin and were grown in pir+ bacteria (EC100D from Epicentre Biotechnologies). To generate bacteria that had the SssI gene integrated into the genome, the plasmid pBSL180‐SssI was electroporated into either Top10 (Invirtogen) or DH10b‐trfA (Genetica), both of which are Mrr‐ strains. After electroporation the cells were placed in SOB medium (no glucose) containing ITPG at a concentration of 100 µM and incubated for 80 min. Cells were then plated on LB agar Kan plates containing 0.2% glucose. Bacterial genomic DNA was isolated from Kan‐resistant cells grown in 0.2% arabinose using lysozyme and detergent lysis, proteinase K treatment, phenol extractions and ethanol precipitation with DNA spooling.
Plasmid DNA (either pGreenLantern, pRSV‐GFP or pMSZ‐CMV‐Neo) was electroporated into these Kan‐resistant cells and selected with either Amp/Kan or Zeo/Kan depending on the plasmid. Routine propagation of these bacteria included antibiotics and 0.2% glucose to prevent plasmid CpG methylation. To stimulate plasmid methylation, cultures grown in 0.2% glucose were diluted (∼1:200 to 1:1000) into fresh growth medium containing either no sugar or various concentrations of arabinose. The degree of plasmid CpG methylation was assessed by restriction endonuclease digestion with the methylation‐sensitive enzyme, HpaII, or the methylation‐insensitive enzyme, MspI.
Mammalian cell transfection
HEK293T cells were grown in Dulbecco's modified Eagle's media (DMEM) supplemented with 10% fetal bovine serum (FBS). Cells were seeded on 6 cm dishes at 1.5 million cells per dish the night prior to transfection. Plasmid DNA was added by standard Hepes calcium phosphate transfection in the presence of chloroquine 50 µM (Luthman and Magnusson, 1983). Eight hours after the addition of the precipitate, the medium was replaced with fresh DMEM containing FBS and changed again at 24 h. Forty‐eight hours after transient transfection, the cells were trypsinized, re‐suspended in deficient RPMI (Roswell Park Memorial Institute) medium at a concentration of 500 000 cells per millilitre. Green fluorescent protein fluorescence was detected using a FACS Caliber (Becton Dickinson) machine with Cell Quest software. Using WinMDI software (Scripps) gates were used to detect the percentage of GFP‐positive cells and the MFI of the gated area. The fluorescence index was calculated as the percentage of GFP‐positive cells multiplied by the MFI.
Transposition assay
HeLa cells grown in DMEM plus 10% FBS were plated at 500 000 cells per 6 cm plate the night prior to transfection. Cells were transiently transfected using Lipofectamine Plus (Invitrogen) as per manufacturers recommendations with 3 µg of DNA (2 µg of transposon and 1 µg of transposase) using either unmethylated or methylated transposon and unmethylated transposase plasmid (pCMV‐HSB16) (Baus et al., 2005). Three days after DNA addition the cells were trypsinized, counted and plated at 10 000 cells per 10 cm plate. Cells were selected in G‐418 (1 mg ml−1) for 2 weeks, with the medium replaced after 7 days. The number of resistant colonies was quantified by fixing, staining and counting the colonies on each plate as has been previously described (Liu et al., 2004).
References
- Alexeyev M.F., Shokolenko I.N. Mini‐Tn10 transposon derivatives for insertion mutagenesis and gene delivery into the chromosome of gram‐negative bacteria. Gene. 1995;160:59–62. doi: 10.1016/0378-1119(95)00141-r. [DOI] [PubMed] [Google Scholar]
- Baus J., Liu L., Heggestad A.D., Sanz S., Fletcher B.S. Hyperactive transposase mutants of the Sleeping Beauty transposon. Mol Ther. 2005;12:1148–1156. doi: 10.1016/j.ymthe.2005.06.484. [DOI] [PubMed] [Google Scholar]
- Bird A.P. CpG islands as gene markers in the vertebrate nucleus. Trends Genet. 1987;3:342–347. [Google Scholar]
- Butler C.A., Gotschlich E.C. High‐frequency mobilization of broad‐host‐range plasmids into Neisseria gonorrhoeae requires methylation in the donor. J Bacteriol. 1991;173:5793–5799. doi: 10.1128/jb.173.18.5793-5799.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Lenert P., Weeratna R., McCluskie M., Wu T., Davis H.L., Krieg A.M. Identification of methylated CpG motifs as inhibitors of the immune stimulatory CpG motifs. Gene Ther. 2001;8:1024–1032. doi: 10.1038/sj.gt.3301482. [DOI] [PubMed] [Google Scholar]
- Cornelie S., Hoebeke J., Schacht A.M., Bertin B., Vicogne J., Capron M., Riveau G. Direct evidence that toll‐like receptor 9 (TLR9) functionally binds plasmid DNA by specific cytosine‐phosphate‐guanine motif recognition. J Biol Chem. 2004a;279:15124–15129. doi: 10.1074/jbc.M313406200. [DOI] [PubMed] [Google Scholar]
- Cornelie S., Poulain‐Godefroy O., Lund C., Vendeville C., Ban E., Capron M., Riveau G. Methylated CpG‐containing plasmid activates the immune system. Scand J Immunol. 2004b;59:143–151. doi: 10.1111/j.0300-9475.2004.01373.x. [DOI] [PubMed] [Google Scholar]
- Guzman L.M., Belin D., Carson M.J., Beckwith J. Tight regulation, modulation, and high‐level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh C.L. Dependence of transcriptional repression on CpG methylation density. Mol Cell Biol. 1994;14:5487–5494. doi: 10.1128/mcb.14.8.5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinman D.M., Barnhart K.M., Conover J. CpG motifs as immune adjuvants. Vaccine. 1999;17:19–25. doi: 10.1016/s0264-410x(98)00151-0. [DOI] [PubMed] [Google Scholar]
- Krieg A.M. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. 2002;20:709–760. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- Krieg A.M., Wu T., Weeratna R., Efler S.M., Love‐Homan L., Yang L. Sequence motifs in adenoviral DNA block immune activation by stimulatory CpG motifs. Proc Natl Acad Sci USA. 1998;95:12631–12636. doi: 10.1073/pnas.95.21.12631. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Sanz S., Heggestad A.D., Antharam V., Notterpek L., Fletcher B.S. Endothelial targeting of the Sleeping Beauty transposon within lung. Mol Ther. 2004;10:97–105. doi: 10.1016/j.ymthe.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Loisel S., Le Gall C., Doucet L., Ferec C., Floch V. Contribution of plasmid DNA to hepatotoxicity after systemic administration of lipoplexes. Hum Gene Ther. 2001;12:685–696. doi: 10.1089/104303401300057405. [DOI] [PubMed] [Google Scholar]
- Luthman H., Magnusson G. High efficiency polyoma DNA transfection of chloroquine treated cells. Nucleic Acids Res. 1983;11:1295–1308. doi: 10.1093/nar/11.5.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichol K., Pearson C.E. CpG methylation modifies the genetic stability of cloned repeat sequences. Genome Res. 2002;12:1246–1256. doi: 10.1101/gr.74502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renbaum P., Abrahamove D., Fainsod A., Wilson G.G., Rottem S., Razin A. Cloning, characterization, and expression in Escherichia coli of the gene coding for the CpG DNA methylase from Spiroplasma sp. strain MQ1(M.SssI) Nucleic Acids Res. 1990;18:1145–1152. doi: 10.1093/nar/18.5.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubeler D., Lorincz M.C., Cimbora D.M., Telling A., Feng Y.Q., Bouhassira E.E., Groudine M. Genomic targeting of methylated DNA: influence of methylation on transcription, replication, chromatin structure, and histone acetylation. Mol Cell Biol. 2000;20:9103–9112. doi: 10.1128/mcb.20.24.9103-9112.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yew N.S., Zhao H., Przybylska M., Wu I.H., Tousignant J.D., Scheule R.K., Cheng S.H. CpG‐depleted plasmid DNA vectors with enhanced safety and long‐term gene expression in vivo. Mol Ther. 2002;5:731–738. doi: 10.1006/mthe.2002.0598. [DOI] [PubMed] [Google Scholar]
- Yusa K., Takeda J., Horie K. Enhancement of Sleeping Beauty transposition by CpG methylation: possible role of heterochromatin formation. Mol Cell Biol. 2004;24:4004–4018. doi: 10.1128/MCB.24.9.4004-4018.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]