

Summary
Mutants were produced in the A‐domain of HbpR, a protein belonging to the XylR family of σ54‐dependent transcription activators, with the purpose of changing its effector recognition specificity from 2‐hydroxybiphenyl (2‐HBP, the cognate effector) to 2‐chlorobiphenyl (2‐CBP). Mutations were introduced in the hbpR gene part for the A‐domain via error‐prone polymerase chain reaction, and assembled on a gene circuitry plasmid in Escherichia coli, permitting HbpR‐dependent induction of the enhanced green fluorescent protein (egfp). Cells with mutant HbpR proteins responsive to 2‐CBP were enriched and separated in a flow cytometry‐assisted cell‐sorting procedure. Some 70 mutants were isolated and the A‐domain mutations mapped. One of these had acquired true 2‐CBP recognition but reacted hypersensitively to 2‐HBP (20‐fold more than the wild type), whereas others had reduced sensitivity to 2‐HBP but a gain of 2‐CBP recognition. Sequencing showed that most mutants carried double or triple mutations in the A‐domain gene part, and were not located in previously recognized conserved residues within the XylR family members. Further selection from a new mutant pool prepared of the hypersensitive mutant did not result in increased 2‐CBP or reduced 2‐HBP recognition. Our data thus demonstrate that a one‐step in vitro‘evolutionary’ adaptation of the HbpR protein can result in both enhancement and reduction of the native effector recognition.
Introduction
Transcription regulatory proteins find increasingly use in artificial genetic circuitry in order to measure and report the presence of specific chemical signals (for recent reviews, see Belkin, 2003; Harms et al., 2006; Sørensen et al., 2006). In its simplest form, such a circuitry consists of a transcription regulator acting on a dedicated promoter/operator which is coupled to a promoterless reporter gene (van der Meer et al., 2004). The DNA construct is subsequently embedded in a suitable bacterial strain or eukaryotic cell line forming the assay ‘vehicle’, hence, the terminology ‘whole cell living biosensors’, ‘bioreporter cells’ or just biosensors (Burlage, 1997). Their use can be manifold and scientific literature on the topic is plentiful. Bacterial bioreporters, for instance, have been applied for toxicity assessment by using known toxicity, cellular damage or stress‐signalling pathways (Belkin, 1998; Van Dyk et al., 2001; Dardenne et al., 2007). They have also been designed for analysing the bioavailable fraction of heavy metals by applying regulators involved in heavy metal detoxification (Corbisier et al., 1999; Tauriainen et al., 2000). Finally, bacterial biosensors for organic compounds were developed by taking regulators and promoters implicated in the compounds' metabolism (King et al., 1990; Sticher et al., 1997; Wise and Kuske, 2000; Stiner and Halverson, 2002; Werlen et al., 2004). More recent gene transcription circuitry examples have solely focused on studying transcriptional noise or oscillation in gene networks without pertinent bioreporting purposes (Elowitz et al., 2002; Rosenfeld et al., 2005).
In most of the circuitry used for bioreporter construction, transcription activators play an essential role as sensing element and as trigger for reporter gene expression. The specificities of the transcription activator to recognize chemical ‘effectors’ determine to a large extent those of the bioreporter ‘assay’ and they are usually taken for granted, having been acquired by mining of existing genes, proteins and organisms (van der Meer et al., 2004). While this was initially justifiable it has to a certain extent limited the applicability of especially bacterial bioreporters, which all require different incubation conditions, depending on the type of transcription activator, reporter protein, vector usage or host strain. One approach to overcome this problem and standardize bioreporter usage for different chemical targets would be to engineer new and different specificities in one type of transcription activator. It has long been known that transcription regulator mutants with different effector recognition properties can be produced (Ramos et al., 1986), but their selection is a cumbersome process and is usually coupled to a series of inducible killing factors, to which escape mutations may arise (Garmendia et al., 2001; Galvao and de Lorenzo, 2005; 2006; Mohn et al., 2006).
Here we describe the use of a non‐selective process of enriching and separating transcription regulator mutants by flow cytometry‐assisted cell sorting (FACS). Cytometry‐assisted cell sorting has previously been demonstrated to be a useful single‐cell screening approach for expression in metagenomic libraries (Uchiyama et al., 2005), or for bacterial promoters with appropriate expression characteristics (Alper et al., 2005). The protein we studied is the NtrC‐type σ54‐dependent transcription activator HbpR, originating in ‘Pseudomonas azelaica’ (Jaspers et al., 2000). HbpR is distantly related to XylR and DmpR, the activators involved in the toluene and phenol pathways, respectively, of Pseudomonas putida (Skärfstad et al., 2000; Garmendia et al., 2001). Proteins of this class are proposed to consist of three major subdomains, one of which, the A‐domain, is necessary for effector recognition (O'Neill et al., 1998). In contrast to XylR, DmpR and other known proteins of this class, HbpR recognizes biaromatic structures, such as 2‐hydroxybiphenyl (2‐HBP), 2,2′‐dihydroxybiphenyl, 2‐aminobiphenyl and 2‐hydroxydiphenylmethane (Jaspers et al., 2000). In the presence of 2‐HBP, HbpR activates the otherwise silent promoters in front of the hbpC and hbpD genes, which code for enzymes involved in 2‐HBP degradation (Jaspers et al., 2001a).
Our goal here was to study whether we could change the specificity of HbpR for 2‐HBP to 2‐chloro‐ and 2‐bromobiphenyl (2‐CBP and 2‐BBP), and perhaps to Aroclor (a technical mixture of higher chlorinated biphenyls) and Triclosan (a widely used disinfectant). No currently known transcription regulators are capable of recognizing those compounds. In order to isolate such mutants, we first constructed a gene circuitry in Escherichia coli, in which HbpR activates the egfp gene transcriptionally fused to the hbpC promoter (Pc). The gene for hbpR was modified in such a way that the part coding for the A‐domain could be easily replaced by mutant A‐domain DNAs, which were produced in vitro by the error‐prone polymerase chain reaction (PCR). Escherichia coli cells containing HbpR mutants inducible by non‐cognate effectors would then ideally produce egfp and be separable by FACS.
Results
Construction of an inducible, HbpR‐dependent and A‐domain cassette vector with egfp as reporter gene
One of the premises for the success of our strategy was a workable genetic circuit in E. coli, consisting of the hbpR gene, a system to replace the hbpR A‐domain part in a single step, and the egfp gene under control of the HbpR‐dependent Pc promoter. The circuit was assembled on the plasmid vector pPROBE‐gfp‐tagless (Fig. 1), a moderate‐copy‐number plasmid in E. coli(Miller et al., 2000). The A‐domain part of hbpR plus a small part of the hbpR promoter was excisable by digestion with SacI and BamHI at the unique sites created for this purpose. In addition, a His6 translation fusion was created upstream of the native hbpR start site to allow protein purification if necessary. Testing of the E. colistrain containing this plasmid pHBP269A0 showed that it was inducible by 2‐HBP; egfp expression measured in flow cytometry (FC) was fourfold higher at 2 µM 2‐HBP and 2.5 h induction time than the negative control (Fig. 2A). Signal saturation occurred at approximately 20 µM, at which point the cells fluoresced up to 100 times more than the negative control. For further experiments, therefore, we limited the 2‐HBP concentration to 20 µM. Egfp fluorescence was not significantly different from the negative control when E. coli (pHBP269A0) was incubated with 2‐CBP, 2‐BBP, biphenyl (BP), naphthalene, Aroclor or Triclosan (Table S1, Fig. 3A).
Figure 1.
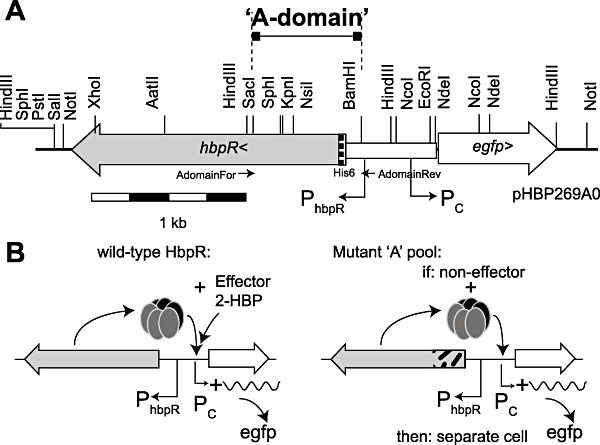
Construction of the gene circuitry and schematic procedure for HbpR mutant selection. A. Location, direction and sizes of hbpR and egfp genes on plasmid pHBP269A0 and the cassette exchanging the A‐domain. Direction of relevant promoters and position of restriction sites are indicated. B. Wild‐type HbpR protein – according to current hypothesis depicted as hexamer – reacting with 2‐HBP as effector to activate egfp expression from the Pc promoter (left). Any mutant in the pool of HbpR mutants with exchanged A‐domain fragments responsive to the new effector will similarly lead to egfp production (right) and can be separated by FACS (see Fig. 4).
Figure 2.
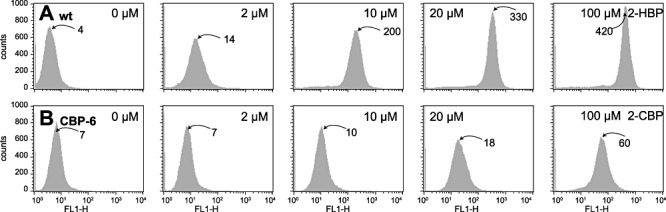
Cell counts as a function of egfp fluorescence (per channel FL1‐H) and 2‐HBP concentration (for E. coli‐expressing wild‐type HbpR protein) (A) and 2‐CBP (for cells expressing mutant CBP‐6) (B). Induction times of cultures 2.5 h at 37°C. Note the logarithmic scale of fluorescence. Values near histogram peaks indicate the corresponding median fluorescence.
Figure 3.
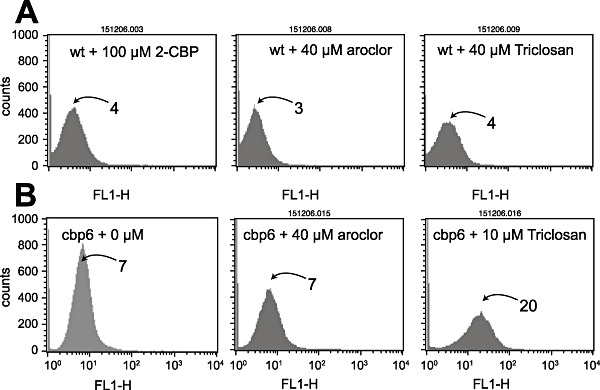
Culture responses as egfp fluorescence per FL1‐H channel after induction with 2‐CBP, Aroclor 1254 or Triclosan. A. Escherichia coli‐expressing wild‐type HbpR from plasmid pHBP269A0. B. Mutant CBP‐6. Note the logarithmic scale of fluorescence values. Panel B1 (CBP‐6 without inducer) is a repetition from Fig. 2 for ease of comparison. Numbers near histogram peaks indicate the corresponding fluorescence value (arbitrary units of the FACS Calibur).
Mutant production and selection
Pools of A‐domain mutants were produced by error‐prone PCR, digested at the unique restriction sites and used to replace the native A‐domain in pHBP269A0. Approximately 106possible mutants in E. coli– not necessarily independent ones – were screened by FC. In order to detect and separate those mutants responsive to effectors other than 2‐HBP we used two strategies (Fig. 4). In the first one, the mutant pool was grown and incubated without any chemical effector with the aim to remove any mutants constitutively producing egfp according to a predefined gating (Fig. 4B). The separated population was then regrown, exposed to the effector and again screened by FC, after which the fluorescent cells were separated (Tables S1 and S2). Our results indicated that in the first mutant pool C around 1% of the cells were highly fluorescent and thus possibly constitutively expressed egfp. On the other hand, induction of the mutant pool C with 2‐HBP produced two to three times as many non‐reacting cells as E. coli with wild‐type HbpR, suggesting that many mutations occurred which completely abolished HbpR functioning (Table S1, Fig. 4). Inducing the separated population with either 2‐CBP, 2‐BBP or BP in a second round produced a clear subpopulation of between 4.6% and 6.7% cells with a high fluorescence (Fig. 4B), suggesting that those were mutants with a gain of effector phenotype (Table S1). On the contrary, no significant subpopulation was detected with gain of responsiveness to naphthalene.
Figure 4.
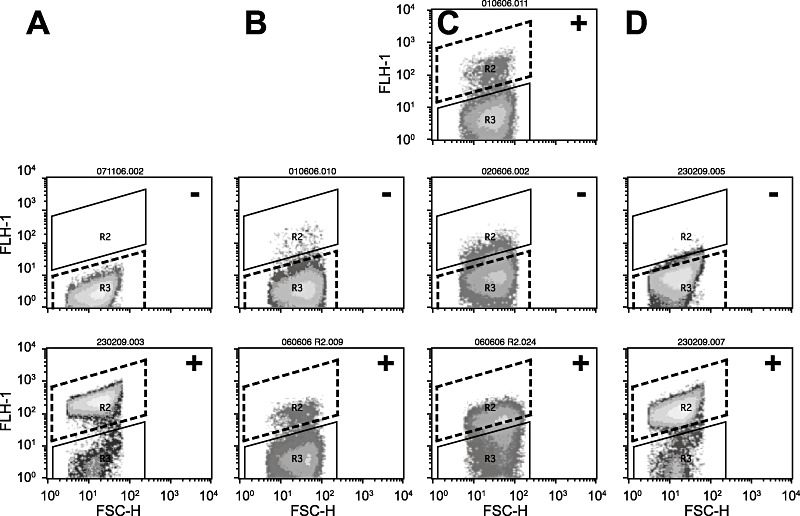
2‐Chlorobiphenyl (2‐CBP) mutant enrichment by flow cytometry‐assisted cell sorting. A. Escherichia coli‐expressing wild‐type HbpR without inducer (middle; ‘−’), and after exposure to 2‐HBP at 20 µM (lower panel; ‘+’). B. Pool C mutants, uninduced – middle; cell recovery (area within dotted line), and induction of recovered cell pool with 100 µM 2‐CBP – bottom; induced part separated (again within area bounded by dotted line). C. Similar as (B), but now first ‘induced’ with 100 µM 2‐CBP – cell sorting, ‘uninduced’– cell sorting, and ‘induced’– cell sorting. D. Example of mutant CBP‐6, uninduced and induced with 100 µM 2‐CBP. Gate definitions correspond to the average fluorescence values listed in Tables S1 and S2. Diagrams express egfp fluorescence per single cell (FLH‐1) versus forward scatter (FSC‐H), both on logarithmic scale.
In the second method, the mutant pool of cells was first induced with a new effector, after which fluorescent cells were sorted according to a predefined gating (Fig. 4C). This population was then regrown and screened again under non‐inducing conditions to sort non‐fluorescent cells. Finally, the non‐fluorescent cells in the second round were again cultivated and induced with the same effector, after which the fluorescent cells were sorted (Fig. 4C). In this case, some 6% of cells seemed responsive to either 2‐CBP, 2‐BBP or BP, but not to naphthalene (Table S2), of which 12–20% were judged constitutively in the second round and finally up to 50% were deemed inducible in the third round. That this was not a general phenomenon is suggested by the screening for naphthalene‐responsive mutants. In this case, no significant enrichment was obtained (Table S2).
Plate screening and isolation of individual mutants
As the FACS machine did not permit isolation of single cells, the finally recovered populations in procedures I and II remained essentially enriched but most likely heterogenous. In the next step, therefore, we separated and cultured the enriched mutant population on agar plates and examined inducibility with the target compound by fluorescent colony formation (Fig. 5A). Via this step, some 1000 mutants were recovered of which 100 were again screened by steady‐state fluorometry (SSF). For 2‐CBP, this resulted in a set of 70 strains with a significantly higher induction ratio than the E. coli carrying the wild‐type HbpR (Fig. 5B). Of these mutants the A‐domain sequence was determined, which showed that multiple different amino acid substitutions gave rise to a 2‐CBP‐responsive phenotype (Table 1). Also A‐domain sequences of various randomly picked constitutively expressing egfp mutants were determined, which demonstrated that various combinations of different amino acid substitutions could produce a constitutive phenotype. Of the enriched and isolated mutants with 2‐CBP phenotype various classes could be discerned: those that gained responsiveness to 2‐CBP while maintaining the same level of inducibility with 2‐HBP (and other native effectors), those that gained 2‐CBP but lost 2‐HBP responsiveness and those that had obtained a higher background expression level on top of which came a gain in responsiveness to 2‐CBP (Table 1, Fig. 5B). One of the mutants with highest response to 2‐CBP (i.e. CBP‐6; I101V, D128N) was reconstructed in two steps to verify if both mutations had indeed been necessary. This showed that the single mutations resulted in only very slightly increased responsiveness to 2‐CBP but that the combination of both acted synergistically. Furthermore, this reconstruction verified that no other changes had been taking place in the HbpR A‐domain which we might have missed (Fig. 6). Mutant CBP‐6 showed approximately linear dose‐dependent egfp induction between 2 and 100 µM of 2‐CBP, at which concentration it became ninefold induced as measured by FC (Figs 2B and 7). Interestingly, mutants separated on the basis of increased responsiveness to 2‐CBP were also more inducible with 2‐BBP or BP, suggesting that these phenotypes go together (not shown). In addition, mutant CBP‐6 showed slightly but significantly higher response in FC to 10 µM Triclosan (Fig. 3B). Higher concentrations of this compound could not be tested because of toxicity to E. coli cells.
Figure 5.
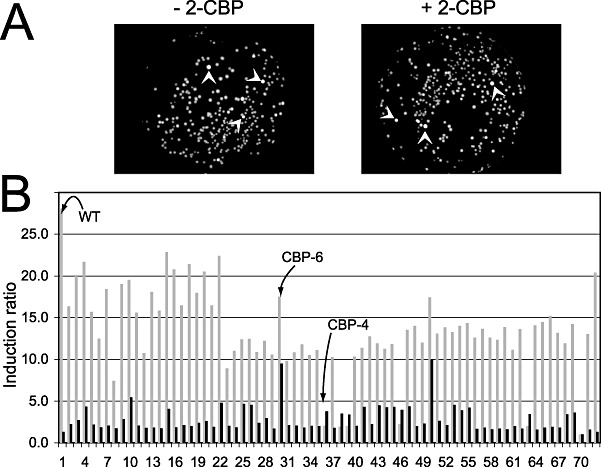
Purification of HbpR mutants responsive to 2‐CBP. A. Fluorescence from E. coliFACS‐enriched mutant pool colonies (left, constitutive mutants) or after exposition to 2‐CBP on filter membrane (right). Arrows point to particularly bright fluorescing colonies such as selected for further study. B. Fluorimetry analysis of some 70 mutants purified via FACS and fluorescence plates after exposure to 2‐HBP (light grey bars) or 2‐CBP (dark grey) at 20 and 100 µM respectively. The ordinate shows induction factor compared with cells in buffer only. Culture number 1 and 73 are expressing HbpR wild type.
Table 1.
Relevant HbpR mutants enriched by FACS.
| Mutationsa | Designation | Fold inductionb with | NI valuec | |
|---|---|---|---|---|
| 2‐HBP | 2‐CBP | |||
| None | Wild type | 22 | 1 | 500 |
| Type: gain of function | ||||
| W3931R | CBP‐1 | 20 | 2 | 400 |
| H2315Q | CBP‐3 | 7 | 1.5 | 400 |
| T8072A, Q198186L, D222226E | CBP‐4 | 2 | 4 | 400 |
| T8072S | CBP 1‐10 | 23 | 2 | 500 |
| I10193V, D128116N | CBP‐6 | 20 | 11 | 500 |
| Type: gain of function, elevated background | ||||
| D4133Y | CBP 10‐II | 19 | 5 | 1200 |
| N7K, H108100Y | CBP 6‐II | 2 | 3 | 2000 |
| Type: elevated background | ||||
| H153141L | CBP 11‐II | 16 | – | 700 |
| W162150R, Q225228L | CBP 12‐II | 16 | – | 600 |
| Type: loss of inducibility, elevated background | ||||
| S3224F, E5143G, H153159Q, S157163P | CBP 2‐II | 5 | – | 1800 |
| K2820M, F7870I, A174162T | CBP 2‐I | 3 | – | 6000 |
| Y5749K | CBP 8‐II | 6 | – | 1800 |
| G6557S, M204192I | CBP 9‐II | 6 | – | 1600 |
| T7971A, A202190G | CBP 1‐II | 3 | – | 3300 |
| D128116Y, P179167L | CBP 5‐II | 5 | – | 3400 |
| W205193R | CBP 3‐II | 4 | – | 3100 |
| A15V, H23R, E9991D, I117106 K, E186174D, A206194K | CBP 4‐I | 1.2 | – | 3000 |
Residue numbering of HbpR; as subscript, corresponding numbering of the XylR amino acid sequence.
Induction factor for 2‐hydroxybiphenyl at 20 µM compared with control in MOPS buffer; for 2‐chlorobiphenyl at 100 µM.
NI value, fluorescence value in steady‐state fluorimetry (arbitrary units given by the instrument) under non‐induced conditions, normalized for the culture turbidity at 600 nm. Values comparable to those in Fig. 7.
Figure 6.
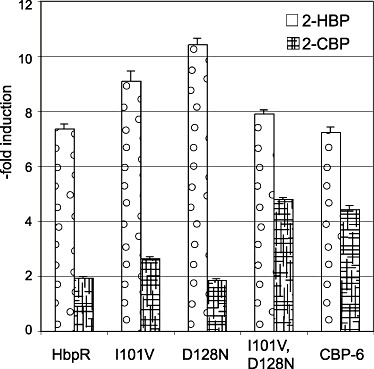
Culture‐induced egfp fluorescence of E. coli‐expressing HbpR wild type, mutants D128N, I101V or CBP‐6 and the double mutant I101V, D128N after 2.5 h exposure at 30°C to 2‐HBP or 2‐CBP at 16 and 80 µM respectively. Ordinate expressed as ‐fold induction, i.e. compared with cultures in buffer only. Error bars represent the additive errors in both sample and blanc measurements.
Figure 7.
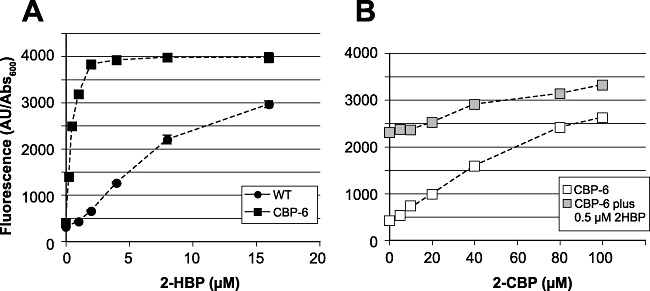
Egfp fluorimetry analysis of E. coli‐expressing HbpR wild type or mutant CBP‐6, as a function of 2‐HBP or 2‐CBP concentration. Culture fluorescence expressed in arbitrary units (given by the machine) divided by the culture turbidity at 600 nm. A. Hypersensitivity response of mutant CBP‐6 compared with wild‐type HbpR. B. Calibration of mutant CBP‐6 to 2‐CBP without and with 0.5 µM 2‐HBP added (grey symbols). Note that error bars may be smaller than the symbol size.
Continued mutagenesis starting from mutant CBP‐6
We then studied whether mutant CBP‐6 would be a good starting point for a second round of error‐prone mutagenesis for selection of improved responsiveness to higher chlorinated biphenyls or for reduced responsiveness to 2‐HBP. Under uninduced conditions, some 3% of the cells in this population (‘pool D’) displayed high fluorescence (hence: probable constitutives), whereas some 35–45% remained non‐inducible with 2‐HBP (Tables S1 and S3). Essentially no improvement was observed in the mutant CBP‐6 pool compared with pure CBP‐6 after exposure to Aroclor at 40 µM (Tables S1 and S3), although the population fluorescence distribution was wider. The same was observed for response of the pool D mutants to 10 µM Triclosan (Table S3), although it cannot be excluded that mutants with higher responsiveness may have been present which we were unable to separate. In order to obtain mutants with possibly further reduced responsiveness to 2‐HBP while retaining recognition of 2‐CBP, we separated non‐induced cells from mixtures induced with 20 µM 2‐HBP and induced cells from mixtures exposed to 100 µM 2‐CBP (Tables S4 and S5). These sorted populations were regrown separately, after which they were exposed to 2‐CBP or 2‐HBP respectively. In this case, a small proportion of cells from the pool of non‐induced cells with 2‐HBP were highly induced with 2‐CBP, and a small proportion of cells previously reacting to 2‐CBP were poorly induced with 2‐HBP. However, when these populations were again separated, regrown and tested for induction with either 2‐HBP or 2‐CBP, we found that they reacted to both (Tables S4 and S5). Therefore, we must assume that no mutants with improved recognition to 2‐CBP and loss of recognition to 2‐HBP were present in the pool D mixture.
Mutant CBP‐6 is hypersensitive to 2‐HBP
Interestingly, although mutant CBP‐6 showed dose‐dependent egfp expression to 2‐CBP (Figs 2 and 7), it appeared to be some 20 times more sensitive to 2‐HBP than HbpR wild type (Fig. 7A). The maximum output level remained the same, however. Exposure of cells with mutant CBP‐6 to mixtures of 2‐CBP and 2‐HBP showed an additive response (although the combined reactivity was less than the sum of each of the individual exposed concentrations), suggesting that 2‐CBP is a true effector for this protein (Fig. 7B). Despite the reported aqueous solubility of 2‐CBP of around 6 mg l−1 (32 µM; Ward, 1986), we could still detect an increase of response of the CBP‐6 mutant above 30 µM up to 100 µM. As these are nominal concentrations (i.e. the calculated concentration from the amount of 2‐CBP added via DMSO), we cannot exclude that some volatilization of 2‐CBP occurred from the assay, which lowered the effective aqueous concentration. The gain of sensitivity at lower concentrations of 2‐HBP is also an interesting improvement compared with the HbpR wild‐type protein.
Discussion
Our results demonstrated conclusively that mutant transcription activator proteins can easily be produced, screened and separated via a FACS procedure exploiting the capability of potential mutants to activate egfp expression in E. coli. As single‐cell screening on FC is rapid and accurate, it was facile to investigate a potential mutant pool of some 106 cells in short time. On the other hand, the cell sorter on this FACS machine did not allow true single‐cell sorting (i.e. separation) and, therefore, a consecutive step had to be designed to separate individual mutants within the enriched mutant pool. We found that induction of colonies grown on agar plates and subsequent imaging of the fluorescence intensities of individual colonies was sufficiently powerful to distinguish non‐inducible, inducible and constitutive phenotypes. Accurate quantification of fluorescence intensities as a function of colony size or age, however, was not successful in our hands. Independent repetitions of the whole procedure (i.e. error‐prone PCR mutagenesis, induction of the mutant pool, FACS and agar plate induction) produced similar frequencies of 2‐CBP and 2‐BBP mutant appearance, indicating that this step in protein evolution is reproducible (Tables S1–S3). Both methods (‘uninduced–induced’ or ‘induced–uninduced–induced’) produced similar results and no difference in the types of mutants obtained via either procedure was obvious. Constitutive egfp‐producing mutants contaminated the final sorted populations via both methods, which via this cell sorter device could not be completely resolved from inducible cells. Partly this is inherent to the heterogenous expression nature of egfp in a population of cells (Tecon and van der Meer, 2006), which results in a normal distribution of egfp intensities around an average (see, for example, Fig. 2), and thus overlapping distribution curves for different mutant populations. All in all the rapidity of the procedure strongly outweighed these limitations and many more potentially interesting mutants were isolated than could be screened in detail later on.
Not unexpectedly for proteins of the XylR family (Skärfstad et al., 2000; Wise and Kuske, 2000; Garmendia et al., 2001; Sarand et al., 2001), but the first time for the HbpR protein, we recovered mutants with various phenotypes (e.g. loss of wild‐type recognition, increased background, gain of new effector recognition, sensitivity gain), which suggests that this type of protein has enormous flexibility in its adaptive landscape. Of importance for our study were those mutants with increased effector recognition and gain of sensitivity. Surprisingly, and in the light of previous mutations of importance found in XylR and DmpR, almost none of the mutants isolated in this study in HbpR mapped in the conserved residues of this protein family, or overlapped with previously known mutations (Fig. S1). Notable exceptions were mutations in the conserved Trp residues at position 39 in HbpR (or 31 in XylR equivalent), at position 162 and 205 (W193 in XylR). Furthermore, the mutation in the Asp residue at position 128 (116 for XylR) which contributed to the 2‐CBP phenotype in mutant CBP‐6 occurred at a relatively conserved position. Those mutations which provided the best 2‐CBP recognition were of conservative nature, replacing Ile101 by a Val, and Asp128 by Asn. This phenomenon of conservative replacements for relaxed recognition has also been noticed for mutants in XylR (Skärfstad et al., 2000; Wise and Kuske, 2000; Garmendia et al., 2001; Sarand et al., 2001). Mutations favouring 2‐CBP responsiveness were more abundant in the so‐called ‘XylR‐N domain’ compared with the V4R domain (Fig. S1). Residue T80 was involved twice in a 2‐CBP gain of phenotype. On the contrary, T79 and F78 mutations were implicated in constitutive behaviour of HbpR. Many of the mutations causing constitutive activation involved non‐conservative amino acid changes (Table 1).
Interestingly, the gain of effector recognition for 2‐CBP in mutant CBP‐6 was accompanied by a hypersensitive reaction to 2‐HBP itself. Although we were unable to detect a response of wild‐type HbpR to 2‐CBP, this may suggest that an initially very low effector recognition was boosted by the 20‐fold increase in overall sensitivity of the protein to its effector. On the other hand, the response to 2‐CBP and 2‐HBP was additive, meaning that 2‐CBP is recognized as a true effector and not as a consequence of an overall relaxation of binding. One possibility to explain hypersensitive nature would be that the effector has a more rapid turnover at the binding site. The triple mutations resulting in the CBP‐4 phenotype (with reduced recognition of 2‐HBP, Table 1) were not related to those of CBP‐6. The mechanism of recognition gain might therefore be different.
To the best of our knowledge, it is the first time that a regulatory protein that recognizes 2‐CBP (and 2‐BBP) as effectors is isolated. Although the mutant CBP‐6 did not measurably respond to Aroclor 1254 as mixture, this may be mainly due to the extremely low aqueous solubility of this compound mixture [∼28 µg l−1 or 0.1 µM (Wiese and Griffin, 1978)], which we could not improve by oversaturating the assay medium (i.e. 40 µM nominal concentration added). No growth inhibitory effect of Aroclor 1254 in this nominal concentration range was observed in the E. colibioassays. It is therefore difficult to assess, but not unlikely, that also other chlorinated biphenyls are recognized by this protein. The E. coli strain with this reporter construct could thus potentially be exploited as a PCB detector, with a current minimal detected concentration of 1.5 µM 2‐CBP (calculated by linear regression from value of the blanc plus three times standard deviation in the blanc, Fig. 7B). Slightly better and significant response was obtained with Triclosan, a widely used disinfectant, at 10 µM nominal concentration. The mutant strain, therefore, could potentially be applied as Triclosan detector. Potentially, this regulatory mutant could also overcome a recurring obstruction in PCB degradation, namely that strains metabolizing PCBs do not recognize PCBs as inducers and have to be co‐induced with other substrates (Cases and de Lorenzo, 2005). For this to work, the HbpR mutant would have to be expressed in a PCB‐degrading bacterium, and the chlorobiphenyl dioxygenase genes brought under the control of the Pc promoter.
Experimental procedures
Strains, media and general growth conditions
Escherichia coli recombinant strains were generally cultivated on Luria–Bertani (LB) medium (Sambrook and Russell, 2001), supplemented with 50 µg ml−1 kanamycin to select for the presence of the reporter plasmid. For induction experiments E. coli strains were incubated in MOPS medium (i.e. per liter, 10 g 3‐(N‐morpholino)propanesulfonic acid, 1 g of NH4Cl, 0.5 g of NaCl, 0.06 g of Na2HPO4·2H2O, 0.045 g of KH2PO4, 20 mM MgCl2, 1 mM CaCl2 and 2 g of glucose, pH 7). Liquid cultures were generally incubated at 37°C with 180 rpm rotary shaking, except for induction experiments that were carried out at 30°C. Bacterial colonies were grown on LB medium solidified with 1.5% agar and incubated at 30°C or 37°C.
Construction of the hbpR‐Pc::egfp circuitry and of the A‐domain cassette
A reporter plasmid for inducible expression of the egfp gene under control of HbpR (or mutant HbpRs) and allowing single‐step exchange of the hbpR A‐domain gene fragment was assembled on the broad‐host range vector pPROBE‐gfp‐tagless (Miller et al., 2000) as follows. To introduce appropriate restriction sites, the full‐length hbpR gene was amplified by using the PCR and primers 040101 (5′‐gtcgacgcggccgcgcactttcgcacg‐3′) and 040102 (5′‐tgcgcatgctcggaggatccggtttca‐3′), and cloned into pGEM‐T‐Easy (Promega, Catalys AG, Wallisellen, Switzerland). From here, hbpR plus a mutated Pc promoter (Tropel and van der Meer, 2005) was assembled into pPROBE‐tagless, digested with SalI and SacI (resulting in pHBP263). Finally, a shorter fragment of the hbpR gene plus the wild‐type Pc promoter region was amplified as a 1395 bp fragment by using the PCR and primers ‘AdomainFor’ (5′‐GTGACGGAGCACCTTGAACACTG‐3′, nucleotide position 825 in hbpR) and ‘hbp2‐Eco’ (5′‐GAGATTTGAAtTCATTGTTATCAC‐3′, position 15 in hbpC, EcoRI site underlined), digested with SacI and EcoRI, and used to replace the same region in pHBP263. This resulted in plasmid pHBP269. Finally, a His6‐tag was introduced before the start of hbpR by PCR amplification. Hereto first two overlapping fragments of hbpR plus its promoter were amplified: (i) with primers hbpRrevHis (5′‐aggagatcgcc ATG cat cat cat cat cat cacatg aaa tca aat aaa aat aat agc gac g‐3′, underlined: new ribosome binding site; in bold: His6‐codons; capitalized ATG: new start site; italicized atg: original hbpR start site) and hbpRRev9 (5′‐tgcgcatgctcggaggatccggtttca‐3′), and (ii) hbpRforHis (5′‐gtg atg atg atg atg atg CAT tggcgatctcctgggtctcgttgggtttaataattttatt‐3′; partially complementary to hbpRrevHis) plus HbpRFor9 (5′‐gtcgacgcggccgcgcactttcgcacg‐3′). Both PCR products were then purified and mixed as template for a third PCR using primers hbpRFor9 and hbpRrev9. The amplified product was purified, cloned into pGEM‐T‐Easy and from there recovered as a BamHI–SacI fragment, which was used to replace the unique BamHI–SacI fragment in pHBP269. The resulting plasmid was named pHBP269A0. The intactness of the hbpC promoter, of hbpR and of the His6‐tag was confirmed by sequencing.
Error‐prone PCR
In order to introduce random mutations in the hbpR A‐domain gene fragment, we used the GeneMorph II Random Mutagenesis Kit (Stratagene, La Jolla, CA, USA) with primers AdomainFor and AdomainRev (5′‐cagcccgatccatgcacctaca‐3′), according to the instructions of the supplier. Approximately 500 ng of supercoiled plasmid template (pHBP269A0, ∼10 kb size) was used in a 30‐cycle reaction, after which the 0.9 kb PCR products were separated on and purified from agarose gel. The pool of PCR products was then digested with BamHI and SacI, and ligated to a batch of pHBP269A0 plasmid digested with the same enzymes and checked for absence of self‐ligation. The ligation mixture was transformed by electroporation to E. coli DH5α competent cells, and 1/10 volume of this pool (100 µl) was plated to check transformation efficiency. To the rest of the transformation mixture glycerol was added at a final concentration of 15% (v/v), after which the cellular mutant pool was stored frozen at −80°C until analysis by flow cytometry. Several such pools were prepared from independently performed Mutazyme reactions, either on pHBP269A0 as template or on pHBP269AM3 (expressing the HbpR mutant CBP‐6 responsive to 2‐CBP, see below).
HbpR activation assays
Single pure bacterial cultures or mutant pool cultures were grown for 16 h at 37°C in LB medium plus kanamycin and diluted 50‐fold in fresh medium of the same. Cells were then allowed to grow until a culture turbidity of 0.4 (at 600 nm), after which they were centrifuged at 2500 g and re‐suspended in the same volume of MOPS buffer. This cell suspension was used for induction assays. For induction, between 1 and 3 ml of E. coli cell suspension was mixed with an aromatic effector [in a concentration range between 0.25 and 100 µM, supplied via dimethylsulfoxide (DMSO) at maximum 1% (v/v) final concentration] in capped 5 or 10 ml glass vials, which were sealed by Teflon‐lined stoppers in the case of volatile effector compounds. Cells were incubated at 30°C with 180 rpm rotary shaking for periods of 2.5–4 h, after which the egfp fluorescence signal was assayed. Incubations with DMSO and water only served as negative, non‐induced controls.
Flow cytometry and cell sorting
Flow cytometry and cell sorting of E. coli cells expressing egfp from the HbpR inducible Pc promoter were performed on a FACS Calibur system (BD Biosciences, Erembodegem, Belgium). The FC was extensively cleaned and washed in 0.22 µm filtered PBS before each run (PBS is phosphate‐buffered saline, per liter, 8 g of NaCl, 0.2 g of KCl, 1.15 g of Na2HPO4·7H2O and 0.2 g of KH2PO4, pH 7.4). Escherichia coli cells exposed to chemical effectors as described above were diluted to approximately 106 cells per millilitre in PBS in a 3 ml sample tube and aspired at ∼12 µl min−1 onto the FC. Counting and detection was set to 100 000 events, using the detectors ‘Forward scatter’ (FSC), ‘Side scatter’ (SSC) and ‘Fluorescence’ (FL1‐H, λex = 488 nm, λem = 530 nm). Data were acquired in the FC‐native program CellQuest Pro (version 4.0.2, BD Biosciences). For cell sorting, a sorting gate was defined on the basis of FSC and FL1‐H parameters, and cells sorted in ‘exclusion’ mode from 100 000 event counts at 300 events per second. Sorted cells were thus collected in sterile PBS, generally between 10 and 20 ml, to which subsequently 1/10 of volume of 10‐fold concentrated LB medium was added, plus kanamycin up to 50 µg ml−1. Sorted cells were then again cultivated for 16 h at 37°C as usual and either passed for a new induction round or plated on LB solid agar plates with kanamycin for colony screening.
Colony induction
Cells sorted from FACS were finally plated for inspection of colony fluorescence without and with exposure to chemical effectors. Colonies were grown for 12–16 h at 37°C at a number of 100–200 per plate. The colonies were then lifted from the plate by placing a nylon membrane (type Hybond‐N, GE Healthcare Europe GmbH, Munich, Germany) on the top and, after 10 s contact time, pealing it off with the help of two forceps. The membrane with colonies facing up was then placed in a glass Petri dish onto a 0.5 ml droplet containing MOPS medium and 10–100 µM of chemical effector. After 4–16 h of contact time at 30°C the colonies were illuminated at 470 nm on a Dark ReaderTM transilluminator (Clare Chemical Research, CO, USA) and digitally photographed. If necessary, fluorescence intensity of colonies was quantified on TIF images with the help of a subroutine of the program Metamorph (version 7.03r, Visitron Systems GmbH, Puchheim, Germany) as described elsewhere (Jaspers et al., 2001b).
Steady‐state fluorometry
Escherichia coli egfp expression from pHBP269A0 or its mutant derivatives were measured in a steady‐state fluorimeter (SSF, FluoStar Galaxy, BMG Labtech GmbH, Offenburg, Germany) at 480 nm excitation and 520 nm emission wavelengths. For SSF, 0.2 ml of induced cell culture was transferred to a 96‐well plate (type µClear 96K, 655090, Greiner Bio‐One Vacuette Schweiz GmbH, St. Gallen, Switzerland) and fluorescence measured with a 56 s intensity cycle (10 flashes, 0.2 s delay, gain 20) against a blank of only MOPS medium. Culture turbidity was measured in the same well at 600 nm and used to standardize the fluorescence emission values.
DNA cloning techniques and DNA sequencing
Recombinant DNA techniques were all carried out according to well‐established procedures (Sambrook and Russell, 2001). Polymerase chain reaction mixtures were prepared as suggested by the suppliers of the Taq Polymerase (Qiagen AG, Hombrechtikon, Switzerland) and run on GeneAmp PCR System thermocyclers (Applied Biosystems, CA, USA). DNA was sequenced using the BigDye Terminator cycling method (version 3.1, Applied Biosystems) and analysed on ABI Prism 3100 capillary sequencers (Applied Biosystems). Kits for purification of PCR products, of DNA fragments from agarose gels, and for isolation of plasmid DNAs from E. coli were used according to the specifications given by the suppliers (Qiagen, Promega).
Chemical substances
All chemicals except Aroclor were obtained from Sigma‐Aldrich and used in the highest‐purity available. Stock solutions were prepared at 20 and 300 mM in DMSO, which were kept at 4°C in the dark. Chemicals tested for induction included 2‐HBP, 2‐CBP, 2‐BBP, BP, naphthalene and Triclosan (CAS RN 3380‐34‐5). A small quantity of Aroclor 1254 was obtained via Christian Schaffner of the Eawag (Dübendorf, Switzerland) and originated from Monsanto (PL2‐124, CAS RN 11097‐69‐1, Cl 54%).
Acknowledgments
The authors thank Alexandra Baehler, Debora Delgado, Nicole Zufferey, Mady Danfaga and Charles Vidoudez for their help in parts of the experiments described here. This work was supported by the European Community fifth Framework Program in which the Swiss participants are funded by the Swiss Federal Office of Education and Sciences (Contract No. QLK3‐CT‐2002‐01923), and by the sixth Framework Program (Contract 018391).
Supplementary material
The following supplementary material is available for this article online:
Graphical positioning of obtained mutants on the HbpR amino add sequence.
Tables S1-S5. Separation percentages of the different mutant pools by flow cytometry.
References
- Alper H., Fischer C., Nevoigt E., Stephanopoulos G. Tuning genetic control through promoter engineering. Proc Natl Acad Sci USA. 2005;102:12678–12683. doi: 10.1073/pnas.0504604102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkin S. A panel of stress‐responsive luminous bacteria for monitoring wastewater toxicity. Methods Mol Biol. 1998;102:247–258. doi: 10.1385/0-89603-520-4:247. [DOI] [PubMed] [Google Scholar]
- Belkin S. Microbial whole‐cell sensing systems of environmental pollutants. Curr Opin Microbiol. 2003;6:206–212. doi: 10.1016/s1369-5274(03)00059-6. [DOI] [PubMed] [Google Scholar]
- Burlage R.S. Emerging technologies: bioreporters, biosensors, and microprobes. In: Hurst C.J., Knudsen G.R., McInerney M.J., Stetzenbach L.D., Walter M.V., editors. ASM Press; 1997. pp. 115–123. [Google Scholar]
- Cases I., De Lorenzo V. Genetically modified organisms for the environment: stories of success and failure and what we have learned from them. Int Microbiol. 2005;8:213–222. [PubMed] [Google Scholar]
- Corbisier P., Van Der Lelie D., Borremans B., Provoost A., De Lorenzo V., Brown N.L. Whole cell‐ and protein‐based biosensors for the detection of bioavailable heavy metals in environmental samples. Anal Chim Acta. 1999;387:235–244. et al. [Google Scholar]
- Dardenne F., Smolders R., De Coen W., Blust R. Prokaryotic gene profiling assays to detect sediment toxicity: evaluating the ecotoxicological relevance of a cell‐based assay. Environ Sci Technol. 2007;41:1790–1796. doi: 10.1021/es062162m. [DOI] [PubMed] [Google Scholar]
- Elowitz M.B., Levine A.J., Siggia E.D., Swain P.S. Stochastic gene expression in a single cell. Science. 2002;297:1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- Galvao T.C., De Lorenzo V. Adaptation of the yeast URA3 selection system to gram‐negative bacteria and generation of a ΔbetCDE Pseudomonas putida strain. Appl Environ Microbiol. 2005;71:883–892. doi: 10.1128/AEM.71.2.883-892.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvao T.C., De Lorenzo V. Transcriptional regulators a la carte: engineering new effector specificities in bacterial regulatory proteins. Curr Opin Biotechnol. 2006;17:34–42. doi: 10.1016/j.copbio.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Garmendia J., Devos D., Valencia A., De Lorenzo V. A la carte transcriptional regulators: unlocking responses of the prokaryotic enhancer‐binding protein XylR to non‐natural effectors. Mol Microbiol. 2001;42:47–59. doi: 10.1046/j.1365-2958.2001.02633.x. [DOI] [PubMed] [Google Scholar]
- Harms H., Wells M.C., Van Der Meer J.R. Whole‐cell living biosensors – are they ready for environmental application? Appl Microbiol Biotechnol. 2006;70:273–280. doi: 10.1007/s00253-006-0319-4. [DOI] [PubMed] [Google Scholar]
- Jaspers M.C., Suske W.A., Schmid A., Goslings D.A., Kohler H.P., Van Der Meer J.R. HbpR, a new member of the XylR/DmpR subclass within the NtrC family of bacterial transcriptional activators, regulates expression of 2‐hydroxybiphenyl metabolism in Pseudomonas azelaica HBP1. J Bacteriol. 2000;182:405–417. doi: 10.1128/jb.182.2.405-417.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspers M.C., Schmid A., Sturme M.H., Goslings D.A., Kohler H.P., Van der Meer J.R. Transcriptional organization and dynamic expression of the hbpCAD genes, which encode the first three enzymes for 2‐hydroxybiphenyl degradation in Pseudomonas azelaica HBP1. J Bacteriol. 2001a;183:270–279. doi: 10.1128/JB.183-1.270-279.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspers M.C.M., Meier C., Zehnder A.J.B., Harms H., Van Der Meer J.R. Measuring mass transfer processes of octane with the help of an alkS‐alkB::gfp‐tagged Escherichia coli. Environ Microbiol. 2001b;3:512–524. doi: 10.1046/j.1462-2920.2001.00218.x. [DOI] [PubMed] [Google Scholar]
- King J.M.H., DiGrazia P.M., Applegate B.M., Burlage R., Sanseverino J., Dunbar P. Rapid, sensitive bioluminescent reporter technology for naphthalene exposure and biodegradation. Science. 1990;249:778–781. doi: 10.1126/science.249.4970.778. , and et al. [DOI] [PubMed] [Google Scholar]
- Van Der Meer J.R., Tropel D., Jaspers M.C.M. Illuminating the detection chain of bacterial bioreporters. Environ Microbiol. 2004;6:1005–1020. doi: 10.1111/j.1462-2920.2004.00655.x. [DOI] [PubMed] [Google Scholar]
- Miller W.G., Leveau J.H., Lindow S.E. Improved gfp and inaZ broad‐host‐range promoter‐probe vectors. Mol Plant Microbe Interact. 2000;13:1243–1250. doi: 10.1094/MPMI.2000.13.11.1243. [DOI] [PubMed] [Google Scholar]
- Mohn W.W., Garmendia J., Galvao T.C., De Lorenzo V. Surveying biotransformations with a la carte genetic traps: translating dehydrochlorination of lindane (gamma‐hexachlorocyclohexane) into lacZ‐based phenotypes. Environ Microbiol. 2006;8:546–555. doi: 10.1111/j.1462-2920.2006.00983.x. [DOI] [PubMed] [Google Scholar]
- O'Neill E., Ng L.C., Sze C.C., Shingler V. Aromatic ligand binding and intramolecular signalling of the phenol‐responsive sigma54‐dependent regulator DmpR. Mol Microbiol. 1998;28:131–141. doi: 10.1046/j.1365-2958.1998.00780.x. [DOI] [PubMed] [Google Scholar]
- Ramos J.L., Stolz A., Reineke W., Timmis K.N. Altered effector specificities in regulators of gene expression: TOL plasmid xylS mutants and their use to engineer expansion of the range of aromatics degraded by bacteria. Proc Natl Acad Sci USA. 1986;83:8467–8471. doi: 10.1073/pnas.83.22.8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld N., Young J.W., Alon U., Swain P.S., Elowitz M.B. Gene regulation at the single‐cell level. Science. 2005;307:1962–1965. doi: 10.1126/science.1106914. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Russell D.W. Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Sarand I., Skärfstad E., Forsman M., Romantschuk M., Shingler V. Role of the DmpR‐mediated regulatory circuit in bacterial biodegradation properties in methylphenol‐amended soils. Appl Environ Microbiol. 2001;67:162–171. doi: 10.1128/AEM.67.1.162-171.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skärfstad E., O'Neill E., Garmendia J., Shingler V. Identification of an effector specificity subregion within the aromatic‐responsive regulators DmpR and XylR by DNA shuffling. J Bacteriol. 2000;182:3008–3016. doi: 10.1128/jb.182.11.3008-3016.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen S.J., Burmølle M., Hansen L.H. Making bio‐sense of toxicity: new developments in whole‐cell biosensors. Curr Opin Biotechnol. 2006;17:11–16. doi: 10.1016/j.copbio.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Sticher P., Jaspers M., Harms H., Zehnder A.J.B., Van Der Meer J.R. Development and characterization of a whole cell bioluminescent sensor for bioavailable middle‐chain alkanes in contaminated groundwater samples. Appl Environ Microbiol. 1997;63:4053–4060. doi: 10.1128/aem.63.10.4053-4060.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiner L., Halverson L.J. Development and characterization of a green fluorescent protein‐based bacterial biosensor for bioavailable toluene and related compounds. Appl Environ Microbiol. 2002;68:1962–1971. doi: 10.1128/AEM.68.4.1962-1971.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauriainen S.M., Virta M.P.J., Karp M.T. Detecting bioavailable toxic metals and metalloids from natural water samples using luminescent sensor bacteria. Wat Res. 2000;34:2661–2666. [Google Scholar]
- Tecon R., Van Der Meer J.R. Information from single‐cell bacterial biosensors: what is it good for? Curr Opin Biotechnol. 2006;17:4–10. doi: 10.1016/j.copbio.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Tropel D., Van Der Meer J.R. Characterization of HbpR binding by site‐directed mutagenesis of its DNA‐binding site and by deletion of the effector domain. FEBS J. 2005;272:1756–1766. doi: 10.1111/j.1742-4658.2005.04607.x. [DOI] [PubMed] [Google Scholar]
- Uchiyama T., Abe T., Ikemura T., Watanabe K. Substrate‐induced gene‐expression screening of environmental metagenome libraries for isolation of catabolic genes. Nat Biotechnol. 2005;23:88–93. doi: 10.1038/nbt1048. [DOI] [PubMed] [Google Scholar]
- Van Dyk T.K., Wei Y., Hanafey M.K., Dolan M., Reeve M.J., Rafalski J.A. A genomic approach to gene fusion technology. Proc Natl Acad Sci USA. 2001;98:2555–2560. doi: 10.1073/pnas.041620498. , and et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward J.S. CRC Press; 1986. [Google Scholar]
- Werlen C., Jaspers M.C.M., Van Der Meer J.R. Measurement of biologically available naphthalene in gas, and aqueous phases by use of a Pseudomonas putida biosensor. Appl Environ Microbiol. 2004;70:43–51. doi: 10.1128/AEM.70.1.43-51.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese C.S., Griffin D.A. The solubility of Aroclor 1254 in seawater. Bull Environ Contam Toxicol. 1978;19:403–411. doi: 10.1007/BF01685817. [DOI] [PubMed] [Google Scholar]
- Wise A.A., Kuske C.R. Generation of novel bacterial regulatory proteins that detect priority pollutant phenols. Appl Environ Microbiol. 2000;66:163–169. doi: 10.1128/aem.66.1.163-169.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Graphical positioning of obtained mutants on the HbpR amino add sequence.
Tables S1-S5. Separation percentages of the different mutant pools by flow cytometry.