

Summary
Bioassays with bioreporter bacteria are usually calibrated with analyte solutions of known concentrations that are analysed along with the samples of interest. This is done as bioreporter output (the intensity of light, fluorescence or colour) does not only depend on the target concentration, but also on the incubation time and physiological activity of the cells in the assay. Comparing the bioreporter output with standardized colour tables in the field seems rather difficult and error‐prone. A new approach to control assay variations and improve application ease could be an internal calibration based on the use of multiple bioreporter cell lines with drastically different reporter protein outputs at a given analyte concentration. To test this concept, different Escherichia coli‐based bioreporter strains expressing either cytochrome c peroxidase (CCP, or CCP mutants) or β‐galactosidase upon induction with arsenite were constructed. The reporter strains differed either in the catalytic activity of the reporter protein (for CCP) or in the rates of reporter protein synthesis (for β‐galactosidase), which, indeed, resulted in output signals with different intensities at the same arsenite concentration. Hence, it was possible to use combinations of these cell lines to define arsenite concentration ranges at which none, one or more cell lines gave qualitative (yes/no) visible signals that were relatively independent of incubation time or bioreporter activity. The discriminated concentration ranges would fit very well with the current permissive (e.g. World Health Organization) levels of arsenite in drinking water (10 µg l−1).
Introduction
In the recent years, new tools for environmental monitoring have been developed on the basis of so‐called bioreporter organisms. Bioreporters are often microbial strains which are genetically modified to produce easily detectable output in response to the presence of specific analytes. Several reviews have been dedicated to this rapidly developing field, exposing details of genetic constructions, type of reporters or analyte detection (Daunert et al., 2000; Leveau and Lindow, 2002; Belkin, 2003; van der Meer et al., 2004; Harms et al., 2006). The detection system of bioreporter microorganisms is in most cases composed of a simple genetic circuitry, comprising a regulatory system (i.e. regulatory gene plus its cognate promoter) and one or more reporter genes. Commonly used reporter proteins are the chromogenic β‐galactosidase (lacZ) or β‐glucuronidase (gusA), luminescent luciferases (luc or luxAB) or autofluorescent proteins (GFP and variants) (Ivask et al., 2002; Paitan et al., 2004; Werlen et al., 2004; DeAngelis et al., 2005).
For quantitative measurements, bioreporter assays need to be calibrated with analyte solutions of known concentrations, which are typically analysed along with the samples of interest (Hakkila et al., 2004; Harms et al., 2005). The reason for this is that the bioreporter output (colour, luminescence or fluorescence) does not only depend on analyte concentration, but also on assay incubation time and the numbers and physiological activities of the cells in the assay (Jansson, 2003; Stocker et al., 2003; van der Meer et al., 2004; Wells et al., 2005). The latter two variables are very difficult to control, in particular when dried or immobilized cells are used (Bjerketorp et al., 2006). Simple applications like bioreporter field test kits or paper test strips are hampered by the difficulty to compare the resulting colour signals with standardized colour tables. Therefore, a robust and reliable system for internal calibration would be highly desirable.
The problem of variable incubation time and uncertain cellular activity could be overcome by the simultaneous use of multiple bioreporter cell constructs, each giving qualitative information about the presence or absence of analyte in a certain concentration range. Bioreporters could be designed and combined in such a way that not the actual individual output but the combination of reacting cell lines indicates the analyte concentration range in unknown samples without the need to measure exact rates or endpoints of colour formation by individual bioreporters (Fig. 1). Although this method would not report exact concentrations, the identification of concentration ranges should be sufficient for many applications, where, for instance, only a distinction of concentrations below and above a predefined permissible level is desired. If in combination with a chromogenic reporter protein this would allow signal interpretation by eye without instrumentation. In the case of arsenic in potable water, a distinction between concentrations below and above the World Health Organization (WHO) drinking water standard of 10 µg l−1 or the local standard of 50 µg l−1 in the most arsenic‐threatened countries of South‐East Asia would be most desirable.
Figure 1.

Principle of the internal calibration approach. (A)–(D) represent bioreporter cell lines differing in their sensitivity to arsenite.
To test this concept, we used two reporter genes to redesign bioreporter bacteria that had previously been developed for the detection of arsenite in drinking water, exploiting the ArsR sensor transcriptional repressor of the natural arsenic resistance of Escherichia coli (Stocker et al., 2003). In the presence of arsenite, ArsR loses affinity for its binding site on the DNA the result of which is depression of the arsR promoter. Equally effective in derepressing the arsR promoter via ArsR is antimonite. In cells that carry the arsenate reductase ArsC, arsenate (at 25% efficiency) and trimethylarsine oxide (at 10% efficiency) will also lead to derepression (Baumann and van der Meer, 2007). In one series of cell lines, the arsR promoter was combined with lacZ for β‐galactosidase. β‐Galactosidase has many advantages and various chromo‐ and fluorogenic substrates exist to assay LacZ activity (Daunert et al., 2000; Lei et al., 2006). In a second series of cell lines we applied the ccp gene. Its product, the cytochrome c peroxidase (CCP), has not frequently been utilized in whole‐cell bioreporter bacteria so far, but a large variety of ccp mutants have been developed which display different catalytic activity for one of its substrates guaiacol (Iffland et al., 2000). The signal outputs of both reporters were thus varied in different ways: for LacZ bioreporters we modified the intervening sequence between arsR and lacZ in order to modulate lacZ expression. For ccp we maintained the same reporter synthesis rate, but hypothesized that because of different catalytic activity of the reporters we would get variable output of the cells at the same arsenite concentration. Our research questions therefore were (i) if both types of reporter constructions would result in different levels of reaction sensitivity to arsenite and (ii) if combinations of such reporter cell lines with variable signal outputs would be suitable to infer arsenite concentration ranges without external calibration and at the required drinking water standards.
Results
β‐Galactosidase‐based bioreporter assay
Bioassays were performed in 96‐well plates to allow spectrophotometrical measurements (Fig. 2) as well as visual signal detection by digital imaging (Fig. 3). For each bioreporter strain the bioassay was performed in triplicate with 15 different arsenite concentrations ranging from 0.2 to 100 µg l−1. Strain 2245 was very sensitive and produced visible colour from the lowest arsenite concentration (0.2 µg l−1) with a linear signal increase up to 6 µg l−1 and a plateau above 10 µg l−1 (Figs 2 and 3). Colour development by strain 1595 began at approximately 10 µg l−1 arsenite and linearly increased at higher concentrations. Both strains had a relatively low background activity in the absence of arsenite. In contrast, strain 1580 was characterized by a high background and little further signal increase towards higher concentrations. Strain 2066 was the least sensitive and gave visible signals only at very high concentrations (around 50 µg l−1). Visual signal differentiation for neighbouring concentrations was difficult with all strains. The combination of qualitative (yes/no) signals of all four strains appeared suitable for the definition of three arsenite concentration ranges: < 6 µg l−1, 10–50 µg l−1 and > 50 µg l−1 (Table 1A). The quality of the responses (visible colour or not) was independent of incubation time in a window between 4 and 8 h after induction. During that time frame the colours gained intensity but the pattern of the signals stayed the same. Signal distribution was also independent of cell density for final cell culture turbidity in the assays of OD600 of between 0.1 and 1.0 (data not shown).
Figure 2.
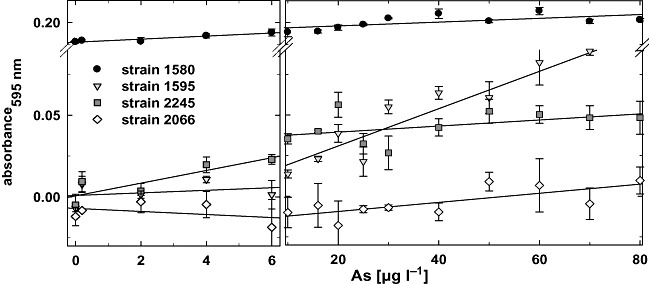
Colour development after 480 min by four bioreporter strains expressing LacZ measured as absorbance at 595 nm. Data points represent the average of three replicates.
Figure 3.
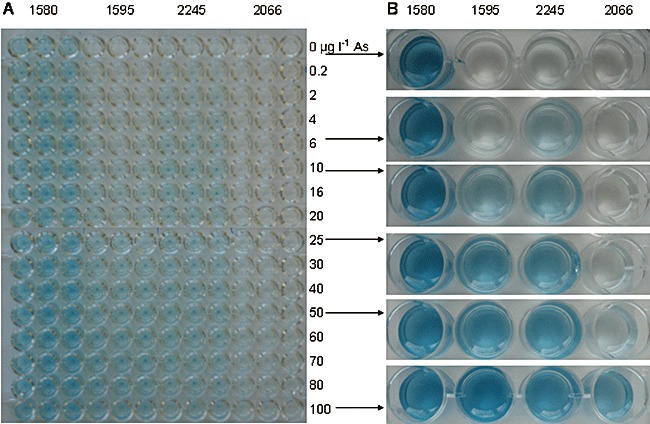
Colour development from X‐gal by four LacZ‐based bioreporter strains as response to different arsenite concentrations. A. Wide concentration range overview of assay carried out in 96‐well plates. The digital image was taken after 8 h, whereas colour development proceeded for 22 h. B. Colour detail in assays performed in 24‐well plates. Image taken after 4 h incubation time at 30°C.
Table 1.
Arsenite concentration ranges distinguished with combinations of LacZ strains (A) and CCP strains (B).
| A | Concentration range (µg l−1) | 1580 | 1595 | 2245 | 2066 | ||
|---|---|---|---|---|---|---|---|
| < 6 | x | x | |||||
| 10–50 | x | x | x | ||||
| > 50 | x | x | x | x | |||
| B | Concentration range (µg l−1) | 1971 | 1981 | 1982 | 2031 | 2332 | 2340 |
| 0 | x | x | x | ||||
| 1–10 | x | x | x | x | |||
| > 20 | x | x | x | x | x | x |
Strains that give a signal in the respective concentration range are marked with x.
CCP‐based bioreporter assay
In this case, the assays were carried out with colonies exposed to arsenite on agar plates and transferred to paper in order to obtain optimal visual signal interpretation. For each strain the bioassay was performed in four replicates with 10 different arsenite concentrations ranging from 1 µg l−1 to 5 mg l−1. The bioreporter strains 1971, 1981 and 1982 displayed relatively similar responses and produced clearly visible, concentration‐dependent signals between 4 and 30 µg l−1 followed by a plateau (Fig. 4). For the strains 1971 and 1982 a further signal increase could be detected after incubation with 1000 µg l−1. The background activity in the absence of arsenite was high enough to result in visible spots. Strain 2031 showed no background activity without arsenite, while colour production was clearly visible at 4 µg l−1 and increased strongly until 20 µg l−1. Strains 2332 and 2340 did not respond to low arsenite concentrations (1–10 µg l−1) and produced slightly increasing signals between 20 µg l−1 and 5 mg l−1 arsenite. The combination of the qualitative signals of these strains appeared to enable the distinction of arsenite‐free controls from the concentrations ranges 1–10 µg l−1 and > 20 µg l−1 (Table 1B). Similar results were obtained with either twice as many cells per spot or shorter (2 h) and longer incubation (16 h) (data not shown).
Figure 4.
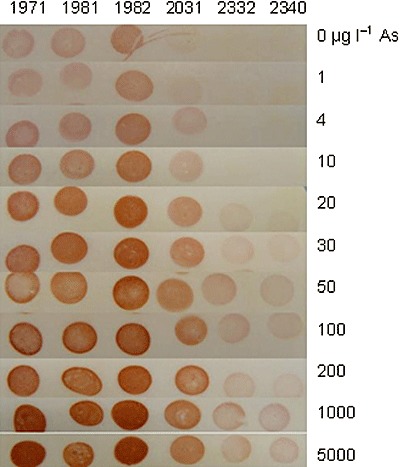
Colour development in the six bioreporter strains expressing CCP as a function of arsenite concentration in the assay. Colonies were spotted on paper after 4 h incubation with arsenite on LB agar plates at 37°C. Digital image was taken 10 min after adding guaiacol and hydrogen peroxide to develop colour.
mRNA content
Reporter mRNA contents of cells exposed to three arsenite concentrations were quantified in order to determine whether differences in signal intensities for the reporters at identical arsenite concentrations were the result of differential transcription or of enzyme synthesis and enzyme activity. Levels of the ccp mRNA in the different CCP bioreporter strains were similar after incubation with the same arsenite concentration, but increased as a function of arsenite concentration (Fig. 5A). An exception was strain 1982, which for unknown reasons contained significantly (P < 0.05) less ccp mRNA at the highest arsenite concentration. The level of lacZ mRNA in the different reporter strains generally increased as a function of arsenite concentration, but varied between all strains. At 100 µg As l−1 the levels of lacZ mRNA differed significantly (P < 0.05) between all strains, while at 10 µg As l−1 the difference was only significant between strain 1580 and the other strains (P < 0.05). Although strains 1595, 2245 and 2066 had similar levels of mRNA at 10 µg As l−1 the colour formation was already visible for the strains 1595 and 2245 but not for 2066. The most likely reason for this difference is the improved lacZ ribosome binding site in strain 2245 and 1595 compared with strain 2066, which catalyses a faster translation rate from the same amount of mRNA (de Smit and van Duin, 1994). Altogether the change of the intergenic region between arsR and lacZ in the different constructs, and the use of two different plasmid vectors for cloning (pMV for 1580 and 1595; pPROBE for 2245 and 2066) did affect the amount of lacZ mRNA produced. Notably, lacZ expression in strain 1580 without and at 10 µg As l−1 did not differ, which is the result of the leaky arsR promoter and read‐through (Fig. 5B). Basal level of arsR expression is required for the natural resistance system to function because of the nature of the arsR transcriptional organization. As ArsR is a transcriptional repressor which binds within its own promoter DNA, no arsR transcription would take place without leakiness.
Figure 5.
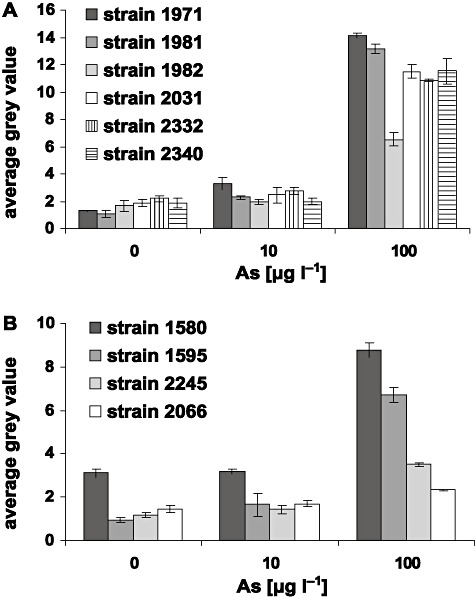
Comparison of signal intensities after dot blot hybridization of mRNA in the six bioreporter strains expressing CCP (A) and in four bioreporter strains expressing LacZ (B) induced with three different arsenite concentrations for 4 h.
Discussion
The aim of this study was to simplify the application of bioreporter bacteria by developing an internal calibration method. The idea was to infer analyte concentrations from combinations of qualitative (yes/no) signals provided by a series of cell lines with different sensitivities for the analyte. The quantitative response of a single cell line used so far in bioreporter assays is replaced by a series of complementary qualitative responses. The advantage of such bioreporter arrays is their relative independence of incubation times and the physiological activity of the cells in the assay. We used two different ways to produce such variable reporter cell lines: (i) by influencing the rate of reporter enzyme synthesis (LacZ) or (ii) by influencing the reporter enzyme specific activity (for CCP). Various other ways of obtaining similar effects can be imagined, for instance, by using different stabilities of reporter proteins (Andersen et al., 1998). We focused on an arsenite‐detecting bioreporter, for which a simple qualitative assay could be useful in field campaigns notably in South‐East Asia (Smith et al., 2000; British Geological Survey, 2001). Several studies have shown that bioreporter strains based on ArsR and the arsR promoter react equally well to antimonite and arsenite (Ramanathan et al., 1997; Scott et al., 1997; Tauriainen et al., 1999). Even wolframite will be able to derepress the system although at a 1000‐fold higher concentration than arsenite. However, we believe that cross‐reaction with antimonite is rather unlikely in groundwater samples, because most arsenite‐contaminated samples contain only negligible concentrations of antimonite (Trang et al., 2005). On the other hand, the bioreporter may be of equal use for the detection of antimonite where it prevails as antimonite is a suspected carcinogen. The presence of phosphate and nitrate in the analyte has not significantly affected the assays as reported by Ramanathan and colleagues (1997), Scott and colleagues (1997) and Harms and colleagues (2005).
LacZ bioreporters
Arsenite measurements have been performed before with LacZ bioreporters under control of the ArsR promoter using classical spectrophotometrical, chemiluminescence and electrochemical detection (Scott et al., 1997; Ramanathan et al., 1998), or by chromogenic visualization using X‐gal (Stocker et al., 2003). We previously showed that leaky expression from the arsR promoter must be reduced to obtain gradually intensifying blue colour formation from X‐gal as a function of arsenite concentration. This was accomplished by placing a second ArsR DNA binding site downstream of arsR (Stocker et al., 2003). Upon long incubation times, however, cells with this bioreporter construct (i.e. strain 1595) will intensify their blue colour at all arsenite concentrations, reducing the visibility of a good colour scale. We therefore produced one variant of strain 1595 in which the background is further reduced (strain 2066) by altering the ribosome binding site in front of lacZ (Fig. S1). Indeed, production of LacZ activity in strain 2066 is very low compared with the others, which was useful for the concept of the multi‐strain assay, because the 2066 bioreporter only produces visible blue colour from X‐gal at arsenite concentrations above 50 µg l−1. At the engineered unique restriction sites in front of lacZ we then replaced fragments of various length originating from the wild‐type lacZ promoter in E. coli, one of which (construct in strain 2245, Fig. S1) appeared to be suitable for the arsenite concentration range below 5 µg l−1. Interestingly, strain 2245 reacted with visibly different blue colour formation from X‐gal already at 0.2 µg l−1 arsenite, compared with the negative control. Thus, the responses of strains 2245 and 1595 complement each other very well in the low (0.2–5 µg l−1) and medium (5–50 µg l−1) concentration ranges. Although strain 1580 is not directly useful in showing colour differences at varying arsenite concentrations, it can be used as positive control for the quality of the test.
By probing lacZ mRNA levels in the four strains, we could provide evidence that the different LacZ activities in the reporter strains as a function of arsenite is due to both differential transcription efficiency or mRNA stability and translational effects. Two transcriptional effects were clearly seen: first, the inclusion of the secondary ArsR binding site in all constructs except 1580, which reduced mRNA formation in the absence of arsenite, and second, the lower lacZ mRNA abundance from the constructs based on plasmid pPROBE (i.e. 2066 and 2245), which might be due to variations in plasmid copy number. However, the strong LacZ activity difference between 2066 and 2245 was not apparent from mRNA levels (Fig. 5B), but the result from the absence (2066) and presence (2245) of a good ribosome binding site. One can conclude that our intention to control the bioreporter sensitivity on the level of reporter enzyme synthesis was successful in that it led to a range of bioreporter strains of quite complementary sensitivities including a nearly constitutive control strain.
CCP bioreporters
The use of CCP as reporter gene resulted in a useful colouration as well, although the assay was a bit less robust than that for β‐galactosidase. The main advantage of ccp was the availability of a well‐characterized set of mutants with variable activity against the classical peroxidase substrate guaiacol (Iffland et al., 2000; 2001). In this case mRNA probing showed that all mutant ccp genes were transcribed with similar efficiencies, and as a function of arsenite concentration (except one; Fig. 5A). However, the colouration resulted in very different intensities at the same arsenite concentration for the various reporters. Those strains containing CCP with improved specific activity (1971, 1981, 1982) compared with the wild type (2031) gave high signals at very low arsenite concentrations, but with a relatively high background in the absence of arsenite. Their increased activity confirms previous findings of Wilming et al., who observed turnover numbers of 102 and 26 s−1 of strain 1971 and 1982, respectively, compared with 0.35 s−1 for the wild‐type enzyme (Wilming et al., 2002). The ccp gene in strain 1981 contains multiple mutations (Y39H–H60R–N184D–D217G–D224Y) and although the substrate turnover was reported to be only slightly higher (0.8 s−1; Iffland et al., 2001) than that of the wild type, a colouration similar to that of 1971 and 1982 for arsenite concentrations up to 100 µg l−1 was observed. The CCP of the bioreporter strains with the lowest observed activity and sensitivity in our tests (2332, 2340) had higher reported turnover numbers (13 s−1 and 5 s−1 respectively) than the wild‐type enzyme (Wilming et al., 2002). The reasons for these discrepancies between reported enzyme activities and signals obtained in our experiments with whole cells are unknown. Measurements of enzyme activities in cell extracts were in accordance with the reported substrate turnover (Fig. S1), which might be an indication for different enzyme activity in extracts and whole cells, or differences in substrate accessibility or transport in whole cells. Our results, however, show that it is possible to receive a set of bioreporter bacteria with varying sensitivity by modifying the catalytic activity of the reporter protein and to use combination of them for internal calibration.
Conclusions
We demonstrated that bioreporter bacteria of various sensitivities can be tailored by modifying different steps of the detection‐signalling chain, i.e. the rate of reporter enzyme synthesis and the catalytic activity of the reporter protein. It appears thus possible to construct sets of bioreporter bacteria that, when combined, cover ranges of target concentrations that could not be covered by a single bioreporter organism due to restrictions of, for example, the induction factor, limitations of spectroscopic signal detection or the insensitivity of visual interpretation. Furthermore, our results demonstrate the potential of reporter protein engineering as opposed to the former focus on analyte sensing and gene regulation for bioreporter improvement (van der Meer et al., 2004).
The accomplished modifications altogether resulted in two sets of bioreporter bacteria that can be used to infer analyte concentration ranges without external calibration. This quantitative information originates from combinations of robust qualitative signals of the individual bioreporter strains. The hypothesized working principle was thus shown to function. A further important result is that permissive concentrations of arsenic could be identified with both bioreporter sets: (i) below the WHO limit only two of the four LacZ bioreporter bacteria produced a visible signal, above 10 µg l−1 three or at concentrations above local arsenic standards (50 µg l−1) all four strains were responding with clear signals, (ii) the CCP bioreporter bacteria indicate a concentration below the WHO limit when four out of six strains show signals. This set could also be reduced to four strains, as three strains gave redundant information.
Our approach promises to simplify bioreporter application by layman as it eliminates time‐consuming and technically demanding calibration. Especially for simple field detection devices such as paper strips (Stocker et al., 2003) our method would be of great value. Besides its higher robustness it has the advantage of the much easier interpretation of sets of qualitative subsignals than of gradual colour changes.
Experimental procedures
Construction of ArsR plasmids
The following reporter plasmids were constructed (Fig. 6) in which ArsR controls expression of either the lacZ gene or the ccp gene [for CCP from Saccharomyces cerevisiae (Iffland et al., 2000)]. In plasmid pMV‐arsR [strain number 1580 (Stocker et al., 2003)] the arsR gene is placed directly upstream of lacZ and in the same direction of transcription, so that expression of arsR and lacZ are under control of the ArsR‐regulated promoter in front of arsR itself (Pars, Fig. 6). To reduce background lacZ expression from the leaky Pars promoter, a second ArsR DNA binding site (ABS) was introduced downstream of arsR in plasmid pMV‐arsR‐ABS [strain number 1595, Fig. 6 (Stocker et al., 2003)]. To change the sequence and ribosome binding site upstream of lacZ, we amplified the entire lacZ gene from the E. coli K12 chromosome while introducing HindIII and NheI restriction sites at the gene extremities. This lacZ DNA was then used to replace the gfp gene in the broad host range vector pPROBE' (Miller et al., 2000). The arsR gene and the downstream secondary ArsR binding site were recovered on a BamHI–SpeI fragment and inserted into pPROBE'–lacZ in front of lacZ to make pPR‐ArsR‐ABS‐LacZ (strain 2066, Fig. 6). The ribosome binding site (RBS) of the original lacZ gene of E. coli was amplified together with the entire lacZ gene by PCR by using primer LacZ Rev NheI (5'‐gctagcttatttttgacaccagaccaactgg‐3') and primer LacZ‐RBS‐For (5′‐gcttgtgagcggataacaatttc acacagg‐3′) and introduced in pPR‐ArsR‐ABS‐LacZ digested by HindIII and NheI (plasmid pPR‐arsR‐ABS‐RBS‐LacZ, strain 2245, Fig. 6). Sequences of the arsR–lacZ intergenic regions are presented in Fig. S2.
Figure 6.
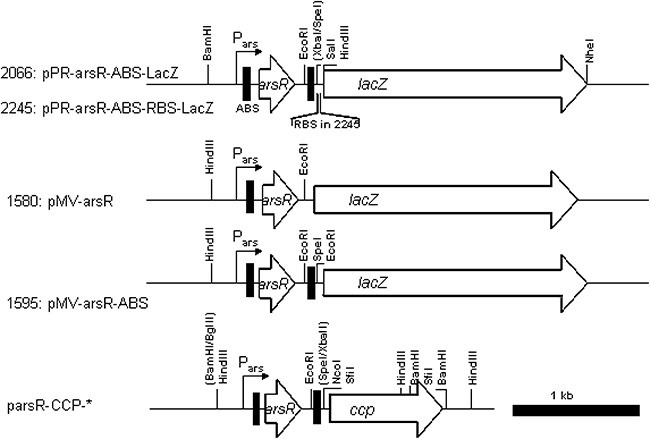
Schematic picture of the plasmids developed in this study. Genes are represented as arrows, drawn to proportion. Relevant restriction sites for cloning are shown in their usual abbreviations. The black vertical bars symbolize the ArsR binding sites (ABS).
The ccp reporter plasmids were all constructed by inserting a BamHI–SpeI fragment containing arsR and the second ArsR binding site from pPR‐arsR‐ABS (Stocker et al., 2003) in a pET15‐based plasmid containing the ccp gene or its variants (Iffland et al., 2000; 2001), pre‐digested with BglII and XbaI (during which the T7 promoter is removed). The arsR gene is thus expressed from Pars and on its turn regulates ccp expression (Fig. 6). In this manner we produced E. coli strains with plasmids pArsR‐CCP‐WT (strain number 2031, wild‐type ccp), pArsR‐CCP‐R48I (strain 2332, R48I mutation in CCP), pArsR‐CCP‐R48T (strain 2340), pArsR‐CCP‐R48Q (strain 1971), pArsR‐CCP‐K2.4 (strain 1981, Y39H‐H60R‐N184D‐D217G‐D224Y mutations) and pArsR‐CCP‐PT1 (strain 1982, R48H mutation). Arsenite bioreporters were constructed in E. coli DH5α.
Cell pre‐culturing
Arsenite in aqueous solution was measured with cell suspensions of E. coli strains (Table 2). Bioreporter strains were plated from –80°C stock cultures on fresh Luria–Broth (LB) plates with appropriate antibiotic selection (ampicillin 100 µg ml−1 or kanamycin 50 µg ml−1) and grown overnight at 37°C. Five millilitres of liquid LB medium plus antibiotic was inoculated with a single colony and incubated for 16 h at 37°C. Cultures were diluted in fresh pre‐heated LB medium without antibiotics at a 1:50 ratio and incubated until an optical density at 600 nm of 0.5 was reached and then used for triplicate bioassays as described below.
Table 2.
Identification and characteristics of E. coli DH5α bioreporter constructs.
| Strain | Plasmid |
|---|---|
| LacZ | |
| 1580 | pMV‐arsR |
| 1595 | pMV‐arsR‐ABS |
| 2245 | pPR‐arsR‐ABS‐RBS‐lacZ |
| 2066 | pPR‐arsR‐ABS‐lacZ |
| CCP | |
| 1971 | pArsR‐ABS‐CCP‐R48Q |
| 1981 | pArsR‐ABS‐CCP‐K2.4 |
| 1982 | pArsR‐ABS‐CCP‐PT1 |
| 2031 | pArsR‐ABS‐CCP‐WT |
| 2332 | pArsR‐ABS‐CCP‐R48I |
| 2340 | pArsR‐ABS‐CCP‐R48T |
ABS, ArsR binding site; RBS, ribosome binding site.
Arsenite assays with E. coliβ‐galactosidase (LacZ) bioreporters
Assay mixtures containing 100 µl of cell suspension (OD600 of 0.5, 1:5 diluted in water), 90 µl of arsenite stock solution and 10 µl of X‐gal substrate solution (5‐bromo‐4‐chloro‐3‐indolyl‐β‐d‐galactosidase, Serva, 0.2 mg ml−1 in dimethylformamid) were prepared directly in 96‐well plates. Cells were exposed to different final arsenite concentrations (0, 0.05, 0.2, 0.5, 2, 4, 6, 10, 20, 25, 30, 40, 50, 60, 70, 80 and 100 µg l−1). Arsenite solutions were prepared by dilution from a 50 mM (3.9 g l−1) sodium arsenite solution commercial stock (Merck) in tap water. The 96‐well plates with the assay mixtures were covered with a lid and incubated at 30°C in a rotary shaker (700 r.p.m.) for 22 h. In order to detect the blue colour formed from X‐gal conversion, the optical density was measured in a microplate reader (Spectramax 250, Molecular Devices) at 595 nm after 0, 1, 2, 3, 4, 5, 6, 7, 8 and 22 h. The turbidity resulting from the cell suspension itself was subtracted from the measured signal by measuring triplicate assay mixtures without X‐gal.
Arsenite measurements with E. coli CCP bioreporters
The CCP bioreporters were cultured as above for the LacZ reporters in small pre‐cultures for 16 h at 37°C after which cells were diluted 50‐fold and again grown until a culture turbidity of ∼0.5 was reached. Cells were then concentrated by centrifugation to achieve a turbidity at 600 nm of ∼6. Ten microlitres of these cell suspensions were then spotted on agar plates containing different concentrations of arsenite (0, 1, 4, 10, 20, 30, 50, 100, 200, 1000 and 5000 µg l−1) and briefly dried. After an incubation of 4 h at 37°C the cell spots were transferred to paper by gently pressing a paper sheet (3M chromatography paper, Whatman) of the right diameter directly on the agar surface. Peroxidase activity of the cells was detected by incubating the paper sheets on filter paper which was wetted with 50 mM potassium phosphate buffer (pH 6.0) containing freshly added guaiacol (final concentration 105 mM, Sigma) and hydrogen peroxide (final concentration 170 µM, Merck) (Iffland et al., 2000). Colour development was allowed to proceed until clearly visible, after which the paper sheets were digitally photographed.
Determination of mRNA contents
Reporter strain cultures grown similarly were exposed to 0, 10 or 100 µg l−1 arsenite for 4 h at 37°C, after which total RNA was isolated from 5 ml of culture using a Macherey Nagel Nucleo Spin Kit. Twenty microlitres from 120 µl of total RNA extract from each sample was blotted on a positively charged nylon membrane (Roche) using a Milliblot system (Millipore). The total RNA on the membrane was fixed with UV (1 min at 120 mJ cm−2) and stored at room temperature until hybridization. DIG‐labelled DNA probes were synthesized using random primed labelling (DIG DNA labelling kit, Roche) from PCR products generated with specific primers (Table 3) for lacZ and ccp respectively. Hybridization was performed for 16 h at 57°C for the ccp probe and 53°C for the lacZ probe using DIG Easy Hyb solution (Roche) and 100 ng µl−1 probe. Subsequent washing was performed according to the manufacturer's protocol (Roche). DIG–DNA–RNA hybrids were detected with the NBT‐CSIB (Roche) approach as described by the manufacturer. Dot blots were scanned and quantitatively analysed using Image J, in which the signal intensities (i.e. the grey intensities of the dots) are expressed as average grey values.
Table 3.
Primers for the synthesis of probes for dot blot hybridization.
| Primer name | Target | Sequence |
|---|---|---|
| sfi_for | ccp | 5′‐GCC ATG GCC AGC ACG GCC ACA CC‐3′ |
| R48Trev | ccp | 5′‐GTG CCA AGC AAG AGT GAC TAA TAC GGG‐3′ |
| LacZfor | lacZ | 5′‐TCG GTT ACG GCC AGG ACA GT‐3′ |
| LacZrev | lacZ | 5′‐CAT CAT TAA AGC GAG TGG CAA CAT‐3′ |
Acknowledgments
The authors thank Kai Johnsson for the generous gift of the ccp variants. Alexandra Baumeyer is thanked for her help in arsR‐ccp cloning and preliminary tests with CCP.
Supplementary material
The following supplementary material is available for this article online:
Specific activity of CCP in cell extracts from CCP bioreporter bacteria incubated for 4 h with 5 different arsenite concentrations.
Intervening sequences in the arsR-lacZ constructions between the end of arsR (taa, underlined) and the beginning of lacZ (atg, underlined). Relevant restriction sites used for cloning are indicated. The ArsR binding site motif is shaded in grey background. Rbs, ribosome binding site. Plasmid pBGD23 is the original construct used by Scott et al., 1997.
This material is available as part of the online article from http://www.blackwell-synergy.com
References
- Andersen J.B., Sternberg C., Poulsen L.K., Bjorn S.P., Givskov M., Molin S. New unstable variants of green fluorescent protein for studies of transient gene expression in bacteria. Appl Environ Microbiol. 1998;64:2240–2246. doi: 10.1128/aem.64.6.2240-2246.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann B., Van Der Meer J.R. Analysis of bioavailable arsenic in rice with whole cell living bioreporter bacteria. J Agric Food Chem. 2007;55:2115–2120. doi: 10.1021/jf0631676. [DOI] [PubMed] [Google Scholar]
- Belkin S. Microbial whole‐cell sensing systems of environmental pollutants. Curr Opin Microbiol. 2003;6:206–212. doi: 10.1016/s1369-5274(03)00059-6. [DOI] [PubMed] [Google Scholar]
- Bjerketorp J., Hakansson S., Belkin S., Jansson J.K. Advances in preservation methods: keeping biosensor microorganisms alive and active. Curr Opin Biotechnol. 2006;17:43–49. doi: 10.1016/j.copbio.2005.12.005. [DOI] [PubMed] [Google Scholar]
- British Geological Survey. 2001.
- Daunert S., Barrett G., Feliciano J.S., Shetty R.S., Shrestha S., Smith‐Spencer W. Genetically engineered whole‐cell sensing systems: coupling biological recognition with reporter genes. Chem Rev. 2000;100:2705–2738. doi: 10.1021/cr990115p. [DOI] [PubMed] [Google Scholar]
- DeAngelis K.M., Ji P.S., Firestone M.K., Lindow S.E. Two novel bacterial biosensors for detection of nitrate availability in the rhizosphere. Appl Environ Microbiol. 2005;71:8537–8547. doi: 10.1128/AEM.71.12.8537-8547.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakkila K., Green T., Leskinen P., Ivask A., Marks R., Virta M. Detection of bioavailable heavy metals in EILATox‐Oregon samples using whole‐cell luminescent bacterial sensors in suspension or immobilized onto fibre‐optic tips. J Appl Toxicol. 2004;24:333–342. doi: 10.1002/jat.1020. [DOI] [PubMed] [Google Scholar]
- Harms H., Rime J., Leupin O., Hug S.J., Van Der Meer J.R. Effect of groundwater composition on arsenic detection by bacterial biosensors. Microchim Acta. 2005;151:217–222. [Google Scholar]
- Harms H., Wells M.C., Van Der Meer J.R. Whole‐cell living biosensors – are they ready for environmental application? Appl Environ Biotechnol. 2006;70:273–280. doi: 10.1007/s00253-006-0319-4. [DOI] [PubMed] [Google Scholar]
- Iffland A., Tafelmeyer P., Saudan C., Johnsson K. Directed molecular evolution of cytochrome c peroxidase. Biochemistry. 2000;39:10790–10798. doi: 10.1021/bi001121e. [DOI] [PubMed] [Google Scholar]
- Iffland A., Gendreizig S., Tafelmeyer P., Johnsson K. Changing the substrate specificity of cytochrome c peroxidase using directed evolution. Biochem Biophys Res Commun. 2001;286:126–132. doi: 10.1006/bbrc.2001.5366. [DOI] [PubMed] [Google Scholar]
- Ivask A., Virta M., Kahru A. Construction and use of specific luminescent recombinant bacterial sensors for the assessment of bioavailable fraction of cadmium, zinc, mercury and chromium in the soil. Soil Biol Biochem. 2002;34:1439–1447. [Google Scholar]
- Jansson J.K. Marker and reporter genes: illuminating tools for environmental microbiologists. Curr Opin Microbiol. 2003;6:310–316. doi: 10.1016/s1369-5274(03)00057-2. [DOI] [PubMed] [Google Scholar]
- Lei Y., Chen W., Mulchandani A. Microbial biosensors. Anal Chim Acta. 2006;568:200–210. doi: 10.1016/j.aca.2005.11.065. [DOI] [PubMed] [Google Scholar]
- Leveau J.H.J., Lindow S.E. Bioreporters in microbial ecology. Curr Opin Microbiol. 2002;5:259–265. doi: 10.1016/s1369-5274(02)00321-1. [DOI] [PubMed] [Google Scholar]
- Van Der Meer J.R., Tropel D., Jaspers M. Illuminating the detection chain of bacterial bioreporters. Environ Microbiol. 2004;6:1005–1020. doi: 10.1111/j.1462-2920.2004.00655.x. [DOI] [PubMed] [Google Scholar]
- Miller W.G., Leveau J.H.J., Lindow S.E. Improved gfp and inaZ broad‐host‐range promoter‐probe vectors. Mol Plant Microbe Interact. 2000;13:1243–1250. doi: 10.1094/MPMI.2000.13.11.1243. [DOI] [PubMed] [Google Scholar]
- Paitan Y., Biran I., Shechter N., Biran D., Rishpon J., Ron E.Z. Monitoring aromatic hydrocarbons by whole cell electrochemical biosensors. Anal Biochem. 2004;335:175–183. doi: 10.1016/j.ab.2004.08.032. [DOI] [PubMed] [Google Scholar]
- Ramanathan S., Shi W.P., Rosen B.P., Daunert S. Sensing antimonite and arsenite at the subattomole level with genetically engineered bioluminescent bacteria. Anal Chem. 1997;69:3380–3384. doi: 10.1021/ac970111p. [DOI] [PubMed] [Google Scholar]
- Ramanathan S., Shi W.P., Rosen B.P., Daunert S. Bacteria‐based chemiluminescence sensing system using beta‐galactosidase under the control of the ArsR regulatory protein of the ars operon. Anal Chim Acta. 1998;369:189–195. [Google Scholar]
- Scott D.L., Ramanathan S., Shi W.P., Rosen B.P., Daunert S. Genetically engineered bacteria: electrochemical sensing systems for antimonite and arsenite. Anal Chem. 1997;69:16–20. doi: 10.1021/ac960788x. [DOI] [PubMed] [Google Scholar]
- De Smit M.H., Van Duin J. Control of translation by mRNA secondary structure in Escherichia coli: a quantitative analysis of literature data. J Mol Biol. 1994;244:144–150. doi: 10.1006/jmbi.1994.1714. [DOI] [PubMed] [Google Scholar]
- Smith A.H., Lingas E.O., Rahman M. Contamination of drinking water by arsenic in Bangladesh: a public health emergency. Bull World Health Organ. 2000;78:1093–1103. [PMC free article] [PubMed] [Google Scholar]
- Stocker J., Balluch D., Gsell M., Harms H., Feliciano J., Daunert S. Development of a set of simple bacterial biosensors for quantitative and rapid measurements of arsenite and arsenate in potable water. Environ Sci Technol. 2003;37:4743–4750. doi: 10.1021/es034258b. et al. [DOI] [PubMed] [Google Scholar]
- Tauriainen S., Virta M., Chang W., Karp M. Measurement of firefly luciferase reporter gene activity from cells and lysates using Escherichia coli arsenite and mercury sensors. Anal Biochem. 1999;272:191–198. doi: 10.1006/abio.1999.4193. [DOI] [PubMed] [Google Scholar]
- Trang P.T.K., Berg M., Viet P.H., Mui N.V., Van Der Meer J.R. Bacterial bioassay for rapid and accurate analysis of arsenic in highly variable groundwater samples. Environ Sci Technol. 2005;39:7625–7630. doi: 10.1021/es050992e. [DOI] [PubMed] [Google Scholar]
- Wells M., Gosch M., Rigler R., Harms H., Lasser T., Van Der Meer J.R. Ultrasensitive reporter protein detection in genetically engineered bacteria. Anal Chem. 2005;77:2683–2689. doi: 10.1021/ac048127k. [DOI] [PubMed] [Google Scholar]
- Werlen C., Jaspers M.C.M., Van Der Meer J.R. Measurement of biologically available naphthalene in gas and aqueous phases by use of a Pseudomonas putida biosensor. Appl Environ Microbiol. 2004;70:43–51. doi: 10.1128/AEM.70.1.43-51.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilming M., Iffland A., Tafelmeyer P., Arrivoli C., Saudan C., Johnsson K. Examining reactivity and specificity of cytochrome c peroxidase by using combinatorial mutagenesis. Chembiochem. 2002;3:1097–1104. doi: 10.1002/1439-7633(20021104)3:11<1097::AID-CBIC1097>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Specific activity of CCP in cell extracts from CCP bioreporter bacteria incubated for 4 h with 5 different arsenite concentrations.
Intervening sequences in the arsR-lacZ constructions between the end of arsR (taa, underlined) and the beginning of lacZ (atg, underlined). Relevant restriction sites used for cloning are indicated. The ArsR binding site motif is shaded in grey background. Rbs, ribosome binding site. Plasmid pBGD23 is the original construct used by Scott et al., 1997.