

Abstract
Biomarkers associated with Alzheimer’s disease (AD)-like brain atrophy in healthy people may identify mechanisms involved in early stage AD. Aside from cerebrospinal fluid (CSF) β-amyloid42 (Aβ42) and tau, no studies have tested associations between CSF proteins and AD-like brain atrophy. We studied 90 healthy elders, who underwent lumbar puncture at baseline, and serial magnetic resonance imaging scans for up to 4 years. We tested statistical effects of baseline CSF proteins (N=70 proteins related to Aβ42-metabolism, microglial activity and synaptic/neuronal function) on atrophy rates in 7 AD-related regions. Besides effects of Aβ42 and phosphorylated tau (P-tau) that were seen in several regions, novel CSF proteins were found to have effects in inferior and middle temporal cortex (including Apolipoprotein CIII, Apolipoprotein D and Apolipoprotein H). Several proteins (including S100β and Matrix Metalloproteinase-3) had effects that depended on the presence of brain Aβ pathology, as measured by CSF Aβ42. Other proteins (including P-tau and Apolipoprotein D) had effects even after adjusting for CSF Aβ42. The statistical effects in this exploratory study were mild and not significant after correction for multiple comparisons, but some of the identified proteins may be associated with brain atrophy in healthy people. Proteins interacting with CSF Aβ42 may be related to Aβ brain pathology, while proteins associated with atrophy even after adjusting for CSF Aβ42 may be related to Aβ-independent mechanisms.
Keywords: cerebrospinal fluid, biomarkers, atrophy, longitudinal, Alzheimer’s disease
1. Introduction
Alzheimer’s disease (AD) leads to severe atrophy of cortical brain tissue and ultimately results in dementia. The disease is believed to have a long pre-symptomatic phase with brain pathology accumulating prior to cognitive symptoms. The major disease hallmarks are plaques composed of β-amyloid (Aβ) peptides and neurofibrillary tangles composed of phosphorylated tau proteins (Braak and Braak, 1991). The view that AD starts prior to any symptoms implies that its earliest changes can only be studied in people who are cognitively normal. However, longitudinal studies on development of cognitive impairment in healthy people are limited by the long follow-up time and large study populations needed to detect significant cognitive changes. An alternative is to study longitudinal atrophy of AD-related brain regions, which are predictive of future cognitive decline (Rusinek et al., 2003). Biomarkers predicting such atrophy in cognitively healthy people may be useful to identify persons at risk for AD, and could inform on biological mechanisms during early disease stages.
Biomarker measurements of Aβ pathology, using positron emission tomography (PET) or cerebrospinal fluid (CSF), likely become abnormal early in AD, even prior to cognitive symptoms (Bateman et al., 2012; Jack et al., 2010). A few studies have tested if Aβ pathology predicts longitudinal brain atrophy in cognitively healthy people (Chételat et al., 2012; Fjell et al.; Henneman et al., 2009; Schott et al., 2010; Sluimer et al., 2010; Tosun et al., 2011). Some (Chételat et al., 2012; Fjell et al., 2010; Schott et al., 2010), but not all (Henneman et al., 2009; Sluimer et al., 2010; Tosun et al., 2011) of these studies found that an AD-like CSF or PET Aβ biomarker pattern predicted longitudinal atrophy. Studies showing that Aβ pathology predicts longitudinal AD-like atrophy in cognitively healthy people are consistent with the “dynamic biomarker model” presented by Jack et al. (Jack et al., 2010), but it should be noted that some studies found Aβ related atrophy also in areas not typically associated with early AD changes (Fjell et al., 2010; Schott et al., 2010).
Early pathological events leading to atrophy in AD may be reflected by other CSF changes, besides altered concentrations of the established biomarkers Aβ42 and tau, but to our knowledge, no previous study has examined if other CSF proteins are associated with longitudinal AD-like atrophy. Here we used CSF proteomics data from a multiplex panel, together with longitudinal structural magnetic resonance imaging (MRI) data to identify novel biochemical predictors of AD-related brain atrophy in cognitively healthy elderly participants in the Alzheimer’s Disease Neuroimaging Initiative (ADNI). We studied brain regions typically shown to be vulnerable in AD (although it should be noted that wide spread atrophy may also be seen in cognitively well-screened and longitudinally stable adults with no signs of Aβ pathology (Oh et al., 2013)). The tested proteins were selected for biological functions possibly altered in early stage AD (Aβ metabolism, microglia activity, and synaptic/neuronal function). Our primary hypothesis was that baseline protein levels were associated with atrophy rates. Although the dominant view in AD research is that Aβ pathology is the initiator in the neuropathological cascade, Aβ-independent processes have also been proposed (Chételat, 2013; Pimplikar et al., 2010). Based on this, we investigated if Aβ influenced the relationship between proteins and atrophy rates. We tested the specific hypotheses that some proteins interact with Aβ pathology to effect atrophy rates (suggesting atrophy mechanism linked to Aβ pathology), and that some proteins effect atrophy rates even when adjusting for the presence of Aβ pathology (suggesting atrophy mechanism that do not depend on Aβ pathology).
2. Methods
2.1 Study design
We examined changes in gray matter volume in a priori specified regions of interest (ROI) in cognitively normal people. Structural magnetic resonance imaging brain scans at multiple time points (up to 6 time points: ADNI screening, and 6, 12, 24, 36 and 48 months, median 5 scans per participant [range 2–6]) were acquired at multiple sites using 1.5 Tesla MRI scanners. Using FreeSurfer longitudinal processing framework, regional gray matter volumes were measured at each time point. Linear mixed effects models were performed to test if baseline CSF protein concentrations (N=70 different proteins tested separately) were associated with rates of brain atrophy (i.e., the rate of change in volume).
2.2 Participants
Data used in the preparation of this article were obtained from the ADNI database (adni.loni.ucla.edu). The ADNI was launched in 2003 by the National Institute on Aging (NIA), the National Institute of Biomedical Imaging and Bioengineering (NIBIB), the Food and Drug Administration (FDA), private pharmaceutical companies and non-profit organizations, as a $60 million, 5-year public-private partnership. The primary goal of ADNI has been to test whether serial MRI, PET, other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early AD. Determination of sensitive and specific markers of very early AD progression is intended to aid researchers and clinicians to develop new treatments and monitor their effectiveness, as well as lessen the time and cost of clinical trials. The Principal Investigator of this initiative is Michael W. Weiner, MD, VA Medical Center and University of California – San Francisco. ADNI is the result of efforts of many co-investigators from a broad range of academic institutions and private corporations, and participants have been recruited from over 50 sites across the U.S. and Canada. The initial goal of ADNI was to recruit 800 participants but ADNI has been followed by ADNI-GO and ADNI-2. To date these three protocols have recruited over 1500 adults, ages 55 to 90, to participate in the research, consisting of cognitively normal older individuals, people with early or late MCI, and people with early AD. For up-to-date information, see www.adni-info.org. The population in this study included ADNI-1 participants with valid results for a CSF multiplex protein panel (described below) and successful longitudinal FreeSurfer processing of MR images from at least two time points.
2.3 Structural MRI acquisition
The participants underwent a standardized 1.5 Tesla MRI protocol, which included T1-weighted MRI scans using a sagittal volumetric magnetization prepared rapid gradient echo (MP-RAGE) sequence, as previously described (Jack et al., 2008).
2.4 FreeSurfer longitudinal MR image processing
Automated cortical volume measures and hippocampal segmentation were performed with FreeSurfer software package, version 4.4 (http://surfer.nmr.mgh.harvard.edu/fswiki), as previously described (Fischl et al., 2004, 2002). To reduce the confounding effect of intra-participant morphological variability, each participant’s longitudinal data series was processed by FreeSurfer longitudinal workflow (http://surfer.nmr.mgh.harvard.edu/fswiki/LongitudinalProcessing). A previous test-retest study validated that the longitudinal processing provides consistent ROI segmentation (Reuter et al., 2012). All images underwent standardized quality control procedures (http://www.loni.ucla.edu/twiki/pub/ADNI/ADNIPostProc/UCS-FFreeSurferMethodsSummary.pdf). Participants with complete segmentation failure or gross errors throughout all brain regions were rated as complete failure. Participants with gross errors in one or more specific brain regions (i.e., temporal lobe regions, superior regions, occipital regions, and insula) were given partial pass rating. Participants with partial pass rating were included in analyses of appropriate brain regions only. For this study, we used volumetric measurements of entorhinal cortex, hippocampus, inferior parietal cortex, inferior temporal cortex, middle temporal cortex, posterior cingulate cortex, and precuneus (combining left and right hemisphere data).
2.5 CSF biomarker concentrations
A CSF sample was collected at study baseline by lumbar puncture, and shipped to the ADNI Biomarker Core laboratory at the University of Pennsylvania Medical Center for long term storage in −80° C. The established CSF AD biomarkers Aβ42, T-tau and P-tau were measured using the multiplex xMAP Luminex platform (Luminex Corp, Austin, TX, USA) with the INNOBIA AlzBio3 kit (Innogenetics, Ghent, Belgium) (Olsson et al., 2005; Shaw et al., 2009). CSF multiplex proteomics was done using an xMAP multiplex panel developed by Rules Based Medicine (MyriadRBM). Previous versions of the MyriadRBM panel had been used to explore CSF or plasma proteins in AD (Craig-Schapiro et al., 2011; Hu et al., 2012, 2010). Details regarding the assay technology, validation and quality control can be found online in a data primer for these CSF analyses (http://adni.loni.ucla.edu/). In brief, the panel included 159 biomarkers. Seventy-six analytes were adequately quantifiable in the CSF samples in this study (the other analytes were mostly below the assay detection limit or had other assay limitations). For these, we conducted a systematic literature review to find previous evidence of associations with Aβ metabolism, microglial activity, synaptic/neuronal function, which identified 67 analytes of interest (Supplementary Table 1). In all, this resulted in 70 proteins (including Aβ42, T-tau and P-tau) available for this particular study. Some analytes were used after log transformation as described in the assay documentation (http://adni.loni.ucla.edu/).
2.6 Statistical analyses
Potentially confounding effects by age, sex, education and APOE genotype on explanatory proteins were evaluated by non-parametric tests (Mann-Whitney test and Spearman Correlation tests, as appropriate). We used generalized linear mixed models to test if proteins were associated with a binary missing indicator for each visit. Effects of explanatory proteins on regional rates of atrophy in brain regions associated with AD were tested by linear mixed-effects models. Regional brain volumes were used as response variables and the time (years) from the first image measurement, protein, protein:time interaction, age, sex and intracranial volume (ICV) were used as fixed effects. Each brain region was tested separately with each explanatory protein. All proteins (including Aβ42) were used as continuous measurements in this analysis. All models included a random intercept and a random slope.
To specifically test if the effects of proteins were influenced by the presence of brain Aβ pathology, all models were run with and without adjusting for Aβ42 as a fixed effect. For this we used Aβ42 dichotomized by the cut-off 192 ng/L, since this cut-off has been found to be highly discriminate of brain Aβ pathology, as evidenced by PET imaging (Weigand et al., 2011) or autopsy (Shaw et al., 2009). For each protein and region, we also tested the protein:Aβ42:time interaction, again using Aβ42 dichotomized by the cut-off 192 ng/L, and including all sub-effects.
After inspection of the correlation within participants, a compound symmetry structure was assumed. We assessed the applicability of the models by evaluating linearity of regional volumes over time within participants, and the normality of the model residuals. Two participants were outliers (in standardized residuals) for models of entorhinal and hippocampal atrophy rates, respectively. The overall results did not change after removing these observations, and we kept all observations in the final models.
Due to the large number of tested proteins and brain regions, we corrected for multiple comparisons using a false discovery rate correction. Details on how the false discovery rate correction was applied are presented in the table legends. Volumes and proteins were standardized (centered and divided by the standard deviation). All tests were two-sided. All statistics were done using R (v. 2.15.2, The R Foundation for Statistical Computing).
3. Results
The study included 90 participants (44 females, 22 APOE ε4 carriers, mean age 76 years [range 62–90]). At the 48 month follow-up visit, 12 participants had converted from NL to MCI and 1 to AD dementia. We used survival analysis to test if any protein was related to conversion. The only proteins significantly associated with conversion were MMP2 (Log MMP2, β=0.76, P=0.048) and PAI1 (Log PAI1, β=−1.27, P=0.049). There was a tendency to significant association for Aβ42 (β=0.0022, P=0.067). No protein was significant after correcting for multiple comparisons (mean levels and ranges for converters and non-converters are available in Supplementary Table 2).
3.1 Potential confounding factors
Age, sex, education and APOE genotype were potential confounders of the relationship between the investigated proteins and regional brain volumes. We tested for imbalance between proteins and these demographic factors, using non-parametric tests (Supplementary Table 3). Age, sex and education were associated with several different proteins. APOE genotype was only associated with Aβ42 and P-tau. Based on these results, we included age, sex and education as covariates in all analyses.
3.2 Missing data
Several participants lacked MRI data for one or more visit (data were missing for 2 participants at the ADNI screening visit, 2 for the month 6 visit, 5 for the month 12 visit, 18 for the month 24 visit, 30 for the month 36 visit, and 73 for the month 48 visit). Using generalized linear mixed models, the protein:time effect was significantly associated with missing visits for 2 proteins (p-values 0.03–0.05). This number of significant associations is expected given the number of tests (N=70), under the null hypothesis.
3.3 CSF proteins associated with longitudinal brain atrophy
During the period of longitudinal observation, there was significant atrophy in all tested regions (Figure 1). Our main objective was to test if baseline measurements of proteins were associated with atrophy rates. We tested this by linear mixed effects models for each protein and brain region. The results of the regression are presented in figure 2 (as T-values for the protein:time interaction). Some proteins were associated with atrophy rates at the significance threshold p<0.01, including P-tau, where high concentrations were associated with atrophy rates in hippocampus and inferior temporal cortex; apolipoprotein D (ApoD), where high concentrations were associated with atrophy rates in middle temporal cortex; and ApoCIII, where high concentrations were associated with atrophy rates in inferior temporal cortex.
Figure 1. Longitudinal atrophy of regional brain volumes.

Thick lines are regression curves calculated by linear mixed effects models, adjusted for age, sex, education and intracranial volume, with random intercepts and slopes. Beta-coefficients and p-values for the fixed time effect are presented in each panel. Volume measurements (y-axes) are standardized. In a few cases, we noted increased volume measurement over time. This likely reflects measurement noise, possibly due to differences in partial volume effects between different scans (these scans were inspected manually to exclude major imaging artifacts).
Figure 2. Heat map of baseline CSF proteins and atrophy rates of regional brain volumes.
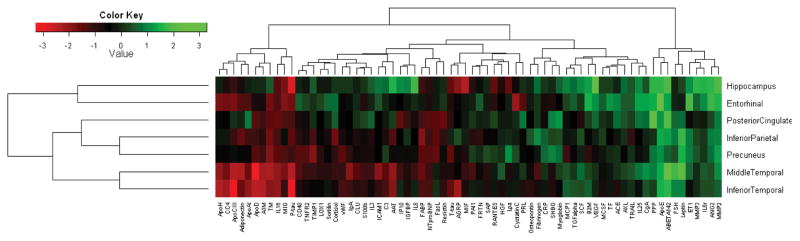
T-values (β-coefficients divided by standard error) for the protein:time interaction for all CSF proteins and regions from linear mixed effects models, irrespective of p-value. The models were adjusted for age, sex, education and intracranial volume. Red colors indicate low t-values (high proteins levels were associated with high atrophy rates) and green colors indicate high t-values (low protein levels were associated with high atrophy rates).
Due to the exploratory nature of the study, we here also present proteins that were significant at a more permissive threshold, p<0.05 (only presenting proteins significant in at least 2 regions, because of suspected spurious findings for proteins that were only significant in one region). At this threshold, several proteins were associated with atrophy rates in hippocampus (low Aβ42), inferior temporal cortex (high ApoD, α-1-Microglobulin [A1M], ApoH and interleukin 16 [IL-16]), middle temporal cortex (low Aβ42, and high A1M, ApoCIII, ApoH and IL-16), and posterior cingulate (low Aβ42).
Results for all proteins significant at p<0.05 are presented in Table 1. Note that the associations were mild and that no protein had a significant effect after correcting for multiple comparisons using false discovery correction.
Table 1.
CSF proteins significantly associated with regional atrophy rates
| Region | CSF protein | protein:time interaction | ||||
|---|---|---|---|---|---|---|
| β | p | Npar | Nobs | Pfdr | ||
| Hippocampus | Aβ42 | 0.020 | 0.045 | 89 | 406 | 0.79 |
| Log P-tau | −0.028 | 0.005 | 89 | 406 | 0.58 | |
| Inferior Temporal Cortex | Log A1M* | −0.017 | 0.017 | 90 | 404 | 0.70 |
| Log ApoCIII* | −0.022 | 0.002 | 90 | 404 | 0.39 | |
| Log ApoD | −0.018 | 0.015 | 90 | 404 | 0.70 | |
| Log ApoH* | −0.018 | 0.017 | 90 | 404 | 0.70 | |
| Log IL16 | −0.015 | 0.040 | 90 | 404 | 0.78 | |
| Log P-tau | −0.022 | 0.002 | 89 | 400 | 0.39 | |
| Middle Temporal Cortex | Aβ42 | 0.021 | 0.019 | 89 | 400 | 0.70 |
| Log A1M | −0.019 | 0.026 | 90 | 404 | 0.71 | |
| Log ApoCIII | −0.021 | 0.016 | 90 | 404 | 0.70 | |
| Log ApoD | −0.027 | 0.001 | 90 | 404 | 0.39 | |
| Log ApoH | −0.017 | 0.045 | 90 | 404 | 0.79 | |
| Log IL16 | −0.018 | 0.040 | 90 | 404 | 0.78 | |
| Log P-tau | −0.021 | 0.016 | 89 | 400 | 0.70 | |
| Posterior Cingulate | Aβ42 | 0.019 | 0.040 | 89 | 406 | 0.78 |
| Inferior Parietal Cortex Precuneus | No significant proteins | |||||
Results from linear mixed effects models, showing tests significant before correcting for multiple comparisons, for proteins significant in at least two regions (13 proteins were significant in one region only and were considered likely spurious findings). The rate of change of regional brain volume was used as response variable. The explanatory protein, time from the baseline visit, interaction protein:time, age, sex, intracranial volume, and education were used as fixed effects. All models had a random intercept and slope. Each brain region was tested separately with each explanatory protein. Results were similar when APOE was included as a fixed effect (data not shown). Npar, number of participants; Nobs, number of observations; Pfdr, corrected using a false discovery rate correction applied on 490 tests (70 proteins tested in 7 regions). Biomarkers and brain volumes were centered and standardized (for example, for Log CSF P-tau levels one SD above the mean, the volume of the hippocampus decreased by 0.028 SD per year).
For these models the protein:Aβ42:time interaction effect was found to be significant, as explained in the results and presented in Table 2.
3.4 CSF proteins associated with longitudinal brain atrophy: effects of Aβ42-interaction
As seen in Table 1, Aβ42 was associated with atrophy rates in hippocampus, middle temporal cortex and posterior cingulate (using continuous Aβ42 data). For the remaining part of the study we examined how Aβ42 influenced associations between other proteins and atrophy rates. We tested if associations between proteins and atrophy rates differed by brain Aβ status, by examining the protein:Aβ42:time interaction (using β42 dichotomized by the cutoff 192 ng/L, as explained in the methods section). For several proteins, we found interactions in at least one brain region (Table 2A and Figure 2). As seen in Figure 3, most of these proteins were associated with atrophy rates mainly in participants with signs of brain Aβ pathology, where high levels some proteins (ferritin [FRTN], S100β, ApoCIII, ApoH, and Hepatocyte Growth Factor [HGF]) were associated with increased atrophy rates. Again, these associations were mild and were not significant after correcting for multiple comparisons.
Table 2.
CSF proteins significantly associated with regional atrophy rates, Aβ42-dependent and Aβ42-independent effects
| Region | CSF protein | A protein:Aβ42:time interaction | B protein:time interaction Unadjusted for Aβ42 |
||||
|---|---|---|---|---|---|---|---|
| β | P | Pfdr | β | P | Pfdr | ||
| Entorhinal | IL25 | 0.042 | 0.024 | 0.40 | NA | NA | NA |
| Log FRTN | −0.039 | 0.043 | 0.51 | NA | NA | NA | |
| Log CC4 | −0.043 | 0.045 | 0.51 | NA | NA | NA | |
| Log HGF | −0.037 | 0.047 | 0.53 | NA | NA | NA | |
| Log Cystatin C | −0.019 | 0.038 | 0.87 | ||||
| Hippocampus | Log P-tau | −0.028 | 0.005 | 0.69 | |||
| Inferior Parietal | Log FRTN | −0.069 | 0.003 | 0.37 | NA | NA | NA |
| Log S100b | −0.061 | 0.009 | 0.37 | NA | NA | NA | |
| ICAM1 | −0.061 | 0.014 | 0.37 | NA | NA | NA | |
| Log HGF | −0.052 | 0.015 | 0.37 | NA | NA | NA | |
| Log NTproBNP | −0.056 | 0.019 | 0.40 | NA | NA | NA | |
| Log Cystatin C | 0.058 | 0.022 | 0.40 | NA | NA | NA | |
| Log MMP3 | −0.052 | 0.029 | 0.41 | NA | NA | NA | |
| Inferior Temporal | Log ApoCIII | −0.045 | 0.002 | 0.37 | NA | NA | NA |
| Log ApoH | −0.041 | 0.004 | 0.37 | NA | NA | NA | |
| Log A1M | −0.040 | 0.011 | 0.37 | NA | NA | NA | |
| Log CC4 | −0.040 | 0.015 | 0.37 | NA | NA | NA | |
| Log IL16 | −0.036 | 0.020 | 0.40 | NA | NA | NA | |
| Log AAT | −0.037 | 0.024 | 0.40 | NA | NA | NA | |
| Log S100b | −0.035 | 0.027 | 0.40 | NA | NA | NA | |
| ICAM1 | −0.034 | 0.041 | 0.50 | NA | NA | NA | |
| Log MMP3 | −0.030 | 0.049 | 0.53 | NA | NA | NA | |
| Log TM | −0.028 | 0.050 | 0.53 | NA | NA | NA | |
| Log P-tau | −0.022 | 0.002 | 0.52 | ||||
| Log ApoD | −0.018 | 0.015 | 0.87 | ||||
| Middle Temporal | Log S100b | −0.047 | 0.009 | 89 | NA | NA | NA |
| Log FRTN | −0.040 | 0.025 | 89 | NA | NA | NA | |
| Log ApoD | −0.027 | 0.001 | 0.51 | ||||
| Log ApoCIII | −0.021 | 0.016 | 0.87 | ||||
| Log P-tau | −0.021 | 0.016 | 0.87 | ||||
| Log TM | −0.020 | 0.020 | 0.87 | ||||
| Log A1M | −0.019 | 0.026 | 0.87 | ||||
| Log IL16 | −0.018 | 0.040 | 0.87 | ||||
| Log AAT | −0.018 | 0.042 | 0.87 | ||||
| Log ApoH | −0.017 | 0.045 | 0.87 | ||||
| Precuneus | Log PPP | 0.052 | 0.015 | 0.37 | NA | NA | NA |
| IL25 | −0.051 | 0.028 | 0.40 | NA | NA | NA | |
| Log NTproBNP | −0.046 | 0.033 | 0.46 | NA | NA | NA | |
| ICAM1 | 0.052 | 0.015 | 0.37 | NA | NA | NA | |
| Posterior Cingulate | Log HGF | −0.045 | 0.005 | 0.37 | NA | NA | NA |
| ICAM1 | −0.047 | 0.015 | 0.37 | NA | NA | NA | |
| Log PPP | 0.035 | 0.027 | 0.40 | NA | NA | NA | |
Results from linear mixed effects models for proteins significantly associated with atrophy rates in at least two regions (23 proteins were significant in one region only and were considered likely spurious findings). Regional atrophy rates were used as response variables. The time from the baseline visit, explanatory protein, age, sex, intracranial volume, and education were used as fixed effects. Each brain region was tested separately with each explanatory protein. Results are given for models that included the protein:Aβ42:time interaction effect (A), protein:time or the interaction effect unadjusted for Aβ42 (B). For proteins listed in (B), coefficients and p-values did not change when including Aβ42 as a predictor in the models (not shown). Results were similar when the models were adjusted for APOE genotype (not shown). The numbers of participants were 84–90, and the numbers of observations 348–406. Pfdr, corrected using a false discovery rate correction (for models of the protein:Aβ42:time interaction effect, the correction was applied on 483 tests [69 proteins tested in 7 regions], for models of the protein:time interaction, the correction was applied on 438 tests [all proteins and regions except the 45 proteins and regions where protein:Aβ42:time was significant]). Biomarkers and brain volumes were centered and standardized.
Figure 3. Interactions between baseline CSF proteins and CSF Aβ42 on atrophy rates.

All proteins and regions where the protein:Aβ42:time interaction was significantly associated with regional atrophy rates at p < 0.01. Y-axes show yearly atrophy rates (standardized), and x-axes show baseline protein concentrations. CSF Aβ42 was dichotomized by the cutoff 192 ng/L. Participants with low CSF Aβ42 are shown in red boxes (dashed regression lines) and participants with normal CSF Aβ42 in black circles (solid regression lines). Regression lines are adjusted for age, sex, education and intracranial volume.
3.5 CSF proteins associated with longitudinal brain atrophy: Aβ42-independent effects
Finally, we tested if proteins were associated with atrophy rates independent of Aβ42. We did this by comparing estimates for the protein:time interaction with and without adjusting for Aβ42 as a covariate (again using Aβ42 dichotomized by the cutoff 192 ng/L, as explained in the methods section). Note that this was not meaningful to do for proteins and regions where the protein:Ab42:time interaction was significant, since we had already concluded that Aβ42 influenced effects of proteins in those regions (Table 2A). For all other proteins and regions, we tested the protein:time interaction with and without adjusting for Aβ42. Several proteins were associated with atrophy rates when the models were not adjusted for Aβ42 (Table 2B). For all these, the effects were unchanged when adjusting the models for Aβ42 (β-coefficients changed only minimally, <6%). Our interpretation of this is that the presence of brain Aβ pathology at baseline did not mediate the detected effects on regional atrophy rates for the proteins listed in Table 2B.
4. Discussion
The key findings of this study were that (1) several CSF proteins obtained at baseline were associated with atrophy rates in AD-related brain regions over a follow-up period of 4 years, suggesting that these proteins may be novel prognostic biomarkers of AD-related atrophy in cognitively normal people; (2) several CSF proteins had interactions with CSF Aβ42, indicating that the presence of Aβ pathology may influence their effects on longitudinal AD-like atrophy; (3) some CSF proteins were associated with atrophy rates even when the models were adjusted for CSF Aβ42, suggesting that these proteins may be associated with atrophy partly independent of Aβ pathology. Several previous studies have been performed on CSF proteomics in AD (Craig-Schapiro et al., 2011; Hu et al., 2010), but to our knowledge this is the first study linking large scale CSF proteomics to imaging measurements of atrophy in healthy older adults. Previous studies integrating CSF proteins and imaging measurements in this field have mainly examined CSF Aβ42, P-tau and T-tau (Fjell et al.; Schott et al., 2010; Tosun et al., 2011). Our results are in agreement with a few previous studies showing that reduced CSF Aβ42 predicts longitudinal brain atrophy in cognitively healthy persons (Fjell et al.; Schott et al., 2010). One earlier study from our group failed to show this association (Tosun et al., 2011), which may be due to shorter follow-up time and/or use of other imaging analysis methods. The present study gives novel information on links between a large number of CSF proteins and longitudinal AD-like atrophy.
Our first major finding was that several CSF proteins obtained at baseline were associated with longitudinal atrophy of hippocampus, inferior temporal cortex, or middle temporal cortex in cognitively healthy people. Elevated P-tau and reduced Aβ42 were associated with atrophy rates in hippocampus, elevated ApoCIII, P-tau, ApoD, A1M, ApoH and IL-16 with atrophy rates in inferior temporal cortex, elevated ApoD, ApoCIII, P-tau, A1M, IL-16 and ApoH and reduced Aβ42 with atrophy rates in middle temporal cortex, and reduced Aβ42 with atrophy rates in posterior cingulate. This is in agreement with previous studies of reduced CSF Aβ42 and elevated CSF P-tau (Blennow et al., 2010) and ApoD (Craig-Schapiro et al., 2011; Terrisse et al., 1998) in patients with AD or other causes of cognitive decline, as well as increased plasma A1M in AD subjects with reduced whole brain volume (Thambisetty et al., 2011) and increased plasma IL-16 in AD (Motta et al., 2007). Several different mechanisms may link these proteins to AD-like brain atrophy. Increased P-tau is widely reported to be associated with AD (Blennow et al., 2010), and probably reflects lysis of neurons containing tau pathology (Buerger et al., 2006). ApoD has been suggested to be involved in neuroprotection, since it reduces effects of excitotoxic injury (He et al., 2009), and is absent from injured human neurons (Navarro et al., 2008). Taken together, our findings are consistent with the hypothesis that several different CSF proteins are related to longitudinal atrophy rates in AD-related brain regions. However, the effects were mild and not significant after correction for multiple comparisons. The present study should be regarded as a pilot study and the identified proteins may be explored further in future studies. To reduce the risk of reporting spurious findings, we only report proteins that were associated with atrophy rates in at least two regions. However, it should be noted that the effects of some proteins still differed between the investigated brain regions, and no protein was significantly associated with atrophy rates in all regions. The reasons for such inconsistencies are unclear. We suggest that proteins with significant effects across several adjacent regions, including P-tau and ApoD for inferior temporal and medial temporal cortex, represent the best candidate protein biomarkers for future studies on proteins related to atrophy rates. Future voxel-wise studies may allow determining in more details the exact brain distribution of these relationships.
The second major finding was that there were significant interactions between CSF Aβ42 and several proteins, in several cases for proteins previously linked to Aβ-metabolism (See supplementary Table 1). Since reduced CSF Aβ42 is a good estimate of brain Aβ load (Fagan et al., 2006), the interactions of these proteins with CSF Aβ42 suggests that brain Aβ pathology influenced the identified proteins’ effects on atrophy rates. In participants with reduced CSF Aβ42, high levels of some proteins (FRTN, S100β, ApoCIII, ApoH and HGF) were associated with increased atrophy rates. For some of the proteins, our findings are in line with previous reports. For example, patients with AD or other types of cognitive decline have been described to have increased CSF levels of ApoH (Abdi et al., 2006; Ohrfelt et al., 2011), S100β (Fagan and Perrin, 2012), and increased brain levels of HGF (Fenton et al., 1998). A possible interpretation of these findings is that the identified proteins mediate neurotrophic effects dependent on and downstream from Aβ pathology. Another possibility is that they reflect biological pathways that are independent from Aβ pathology, but which only results in neuronal atrophy if they are present together with Aβ pathology, constituting a “dual hit” mechanism, where Aβ pathology is a necessary prerequisite for damaging effects of other biological pathways. A third possibility is that they reflect pathways upstream of Aβ pathology, and which Aβ pathology to some extends depends on. Studies simultaneously examining protein levels and longitudinal development of Aβ pathology and atrophy may help to reveal the precise relationships between the identified proteins, atrophy and Aβ pathology.
The third major finding of this study was that several CSF proteins obtained at baseline were associated with atrophy rates independent of Aβ42. We identified such proteins for hippocampus (P-tau), inferior temporal cortex (P-tau, ApoD), and middle temporal cortex (P-tau, ApoCIII, ApoD, TM, A1M, IL-16, AAT and ApoH). The finding that P-tau was associated with longitudinal atrophy of several AD-related regions independently of Aβ42 is intriguing. Both Aβ and tau pathology are common in elderly people without cognitive impairment, indicating that both types of pathology are well established in AD prior to symptom debut (Savva et al., 2009). Tau pathology may also be present in the brain in very young age, although it is not known if such deposits constitute initial stages of AD (Braak et al., 2011). Our results could be taken to support that tau pathology, as reflected by CSF P-tau, modulates AD-like atrophy partly independent of Aβ pathology. Likewise, ApoCIII, ApoD A1M, IL-16, AAT, ApoH and TM levels may also represent mechanisms of atrophy that are partly independent of Aβ pathology. Currently it is not clear to what extent these mechanisms of atrophy are relevant specifically for AD, or whether they rather represent atrophy processes of normal aging or other degenerative processes.
Limitations of the study include the examination of a small number of brain regions, which was done to reduce the problem of multiple comparisons. The regions were chosen a priori to any analysis, and it is possible that examination of other regions would have produced different results. It should also be noted that wide-spread atrophy is seen in normal aging, which makes it difficult to define “preserved regions” for comparison to the regions included in this study (for example, in this cohort, atrophy rates in the lateral occipital lobe was of a similar magnitude as in entorhinal cortex or precuneus). Another limitation of the study is that the results may be biased by the selection of the examined proteins, which were included mainly due to known associations with Aβ metabolism, microglia activity or synaptic function, limiting our capacity to detect proteins associated with unexpected pathways. Future studies may include larger sets of brain regions (or be done using a voxel-based approach), and/or include larger unbiased proteomics data sets, but such studies will encounter statistical difficulties due to problems with multiple comparisons. Finally, we acknowledge the need for replication of these findings in another cohort, especially since the detected statistical effects were small and not significant when correcting for multiple comparisons.
5. Conclusion
We have identified several CSF proteins, primarily linked to microglia activity and/or Aβ-metabolism, where baseline levels are associated with atrophy rates in AD-related brain regions over a follow-up period of 4 years in cognitively healthy elders. These proteins may be involved in biological pathways involved in early stage AD, and may be explored further for prediction of future AD risk. Several proteins interacted with baseline CSF Aβ42, indicating that Aβ pathology moderated their effects on atrophy rates. These proteins may represent key pathological mechanisms linking Aβ pathology to neuronal loss. Some proteins, including P-tau, were related to atrophy rates even when the models were adjusted for Aβ42, and may thus represent mechanisms of neuronal loss that are at least partly independent of Aβ pathology. However, the effects were mild and no proteins had significant effects when correcting for multiple comparisons. The findings thus need to be replicated in other cohorts.
Supplementary Material
Acknowledgments
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from Abbott, AstraZeneca AB, Bayer Schering Pharma AG, Bristol-Myers Squibb, Eisai Global Clinical Development, Elan Corporation, Genentech, GE Healthcare, Glaxo-SmithKline, Innogenetics, Johnson and Johnson, Eli Lilly and Co., Medpace Inc., Merck and Co. Inc., Novartis AG, Pfizer Inc., F. Hoffman-La Roche, Schering-Plough, Synarc Inc., as well as non-profit partners the Alzheimer’s Association and Alzheimer’s Drug Discovery Foundation, with participation from the U.S. Food and Drug Administration. Private sector contributions to ADNI are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of California, Los Angeles. This research was also supported by NIH grants P30 AG010129 and K01 AG030514, the Swedish Research Council, Goteborgs Lakaresallskap, Svenska Lakaresallskapet, Sahlgrenska Universitetssjukhuset, Carl-Bertil Laurells fond, and Klinisk Biokemi i Norden.
Footnotes
Conflicts of interest
NM, PI, RN, and DT report no conflicts of interest. LMS previously was consultant for Innogenetics and collaborates on quality assessment activities as part of the Alzheimer’s Disease Neuroimaging Initiative; serves as a consultant to Janssen AI R & D on biomarker studies. JQT may accrue revenue in the future on patents submitted by the University of Pennsylvania wherein he is co-Inventor and he received revenue from the sale of Avid to Eli Lily as co-inventor on imaging related patents submitted by the University of Pennsylvania; and is the William Maul Measey-Truman G. Schnabel, Jr., M.D. Professor of Geriatric Medicine and Gerontology. MW has been on scientific advisory boards for Pfizer and BOLT Inter-national; has been a consultant for Pfizer Inc., Janssen, KLJ Associates, Easton Associates, Harvard University, inThought, INC Research, Inc., University of California, Los Angeles, Alzheimer’s Drug Discovery Foundation and Sanofi-Aventis Groupe; has received funding for travel from Pfizer, AD PD meeting, Paul Sabatier University, Novartis, Tohoku University, MCI Group, France, Travel eDreams, Inc., Neuroscience School of Advanced Studies (NSAS), Danone Trading, BV, CTAD ANT Congres; serves as an associate editor of Alzheimer’s & Dementia; has received honoraria from Pfizer, Tohoku University, and Danone Trading, BV; has research support from Merck, Avid, DOD and VA; and has stock options in Synarc and Elan. CRJ provides consulting services for Siemens Healthcare and Janssen Research and Development L.L.C. He receives research funding from the National Institutes of Health (R01-AG011378, RO1-AG041851, RO1-AG037551, U01-HL096917, U01-AG032438, U01-AG024904), and the Alexander Family Alzheimer’s Disease Research Professorship of the Mayo Foundation.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdi F, Quinn JF, Jankovic J, McIntosh M, Leverenz JB, Peskind E, Nixon R, Nutt J, Chung K, Zabetian C, Samii A, Lin M, Hattan S, Pan C, Wang Y, Jin J, Zhu D, Li GJ, Liu Y, Waichunas D, Montine TJ, Zhang J. Detection of biomarkers with a multiplex quantitative proteomic platform in cerebrospinal fluid of patients with neurodegenerative disorders. J Alzheimers Dis. 2006;9:293–348. doi: 10.3233/jad-2006-9309. [DOI] [PubMed] [Google Scholar]
- Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nature reviews. Neurology. 2010;6:131–44. doi: 10.1038/nrneurol.2010.4. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70:960–969. doi: 10.1097/NEN.0b013e318232a379. [DOI] [PubMed] [Google Scholar]
- Buchhave P, Minthon L, Zetterberg H, Wallin AK, Blennow K, Hansson O. Cerebrospinal fluid levels of beta-amyloid 1-42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Archives of general psychiatry. 2012;69:98–106. doi: 10.1001/archgenpsychiatry.2011.155. [DOI] [PubMed] [Google Scholar]
- Buerger K, Ewers M, Pirttila T, Zinkowski R, Alafuzoff I, Teipel SJ, DeBernardis J, Kerkman D, McCulloch C, Soininen H, Hampel H. CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain. 2006;129:3035–41. doi: 10.1093/brain/awl269. [DOI] [PubMed] [Google Scholar]
- Chételat G. Alzheimer disease: Aβ-independent processes—rethinking preclinical AD. Nat Rev Neurol. 2013;9:123–124. doi: 10.1038/nrneurol.2013.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chételat G, Villemagne VL, Villain N, Jones G, Ellis KA, Ames D, Martins RN, Masters CL, Rowe CC. Accelerated cortical atrophy in cognitively normal elderly with high β-amyloid deposition. Neurology. 2012;78:477–484. doi: 10.1212/WNL.0b013e318246d67a. [DOI] [PubMed] [Google Scholar]
- Craig-Schapiro R, Kuhn M, Xiong C, Pickering EH, Liu J, Misko TP, Perrin RJ, Bales KR, Soares H, Fagan AM, Holtzman DM. Multiplexed immunoassay panel identifies novel CSF biomarkers for Alzheimer’s disease diagnosis and prognosis. PLoS One. 2011;6:e18850. doi: 10.1371/journal.pone.0018850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA, DeKosky ST, Morris JC, Holtzman DM. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59:512–9. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Perrin RJ. Upcoming candidate cerebrospinal fluid biomarkers of Alzheimer’s disease. Biomark Med. 2012;6:455–76. doi: 10.2217/bmm.12.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton H, Finch PW, Rubin JS, Rosenberg JM, Taylor WG, Kuo-Leblanc V, Rodriguez-Wolf M, Baird A, Schipper HM, Stopa EG. Hepatocyte growth factor (HGF/SF) in Alzheimer’s disease. Brain Res. 1998;779:262–70. doi: 10.1016/s0006-8993(97)00958-x. [DOI] [PubMed] [Google Scholar]
- Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, van der Kouwe A, Killiany R, Kennedy D, Klaveness S, Montillo A, Makris N, Rosen B, Dale AM. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33:341–355. doi: 10.1016/s0896-6273(02)00569-x. [DOI] [PubMed] [Google Scholar]
- Fischl B, van der Kouwe A, Destrieux C, Halgren E, Ségonne F, Salat DH, Busa E, Seidman LJ, Goldstein J, Kennedy D, Caviness V, Makris N, Rosen B, Dale AM. Automatically parcellating the human cerebral cortex. Cereb Cortex. 2004;14:11–22. doi: 10.1093/cercor/bhg087. [DOI] [PubMed] [Google Scholar]
- Fjell AM, Walhovd KB, Fennema-Notestine C, McEvoy LK, Hagler DJ, Holland D, Blennow K, Brewer JB, Dale AM. Brain Atrophy in Healthy Aging Is Related to CSF Levels of A{beta}1-42. Cereb Cortex. doi: 10.1093/cercor/bhp279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Walhovd KB, Fennema-Notestine C, McEvoy LK, Hagler DJ, Holland D, Blennow K, Brewer JB, Dale AM. Brain Atrophy in Healthy Aging Is Related to CSF Levels of A{beta}1-42. Cereb Cortex. 2010 doi: 10.1093/cercor/bhp279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Jittiwat J, Kim J-H, Jenner AM, Farooqui AA, Patel SC, Ong W-Y. Apolipoprotein D modulates F2-isoprostane and 7-ketocholesterol formation and has a neuroprotective effect on organotypic hippocampal cultures after kainate-induced excitotoxic injury. Neurosci Lett. 2009;455:183–186. doi: 10.1016/j.neulet.2009.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneman WJP, Vrenken H, Barnes J, Sluimer IC, Verwey NA, Blankenstein MA, Klein M, Fox NC, Scheltens P, Barkhof F, van der Flier WM. Baseline CSF p-tau levels independently predict progression of hippocampal atrophy in Alzheimer disease. Neurology. 2009;73:935–940. doi: 10.1212/WNL.0b013e3181b879ac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu WT, Chen-Plotkin A, Arnold SE, Grossman M, Clark CM, Shaw LM, Pickering E, Kuhn M, Chen Y, McCluskey L, Elman L, Karlawish J, Hurtig HI, Siderowf A, Lee VM, Soares H, Trojanowski JQ. Novel CSF biomarkers for Alzheimer’s disease and mild cognitive impairment. Acta Neuropathol. 2010;119:669–78. doi: 10.1007/s00401-010-0667-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu WT, Holtzman DM, Fagan AM, Shaw LM, Perrin R, Arnold SE, Grossman M, Xiong C, Craig-Schapiro R, Clark CM, Pickering E, Kuhn M, Chen Y, Van Deerlin VM, McCluskey L, Elman L, Karlawish J, Chen-Plotkin A, Hurtig HI, Siderowf A, Swenson F, Lee VM, Morris JC, Trojanowski JQ, Soares H. Plasma multianalyte profiling in mild cognitive impairment and Alzheimer disease. Neurology. 2012;79:897–905. doi: 10.1212/WNL.0b013e318266fa70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Bernstein MA, Fox NC, Thompson P, Alexander G, Harvey D, Borowski B, Britson PJ, Whitwell LJ, Ward C, Dale AM, Felmlee JP, Gunter JL, Hill DLG, Killiany R, Schuff N, Fox-Bosetti S, Lin C, Studholme C, DeCarli CS, Krueger G, Ward HA, Metzger GJ, Scott KT, Mallozzi R, Blezek D, Levy J, Debbins JP, Fleisher AS, Albert M, Green R, Bartzokis G, Glover G, Mugler J, Weiner MW. The Alzheimer’s Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging. 2008;27:685–691. doi: 10.1002/jmri.21049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9:119–28. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motta M, Imbesi R, Di Rosa M, Stivala F, Malaguarnera L. Altered plasma cytokine levels in Alzheimer’s disease: correlation with the disease progression. Immunol Lett. 2007;114:46–51. doi: 10.1016/j.imlet.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Navarro A, Ordóñez C, Martínez E, Pérez C, Astudillo A, Tolivia J. Apolipoprotein D expression absence in degenerating neurons of human central nervous system. Histol Histopathol. 2008;23:995–1001. doi: 10.14670/HH-23.995. [DOI] [PubMed] [Google Scholar]
- Oh H, Madison C, Villeneuve S, Markley C, Jagust WJ. Association of Gray Matter Atrophy with Age, β-Amyloid, and Cognition in Aging. Cereb Cortex. 2013 doi: 10.1093/cercor/bht017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohrfelt A, Andreasson U, Simon A, Zetterberg H, Edman A, Potter W, Holder D, Devanarayan V, Seeburger J, Smith AD, Blennow K, Wallin A. Screening for new biomarkers for subcortical vascular dementia and Alzheimer’s disease. Dement Geriatr Cogn Dis Extra. 2011;1:31–42. doi: 10.1159/000323417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson A, Vanderstichele H, Andreasen N, De Meyer G, Wallin A, Holmberg B, Rosengren L, Vanmechelen E, Blennow K. Simultaneous measurement of betaamyloid(1-42), total tau, and phosphorylated tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clin Chem. 2005;51:336–45. doi: 10.1373/clinchem.2004.039347. [DOI] [PubMed] [Google Scholar]
- Pimplikar SW, Nixon RA, Robakis NK, Shen J, Tsai L-H. Amyloid-independent mechanisms in Alzheimer’s disease pathogenesis. J Neurosci. 2010;30:14946–14954. doi: 10.1523/JNEUROSCI.4305-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter M, Schmansky NJ, Rosas HD, Fischl B. Within-subject template estimation for unbiased longitudinal image analysis. Neuroimage. 2012;61:1402–1418. doi: 10.1016/j.neuroimage.2012.02.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusinek H, De Santi S, Frid D, Tsui WH, Tarshish CY, Convit A, de Leon MJ. Regional brain atrophy rate predicts future cognitive decline: 6-year longitudinal MR imaging study of normal aging. Radiology. 2003;229:691–696. doi: 10.1148/radiol.2293021299. [DOI] [PubMed] [Google Scholar]
- Savva GM, Wharton SB, Ince PG, Forster G, Matthews FE, Brayne C. Age, neuropathology, and dementia. N Engl J Med. 2009;360:2302–9. doi: 10.1056/NEJMoa0806142. [DOI] [PubMed] [Google Scholar]
- Schott JM, Bartlett JW, Fox NC, Barnes J. Increased brain atrophy rates in cognitively normal older adults with low cerebrospinal fluid Aβ1-42. Ann Neurol. 2010;68:825–834. doi: 10.1002/ana.22315. [DOI] [PubMed] [Google Scholar]
- Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, Dean R, Siemers E, Potter W, Lee VM, Trojanowski JQ. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403–13. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluimer JD, Bouwman FH, Vrenken H, Blankenstein MA, Barkhof F, van der Flier WM, Scheltens P. Whole-brain atrophy rate and CSF biomarker levels in MCI and AD: a longitudinal study. Neurobiol Aging. 2010;31:758–764. doi: 10.1016/j.neurobiolaging.2008.06.016. [DOI] [PubMed] [Google Scholar]
- Terrisse L, Poirier J, Bertrand P, Merched A, Visvikis S, Siest G, Milne R, Rassart E. Increased levels of apolipoprotein D in cerebrospinal fluid and hippocampus of Alzheimer’s patients. J Neurochem. 1998;71:1643–1650. doi: 10.1046/j.1471-4159.1998.71041643.x. [DOI] [PubMed] [Google Scholar]
- Thambisetty M, Simmons A, Hye A, Campbell J, Westman E, Zhang Y, Wahlund LO, Kinsey A, Causevic M, Killick R, Kloszewska I, Mecocci P, Soininen H, Tsolaki M, Vellas B, Spenger C, Lovestone S. Plasma biomarkers of brain atrophy in Alzheimer’s disease. PLoS One. 2011;6:e28527. doi: 10.1371/journal.pone.0028527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosun D, Schuff N, Shaw LM, Trojanowski JQ, Weiner MW Alzheimer’s Disease NeuroImaging I. Relationship between CSF biomarkers of Alzheimer’s disease and rates of regional cortical thinning in ADNI data. Journal of Alzheimer’s disease: JAD. 2011;26(Suppl 3):77–90. doi: 10.3233/JAD-2011-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigand SD, Vemuri P, Wiste HJ, Senjem ML, Pankratz VS, Aisen PS, Weiner MW, Petersen RC, Shaw LM, Trojanowski JQ, Knopman DS, Jack CR. Transforming CSF A?42 measures into calculated Pittsburgh Compound B (PIBcalc) units of brain A? amyloid. Alzheimers Dement. 2011;7:133–141. doi: 10.1016/j.jalz.2010.08.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.