

Summary
Conserved from yeast to humans, TFIIH is essential for RNA polymerase II transcription and nucleotide excision repair (NER). TFIIH consists of a core that includes the DNA helicase Xeroderma pigmentosum B (XPB) and a kinase subcomplex. Trypanosoma brucei TFIIH harbors all core complex components and is indispensable for RNA polymerase II transcription of spliced leader RNA genes (SLRNAs). Kinetoplastid organisms, however, possess two highly divergent XPB paralogs with only the larger being identified as a TFIIH subunit in T. brucei. Here we show that a knockout of the gene for the smaller paralog, termed XPB-R (R for repair) resulted in viable cultured trypanosomes that grew slower than normal. XPB-R depletion did not affect transcription in vivo or in vitro and XPB-R was not found to occupy the SLRNA promoter which assembles a RNA polymerase II transcription pre-initiation complex including TFIIH. However, XPB-R−/− cells were much less tolerant than wild-type cells to UV light- and cisplatin-induced DNAdamage, which require NER. Since XPB-R−/− cells were not impaired in DNA base excision repair, XPB-R appears to function specifically in NER. Interestingly, several other protists possess highly divergent XPB paralogs suggesting that XPBs specialized in transcription or NER exist beyond the Kinetoplastida.
Keywords: Xeroderma pigmentosum B, DNA repair, nucleotide excision repair, helicase, TFIIH, trypanosome
Introduction
The multi-subunit transcription factor TFIIH is of central importance to transcription initiation by RNA polymerase (pol) II and DNA nucleotide excision repair (NER) in yeast and mammalian systems (Feaver et al., 1993; Schaeffer et al., 1993; Compe and Egly, 2012). It consists of two sub-complexes: the core and the cyclin activating kinase (CAK) complex. The core complex includes the two ATP-dependent DNA helicases Xeroderma pigmentosum B (XPB, also known as ERCC3, RAD25, SSL2), and XPD (also known as ERCC2, RAD3), their respective regulators p52 and p44 along with p62, p34 and p8 (also known as TTDA, TFB5). XPD connects the core complex to the CAK complex which consists of cyclin-dependent kinase 7 (CDK7), cyclin H and MAT1. Both XPB and XPD belong to the SF2 superfamily of helicases and contain seven characteristic helicase motifs, namely Walker motifs I, Ia, II, III, IV, V, and VI. While XPB is a 3′ – 5′ helicase, XPD exhibits 5′ - 3′ activity. Recent studies in human cells have demonstrated that these helicases have distinct roles in NER and transcription. The ATPase activity of XPB is essential for DNA opening in both repair and transcription while its helicase activity is specifically important for transcription: by unwinding DNA it facilitates RNA pol II escape from the promoter (Lin et al., 2005; Coin et al., 2007). The TFIIH subunit p52 directly interacts with an N-terminal region of XPB, termed XPB domain, and regulates its ATPase activity (Jawhari et al., 2002; Coin et al., 2007). In contrast, XPD helicase activity is essential for NER whereas it appears to be dispensable for transcription initiation (Tirode et al., 1999; Coin et al., 2007). Given the importance of these helicases in NER, it is not surprising that mutations in XPB and XPD cause deficiency in NER leading to the human diseases Xeroderma pigmentosum, Cockayne Syndrome and Trichothiodystrophy (Compe and Egly, 2012).
XPB homologs have been identified in both bacteria and archaea (Richards et al., 2008; Biswas et al., 2009; Balasingham et al., 2012). Recent data suggest that archaeal XPB functions in NER. Crystal structure data of XPB from the archaeon Archaeoglobus fulgidus revealed a DNA damage recognition domain (Fan et al., 2006), and a functional analysis of the association between Sulfolobus solfataricus XPB and BAX1 endonuclease showed that this complex can recognize, unwind and cleave model NER substrates in vitro (Rouillon and White, 2010). Moreover, homologs of other eukaryotic NER proteins have been identified in archaeal organisms as well (Rouillon and White, 2011). Conversely, the bacterial NER pathway is controlled by so-called Uvr proteins whose amino acid sequences are different from those of eukaryotic NER proteins (Truglio et al., 2006). Interestingly, bacterial and archaeal XPB appears to function independently of a TFIIH complex since, except for XPD, TFIIH subunit homologs have remained elusive in these organisms (Rouillon and White, 2011).
In eukaryotes, XPB exerts its dual function in transcription and DNA repair exclusively within a TFIIH complex (Compe and Egly, 2012). Notably, the completed genomes of members of the protistan family Trypanosomatidae, which include the human parasites Trypanosoma brucei, Trypanosoma cruzi and Leishmania major, harbor two distinct XPB genes that encode two divergent XPB paralogs (Ivens et al., 2005). The T. brucei TFIIH complex has been functionally, biochemically and structurally characterized, and has been shown to have a basal function in RNA polymerase (pol) II transcription as in other eukaryotes (Lee et al., 2007; Lecordier et al., 2007). This function, however, may be restricted to a single gene, namely the spliced leader (SL) RNA gene (SLRNA). In trypanosomatids, protein coding genes are arranged in long tandem arrays that are transcribed polycistronically. Individual mRNAs are then processed from precursor RNA by polyadenylation and SL trans splicing (Günzl, 2010). Transcription of the gene arrays initiates predominantly in divergent strand-switch regions (dSSRs) in which the arrays are arranged head-to-head (Martinez-Calvillo et al., 2003; Martinez-Calvillo et al., 2010). While dSSRs typically harbor two peaks of open chromatin marks, it remains unclear whether basal transcription factors such as TFIIB or TFIIH are required for RNA pol II transcription initiation in these regions (see reviews by Alsford et al., 2012; Günzl, 2012). In contrast, SLRNA genes, which are tandemly linked on chromosome 9 and encode the SL donor in the trans splicing process, are transcribed monocistronically by RNA pol II from a concrete transcription initiation site. Accordingly, the SLRNA promoter assembles a conventional, albeit highly divergent, transcription pre-initiation complex which includes TFIIH (Günzl, 2012). The T. brucei TFIIH was characterized biochemically by tandem affinity purification revealing a full core of seven subunits and two additional subunits, termed TSP1 and TSP2, which most likely represent divergent subunit orthologs of the factor TFIIE (Lecordier et al., 2007; Lee et al., 2009). Importantly, only the larger of the two XPB paralogs was identified in the TFIIH complex regardless of whether the complex was purified through tagging XPD or TSP2 (Lecordier et al., 2007; Lee et al., 2009).
Here, we functionally characterized the smaller T. brucei XPB paralog which has previously been named XPBz (Lecordier et al., 2007). Our results showed that this helicase was neither important for transcription nor essential for the viability of the insect stage, procyclic form of the parasite in culture although the gene knockout did retard trypanosome proliferation. Instead, we found that the helicase specifically functioned in NER and, correspondingly, was localized throughout the nucleus. We therefore renamed this helicase XPB-R (R for repair). Furthermore, our results strongly indicate that XPB-R does not assemble into a TFIIH complex yet forms a repair complex with the trypanosome p52 ortholog. Interestingly, comparative genomics showed that the two divergent XPB orthologs are present in all kinetoplastids including the bodonid Bodo saltans and a phylogenetic analysis revealed that highly divergent XPB paralogs are present in distantly related protistan organisms suggesting that a bifunctional TFIIH complex is not the norm among deep branching eukaryotes.
Results
XPB-specific domains are conserved in both trypanosome XPB and XPB-R
Genome annotation of T. brucei identified two genes, Tb927.3.5100 and Tb927.11.16270 (accession numbers are from www.genedb.org; Logan-Klumpler et al., 2012), that encode XPB proteins with calculated masses of 105 kDa and 89 kDa, respectively (Berriman et al., 2005). The sequences of these two XPB paralogs are divergent, exhibiting only 24.2% identity and 37.1% similarity between them according to a pair-wise alignment using the EMBOSS Needle server (http://www.ebi.ac.uk/Tools/psa/emboss_needle/). Despite this divergence both proteins are clearly XPB helicases: they harbor all seven Walker motifs of the SF2 superfamily of helicases and they possess an N-terminal XPB domain that interacts with the regulatory TFIIH subunit p52 (Jawhari et al., 2002; Coin et al., 2007), an XPB-specific R-E-D residue loop implicated in recognition of damaged DNA sites (Fan et al., 2006; Oksenych et al., 2009), and an XPB signature sequence motif that was recently identified by a comparative genomics analysis (Figs. 1A and 1B) (Bedez et al., 2013). Both XPB paralogs are present in all sequenced kinetoplastid genomes, including that of the bodonid B. saltans, and a multiple sequence alignment showed that XPB domains are equally well conserved among the paralogous sequences (Fig. S1). Moreover, the archaeal XPB structure revealed the presence of a Thumb-like domain (ThM) between Walker motifs III and IV that is structurally conserved in human XPB and thought to cooperate with the R-E-D loop in anchoring XPB to sites of DNA damage (Fan et al., 2006; Oksenych et al., 2009). While sequence conservation between human and archaeal ThM domains is very limited, there is clear sequence conservation between the human ThM sequence and the aligned kinetoplastid XPB sequences further supporting the notion that kinetoplastid organisms harbor two functional XPB paralogs (Fig. S1).
Fig. 1. Trypanosomatids possess two different XPB helicases.

A. Schematic (to scale) of human XPB (Hs XPB) and of T. brucei (Tb) XPB-R and XPB showing the XPB and RED domains, Walker motifs I, Ia, II-VI, and an XBP-specific sequence signature motif (SSM).
B. Alignment of XPB and RED domain sequences from humans (Hs), Drosophila melanogaster (Dm), Caenorhabditis elegans (Ce), Saccharomyces cerevisiae (Sc), and T. brucei. Positions that are identical and similar across the model organisms are shaded in black and gray, respectively. Specific conservations in either TbXPB-R or TbXPB are shaded in pink and positions that are identical in trypanosome XPBs and different from those of the model organisms are shaded in cyan. Stars in the upper panel identify the phenylalanine and threonine residues found mutated in many Xeroderma pigmentosum patients. Stars in the lower panel identify Walker motif III and the R-E-D residue loop.
C. Immunoprecipitation (IP) of either XPB-R-PTP (left panels) or p52-HA (right panels). Equivalent amounts of the input extract (Inp) and the supernatant (S) were analyzed whereas four times of the precipitate (4xP) were loaded. XPB-R-PTP was detected with the monoclonal anti-protein C epitope HPC4 antibody, p52-HA with a monoclonal anti-HA antibody, and the TFIIH subunit TSP2 with a polyclonal immune serum. Precipitated XPB-R-PTP was released from beads by TEV protease digest which removed the protein A domains, resulting in the smaller XPB-R-P protein. Asterisks identify the N-terminally shortened XPB-R species seen in extract.
To further verify the nature of XPB-R, we tested whether this protein interacts with its functional partner p52. We created an insect-stage, procyclic T. brucei cell line in which XPB-R and p52 (accession number Tb927.10.5210) were C-terminally tagged with the composite PTP tag and the HA tag, respectively. Previously, we have shown that p52-HA interacted with XPD as part of the TFIIH complex and correctly localized to the nucleus, indicating that the tag does not interfere with p52 function (Lee et al., 2007). Similarly, cell proliferation assays (see below) indicated that XPB-R-PTP was functional as well. In whole cell lysates XPB-R-PTP was expressed as a single protein of correct size in two independently derived cell lines. However, in the extract sample, the appearance of a second band, which was 15 kDa smaller than the full length protein, suggested that some of XPB-R-PTP was cleaved at the N-terminus during extract preparation (Fig. S4). Both bands were precipitated with IgG beads which bind to the protein A domains of the PTP tag (Fig. 1C). p52-HA was co-precipitated but TSP2, a trypanosome TFIIH subunit partner of p52, was not. In the reciprocal anti-HA immunoprecipitation experiment, full length XPB-R-PTP was efficiently co-precipitated confirming the XPB-R/p52 interaction. The smaller, N-terminally degraded XPB-R-PTP form, however, did not interact with p52-HA which indicated that trypanosome XPB-R does possess a functional N-terminal p52 interaction domain. This negative co-precipitation result confirmed our previous finding (Nguyen et al., 2006) that protein G-bound anti-HA antibody does not co-precipitate PTP-tagged protein non-specifically. As expected, TSP2 did co-precipitate with p52, verifying that both proteins are part of the same TFIIH complex. Together, these results showed that kinetoplastid organisms harbor two highly divergent XPB paralogs with conserved functional domains. Although T. brucei XPB-R was not detected in TFIIH complexes that were purified via tagged XPD or TSP2 (Lee et al., 2009), its interaction with p52 suggested that it is a functional enzyme in this parasite.
Knockout of XPB-R retards trypanosome proliferation in culture
Previously, XPB-R silencing in procyclic trypanosomes did not affect cell proliferation (Lecordier et al., 2007). We confirmed this result (data not shown) and, in addition, analyzed XPB-R silencing in bloodstream form trypanosomes by inducing synthesis of XPB-R dsRNA from a tetracycline operator-controlled stem-loop construct that had been integrated in tetracycline repressor-expressing single marker cells (Wirtz et al., 1999). Although semi-quantitative reverse transcription (RT)-PCR showed that XPB-R mRNA was reduced after one and two days of induction, XPB-R silencing did not affect trypanosome proliferation suggesting that XPB-R is not an essential gene in either procyclic or bloodstream form trypanosomes (Fig. S2). Accordingly, we succeeded in generating a procyclic XPB-R−/− knockout cell line in which the two XPB-R alleles were replaced by the hygromycin (HYGR) and blasticidin (BLAR) resistance markers (Fig. 2A). After the second transfection of a single allele knockout XPB-R+/− cell line, we obtained two independently derived, double resistant XPB-R−/− cell lines which we termed KO-1 and KO-2. The gene knockouts were confirmed by a PCR of genomic DNA prepared from wild-type, XPB-R+/− and KO cell lines using primers that annealed to the 5′ and 3′ XPB-R gene flanks outside of the transfected, linear DNA constructs. While the KO cells gained HYGR- and BLAR-specific amplification products, they completely lost the wild-type product (Fig. 2B). Correspondingly, an RT-PCR analysis with oligonucleotides specific to the XPB-R coding region showed that the KO cells did not express XPB-R mRNA any longer (Fig. 2C). The successful XPB-R knockout demonstrated that this gene was dispensable for cultured procyclics. However, the KO cell lines clearly grew slower than wild-type cells. While our wild-type strain had a doubling time of 8.5 hours, KO-1 and KO-2 cells doubled every 13.8 and 15.6 hours, respectively (Fig. 2D). This slow growth phenotype remained constant over a period of three months and was not affected by the presence or absence of antibiotics (data not shown). To determine whether the knockout delayed the cell cycle in a particular stage, we determined the number of kinetoplasts and nuclei in DAPI-stained wild-type and XPB-R−/− cells (n=100 for each cell line). Compared to the wild-type cell line, we found a strong accumulation of 2K2N cells (25% versus 6%) and a concomitant decrease of 1K1N trypanosomes (44% versus 68%) indicating a cell cycle-relevant function of XPB-R in late mitosis/cytokinesis (Fig. S3).
Fig. 2. A knockout of XPB-R increases the doubling time of trypanosomes in culture.

A. Schematic (not to scale) of the XPB-R locus in wild-type and XPB-R−/− (KO) cells. In the latter, the XPB-R coding regions of the two alleles were replaced with those of hygromycin phosphotransferase (HYGR) and blasticidin-S deaminase (BLAR). Oligonucleotides used for competitive PCR analysis of XPB-R, HYGR and BLAR alleles are indicated by arrows.
B. Corresponding competitive PCR analysis of total DNA prepared from wild-type cells, cells in which one XPB-R allele was knocked out (XPB-R+/−), and two independently obtained XPB-R−/− knockout cell lines (KO-1, KO-2)
C. RT-PCR of XPB-R and, as a control, of U2A′ mRNA. A control reaction with wild-type RNA in the absence of reverse transcriptase (−RT) demonstrated effective removal of genomic DNA from the RNA preparation.
D. KO-1 and KO-2 cells have a prolonged doubling time versus wild-type cells.
XPB-R does not function in transcription
To our knowledge eukaryotic XPB helicases function exclusively in transcription and NER. It was therefore possible that XPB-R had an important, albeit non-essential, function in transcription, possibly in a regulatory role. Since the SLRNA promoter is the only trypanosome promoter known to assemble an RNA pol II transcription pre-initiation complex including TFIIH (Lee et al., 2010), we first tested whether XPB-R occupies the SLRNA promoter by chromatin immunoprecipitation (ChIP). We previously showed that chromatin bound by PTP-tagged proteins can be highly enriched by a ChIP-grade, anti-protein A antibody (Lee et al., 2010). Accordingly, when we used a cell line that exclusively expressed functional XPD-PTP (Lee et al., 2007), we observed a 21 fold enrichment of the SLRNA promoter over a precipitation with a comparable non-specific immune serum (Fig. 3A). This occupancy was strongly reduced in the SLRNA intergenic spacer approximately 500 bp downstream of the promoter, revealing the specificity of the ChIP assay. To analyze XPB-R in an equivalent way, we generated a cell line that exclusively expressed XPB-R-PTP. This was achieved by targeted integration of plasmid XPB-R-PTP-NEO into the remaining XPB-R allele of the single knockout XPB-R+/− cell line (Fig. S4). To assess whether the PTP tag interferes with XPB-R function, we determined the doubling time of the XPB-R-PTP-expressing cells which, with 11.1 hours, was intermediate between wild-type and XPB-R−/− cells. Interestingly, single knockout XPB-R+/− cells exhibited a similarly prolonged doubling time (12 h) suggesting that maximal trypanosome proliferation requires a sufficiently high level of XPB-R and that the slower proliferation rate of XPB-R-PTP-expressing cells is due to haplo-insufficiency rather than a functionally deleterious PTP tag. Nevertheless, XPB-R-PTP did not occupy the SLRNA promoter, indicating that this helicase has no function in pre-initiation complex formation and RNA pol II transcription initiation (Fig. 3A).
Fig. 3. XPB-R does not function in transcription.

A. Anti-XPD-PTP and anti-XPB-R-PTP ChIP with a ChIP-grade, polyclonal anti-protein A antibody (α ProtA) and a comparable non-specific immune serum (nonsp. IS). Top panels, semiquantitative PCR of the SLRNA promoter (SL prom), the SLRNA intergenic region (SL intgen), and the α tubulin coding region (α tub cod). Lower panel, corresponding qPCR analysis showing the corrected fold difference of precipitated DNA over the nonspecific immunoprecipitation control.
B. Co-transcription of the RNA pol I-recruiting GPEET-trm template DNA and the SLRNA promoter template SLins19 in extract prepared from cells that exclusively express XPB-R-PTP. The extract was either mock-treated or depleted of XPB-R-PTP by IgG affinity chromatography (depl). In a third reaction, tandem affinity-purified XPB-R was added to the XPB-R-PTP-depleted extract (depl + XPB-R elu). Main panel, transcription signals were obtained by primer extension of newly synthesized RNA with radiolabeled oligonucleotides specific for GPEET-trm or for SLins19 RNA. Left panel, detection of XPB-R-PTP including its partially degraded product (degrad prod) and, as a loading control, of U2A′ in mock-treated and XPB-R-PTP-depleted extract by immunoblotting.
C. Nascent RNA was radiolabeled in permeabilized wild-type (WT), KO-1 and KO-2 cells, separated by denaturing PAGE, and visualized by autoradiography. Signals that correspond to tRNAs, the SL RNA, and pre-mRNA/pre-rRNA are indicated on the right.
D. Labeled nascent RNA was hybridized to dot blots of plasmids containing the complete coding regions of GPEET procyclin, 18S rRNA, heat shock protein 70 (HSP70), actin and α tubulin. In addition, a plasmid harboring the coupled genes of threonine tRNA and U6 snRNA (tRNATHR/U6 snRNA) and a control plasmid without insert (ctrl) were analyzed.
E. Quantification of RNA signals from three independent experiments. In each case the wild-type signal was set to 100. Error bars represent standard deviations.
To further validate this notion, we prepared transcriptionally active extract from XPB-R-PTP-expressing cells and depleted the helicase by IgG affinity chromatography. Although the extract was almost fully depleted of XPB-R, it supported transcription of the SLRNA promoter template SLins19 equally well as mock-depleted extract did (Fig 3B). Accordingly, adding back tandem affinity-purified XPB-R (see below) to the extract had no impact on transcription efficiency.
Finally, we employed permeabilization of wild-type and XPB-R−/− cells to label nascent RNA which, in this system, increases linearly for up to 20 minutes (Ullu and Tschudi, 1990). Due to a very strong synthesis rate, newly synthesized SL RNA can be directly visualized after separating labeled RNA by denaturing PAGE (Fig. 3C). To assess protein coding gene transcription in these experiments, we hybridized the labeled RNA to dot blots of plasmids that contained whole coding regions (Fig. 3D). Quantification of SL RNA bands by densitometry and of dots by scintillation counting in three independent experiments did not reveal any nascent RNA synthesis defect in XPB-R−/− cells in either KO cell line (Fig. 3E). This was true for the RNA pol I transcripts 18S rRNA and GPEET procyclin mRNA, for the RNA pol II-synthesized heat shock protein 70, actin and α tubulin mRNAs, and for the RNA pol III-derived threonine tRNA and U6 snRNA. Therefore, we concluded that XPB-R does not function in transcription.
XPB-R is important for Nucleotide Excision Repair
NER is essential for the removal of covalently cross-linked bulky DNA adducts that cause a severe distortion of the DNA double helix. Formation of such DNA adducts can be induced by exposure to UV light or by treatment with cisplatin, strategies that have been employed in various systems (Kamileri et al., 2012) including trypanosomes (Sheader et al., 2004). Ultraviolet (UV) radiation causes two major types of DNA lesions, namely cis-syn cyclobutane-pyrimidine dimers and 6,4 pyrimidine-pyrimidine photoproducts, both of which distort the helical structure of DNA (Thoma, 1999). To determine the role of XPB-R in the repair of UV-induced DNA lesions, wild-type and XPB-R−/− cells were treated with increasing doses of UV radiation. Two days after the exposure, cell densities were determined and compared to those of untreated control cells. Even at the lowermost dose of 300 J/m2, XPB-R−/− cells showed only 17.4% survival as compared to untreated XPB-R−/− cells while 75.3% of wild-type cells that had received the same dose survived relative to the untreated wild-type control. This indicated that the repair of DNA lesions that require NER is strongly impaired in XPB-R−/− cells. To validate this result, wild-type and XPB-R−/− cells were exposed to increasing concentrations of cisplatin, a chemical that induces the formation of bulky DNA lesions. The majority of cisplatin-induced lesions are 1,2-intrastrand crosslinks between adjacent guanines or between guanine and adenine both of which are efficiently removed by NER (Kartalou and Essigmann, 2001). As shown in figure 4B, XPB-R−/− cells were more sensitive towards cisplatin treatment than wild-type cells. For example, while treatment with 1 μM of cisplatin reduced wild-type cell survival to 65.5%, only 33.5% of XPB-R−/− cells survived the same treatment. Taken together, the increased sensitivity of XPB-R−/− cells towards UV radiation and cisplatin strongly indicated direct involvement of XPB-R in NER.
Fig. 4. XPB-R−/− cells are specifically sensitive to DNA lesions requiring NER.
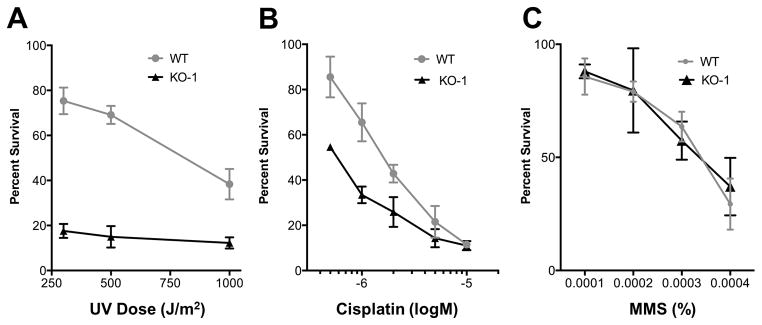
Wild-type (WT) and KO-1 cell survival after treatment with (A) UV light, (B) cisplatin or (C) methyl methanesulfonate (MMS) was determined relative to untreated wild-type and KO-1 cells, respectively. The results were obtained from three independent experiments. Error bars represent standard deviations.
To assess whether XPB-R’s role in DNA repair is specific to NER, cells were treated with methylmethane sulfonate (MMS), an alkylating agent that methylates DNA on N7-deoxyguanine and N3-deoxyadenine. MMS causes formation of DNA base lesions that are repaired by the base excision repair (BER) pathway. If left unrepaired, these MMS-induced lesions can cause stalling of replication forks and lead to single or double strand DNA breaks (Brem et al., 2008). Despite using a range of different MMS concentrations, we did not detect a significant difference between XPB-R−/− cells and wild-type cells in their sensitivity towards MMS (Fig. 4C). We therefore concluded that XPB-R is specifically important for NER and not required for BER.
An XPB-R/p52 complex functions independently of a TFIIH complex
Previously, the T. brucei TFIIH complex was isolated and mass spectrometrically analyzed in four independent experiments: it was purified using XPD as bait by two different research groups (Lecordier et al., 2007; Lee et al., 2009) or through tagging TSP2 (Lee et al., 2009), and it was co-purified as an associated complex of the multi-subunit mediator complex (Lee et al., 2010). In each case, XPB was identified as a TFIIH subunit whereas XPB-R was never found. Although unlikely, it is possible that XPB-R is part of a distinct TFIIH complex of minor abundance that was not detected in these biochemical protein complex characterizations. To facilitate XPB-R detection in isolated TFIIH complexes, we tried unsuccessfully to raise a specific polyclonal immune serum in rats against two partial, recombinant XPB-R proteins that were fused to the GST tag and purified from Escherichia coli (data not shown). Next, we carried out a tandem affinity purification of XPB-R-PTP (Fig. S4). Although the purification was efficient and the final eluate revealed several distinct protein bands, mass spectrometry identified only proteins that typically co-purify as contaminants such as α/β tubulin and HSP70 but no other proteins (Figs. S4C to S4E). Since p52, which did interact with XPB-R-PTP in reciprocal co-IPs (Fig. 1C), was not detected in this analysis, it was possible that tagging XPB-R at the C-terminus had a deleterious effect on the XPB-R/p52 interaction. It therefore appeared that either XPB-R tagging partially interfered with the XPB-R/p52 interaction, or that this interaction is weak and does not withstand purification. To obviate the latter concern, we sedimented crude extract, freshly prepared from cells expressing XPB-R-PTP and p52-HA, through a linear sucrose gradient by ultracentrifugation, a process which is much less stringent than tandem affinity purification. An immunoblot analysis of proteins collected from gradient fractions that were taken from top to bottom showed that p52-HA sedimentation peaked in fractions 6/7 and in fractions 14–16, between the 17S apoferritin and the 19S thyroglobulin markers (Fig. 5A). The latter sedimentation peak corresponded to the previously determined ~17.5 S sedimentation coefficient of the TFIIH complex (Lee et al., 2009) and, accordingly, the TFIIH subunit TSP2 did co-peak with p52-HA in these fractions. The additional peak of TSP2 in fraction 11 likely corresponds to a XPB/TSP1/TSP2 subcomplex that we previously found missing from a partial TFIIH complex (Lee et al., 2009). Importantly though, neither band of XPB-R-PTP showed a peak in fractions 14–16 and they were, in fact, hardly detectable in these fractions, further supporting the notion that XPB-R does not function within a TFIIH complex. Its sedimentation in fractions 5 and 6 suggested that it peaks there by itself and/or in a complex with p52-HA. Together, these data strongly indicated that XPB-R is not part of a TFIIH complex comprising XPD and the other trypanosome TFIIH orthologs of p62, p44, and p34.
Fig. 5. XPB-R and p52 but not XPD function together in NER.

A. Crude extract from XPB-R-PTP-expressing trypanosomes was sedimented through a linear 10–40% sucrose gradient by ultracentrifugation. Fractions were taken from top to bottom and analyzed by immunoblotting as described above. As sedimentation markers IgG (6.6S), appoferritin (17S) and thyroglobulin (19S) were co-analyzed.
B. Upper panel, growth curve of a representative, procyclic cell line (one out of three) in which doxycycline induces p52 silencing. Lower panel, semi-quantitative RT-PCR analysis of p52 mRNA and, as a control, of U2A′ mRNA in total RNA prepared from non-induced cells and from cells that were induced for one or two days.
C. Diagrams showing the survival of cells treated with UV relative to untreated cells. Three cell lines were investigated in which XPD (upper panel), XPB-R (middle panel) or p52 (lower panel) were silenced (+ dox) or not (− dox). Each experiment was conducted three times, and the results were statistically analyzed by an unpaired, two-tailed student’s t test assuming unequal variances. One and two asterisks indicate p values that are smaller than 0.05 and 0.01, respectively.
To corroborate these findings at the functional level, we generated procyclic cell lines for conditional p52 silencing by RNAi analogously to the XPB-R RNAi cell lines described above. Examination of three independently derived cell lines demonstrated that cultures stopped growing 3 days after induction of p52 dsRNA synthesis (Fig. 5B). These growth defects closely resembled those observed previously upon silencing of XPD (Lee et al., 2007), indicating that p52 is an essential component of TFIIH. To assess whether RNAi-mediated depletion of either XPD, XPB-R or p52 in cells led to increased UV susceptibility, we silenced the expression of the respective genes in procyclic cell lines, exposed induced and non-induced cells to 500 or 1000 J/m2 UV light, and recorded cell survival one day after the treatment. While XPD silencing was as efficient as previously published (data not shown), UV exposure did not affect the survival rate of XPD-silenced cells (Fig. 5C, top panel), strongly indicating that trypanosome TFIIH is specialized in transcription and, unlike its bifunctional counterparts in yeast and mammals, not involved in DNA repair. Conversely and as expected, XPB-R silencing significantly increased the UV susceptibility of trypanosomes (Fig. 5C, middle panel). Since UV treatment was similarly lethal to p52-silenced cells (Fig. 5C, bottom panel), these results identify p52 as a functional partner of XPB-R in DNA repair and as the only trypanosome protein so far that is likely involved in both RNA pol II transcription and NER. They also confirm our co-IP results (Fig. 1C).
XPB-R-PTP is localized in the nucleus and partially co-localizes with p52-HA
NER factors including various TFIIH subunits typically exhibit a diffuse nuclear staining pattern (Vermeulen et al., 2000; Hoogstraten et al., 2008). We have previously shown that in T. brucei p52-HA localizes to the nucleus with the strongest accumulation in one or two perinucleolar foci. These foci most likely represent the ~100 tandemly linked SLRNA copies on chromosomes 9 that are transcribed monocistronically, thereby requiring the independent formation of transcription pre-initiation complexes on each and every SLRNA promoter (Lee et al., 2010). Accordingly, when we used a cell line in which the TFIIH helicase XPD and p52 were PTP- and HA-tagged, respectively (Lee et al., 2007), we observed that XPD-PTP predominantly localized to one or two perinucleolar foci in the nucleus with additional faint staining of other nuclear regions (Figs. 6 and S5, left panels). p52-HA invariably co-localized with XPD-PTP in these foci presumably as part of the TFIIH complex, yet it also appeared to be more widely distributed than XPD within the nucleus. When we co-localized XPB-R-PTP with p52-HA in the cell line described above (Figs. 6 and S5, right panels), we detected a partial overlap of the two proteins which supports our finding that XPB-R and p52 do interact. On the other hand, the clearest difference from the XPD analysis was that XPB-R-PTP most often did not co-localize with the brightest p52-HA spots, supporting our finding that XPB-R does not occupy the SLRNA promoter and is not required for RNA pol II transcription initiation at SLRNA genes. These results support a model in which trypanosomes possess a TFIIH complex with a basal function in RNA pol II transcription initiation, and an XPB-R/p52 complex specifically functioning in NER.
Fig. 6.

XPB-R-PTP co-localizes with p52-HA outside putative SLRNA expression foci in the nucleus. p52-HA (green) was co-localized with XPD-PTP (red; left panels) or with XPB-R-PTP (red, right panels). White bars represent 10 μm.
Highly divergent XPB paralogs are present in other protistan lineages
We have shown that T. brucei possesses two highly divergent XPB paralogs. One participates in the transcriptionally active TFIIH complex while the other is important for DNA repair and, likely, functions independently of TFIIH. Since TFIIH analyses have mainly been restricted to yeast and higher eukaryotes, we wanted to know whether divergent XPB paralogs could be found in other eukaryotes. We carried out a phylogenetic analysis of XPB sequences of 33 species whose genomes were completely sequenced (Fig. S6). Each XPB sequence that was retrieved gave an E value smaller than e−95 during a protein-protein BLAST analysis against the human XPB sequence and they all harbored the R-E-D motif, except for one of the two sequences found in the ciliate Paramecium tetraurelia that contained an R-Q-D motif instead (Fig. S1). Despite the small E-values, multiple sequence alignment revealed several blocks of ambiguous alignment due to poorly conserved regions. To restrict our analysis to appropriately aligned sequences we applied the Gblock algorithm (Talavera and Castresana, 2007) in combination with maximum likelihood calculations. As expected, metazoa, yeasts and fungi have only a single XPB sequence which correlates with the dual function of TFIIH described in these organisms. The two plant species investigated, Oryza sativa and Arabidopsis thaliana, do possess two XPBs but their encoded sequences are nearly identical and more closely conserved within each species. This strongly suggests that these XPB genes stem from recent gene duplications that have not functionally diverged. In contrast, all kinetoplastid organisms possess two highly divergent XPBs that clearly segregate into two separate branches, one with T. brucei XPB and one with T. brucei XPB-R. Even the only bodonid species investigated, B. saltans, encoded unambiguously identifiable orthologs of these two helicases. The root of the two clusters is very deep, raising the possibility that the presence of two functionally distinct XPB helicases is an ancient eukaryotic trait. This hypothesis is supported by the presence of similarly divergent XPB paralogs in the ciliates Tetrahymena thermophila and P. tetraurelia although the ciliate XPB sequences do not form two separate clusters. Ciliates are grouped together with Apicomplexa in the supergroup Chromalveolata. However, all apicomplexan species investigated have only a single XPB gene. On the other hand, Entamoeba histolytica and Entamoeba dispar again have two highly divergent XPB genes that form two deeply rooted branches, while other members of the supergroup Amoebozoa, Dictyostelium discoideum and Acanthamoeba castellanii, have only a single XPB gene. Finally, among early diverged protists, the Giardia lamblia genome revealed only a single XPB gene whereas Trichomonas vaginalis and Naegleria gruberi encode two divergent XPB paralogs, although their roots are not as deep as those of ciliates and Entamoeba.
If distinct XPBs for transcription and DNA repair were a plesiomorphic trait in eukaryotes, we would expect a phylogenetic tree with separate XPB-R and XPB branches. This was never observed despite applying several different phylogenetic approaches (data not shown). On the other hand, the bootstrap values at the base of the tree in figure S6 are not high and it is possible that specific XPB-R and XPB branches do exist. Accordingly, when we carried out the same phylogenetic analysis with an archaeal sequence as an outgroup, the basal branching of the tree changed while the XPB and XPB-R dichotomies described here remained (Fig. S7). Therefore, it will require more whole genome sequences from protistan organisms and a more comprehensive approach to solve the basal eukaryotic branching of the XPB tree unambiguously. Until then our data suggest that the presence of two functionally distinct XPBs is a phenomenon that exists beyond the Kinetoplastida.
Discussion
This is the first report of a eukaryotic XPB paralog that is specialized in DNA repair while having no role in transcription. We showed that transcription of genes by all three nuclear RNA pols was not impaired in XPB-R−/− cells. Moreover and in contrast to TFIIH subunits, XPB-R did not occupy the SLRNA promoter, the only trypanosome promoter known to assemble a RNA pol II transcription pre-initiation complex, and depletion of XPB-R from extract did not affect SLRNA transcription in vitro. On the other hand, removal of the XPB-R gene rendered trypanosomes highly sensitive to UV irradiation and cisplatin, but not to methylmethane sulfonate, strongly indicating that XPB-R, like its XPB homologs from yeast to humans, specifically functions in NER and not in other DNA repair pathways. Interestingly, although not absolutely required for cell viability, the expression level of XPB-R directly influenced the trypanosome doubling time. Knockout of a single XPB-R allele increased the 8.5 h doubling time of wild-type to 12 h and a complete knockout to ~14.7 h. Given that a specific down-regulation of XPB-R would result in slow growth and impaired DNA repair, we speculate that regulation of its expression may play a role in pathways of programmed cell death that have been described in trypanosomes (Welburn et al., 2006; Michaeli, 2012).
An interesting question emanating from our study is why XPB-R−/− cells take longer to double than wild-type cells. One possibility is that XPB-R−/− cells accumulate mutations that lead to increased cell death thus slowing culture growth. If this was the case, one would expect that XPB-R−/− cell lines would grow slower over time because all cells would accumulate mutations simultaneously. However, the doubling time of the knockout cells did not change over a period of ~150 generations. In addition, we did not detect a substantial increase of aberrant or dead cells in XPB-R−/− cell cultures when compared to wild-type cultures suggesting that accumulation of mutations is not the major cause of the growth defect (Fig. S3 and data not shown). Instead, our results indicated that XPB-R−/− cells take substantially longer to complete mitosis and/or initiate cytokinesis because a quarter of XPB-R−/− cells, compared to 6% of wild-type cells, was found to be in the precytokinesis stage with two kinetoplasts and two nuclei. Given that XPB-R is a DNA-dependent helicase, this finding suggests that XPB-R has a role in late mitosis. While this could be a trypanosome-specific function, a recent study did find human XPB but not other TFIIH subunits to be associated with centrosomes and adjacent parts of the mitotic spindle during mitosis (Weber et al., 2010).
Thus far, eukaryotic XPB function has exclusively been described in the context of the TFIIH complex (Naegeli and Sugasawa, 2011; Compe and Egly, 2012). The two TFIIH subunits that interact with XPB and modify its enzymatic activities are p52 and p8 (Fregoso et al., 2007; Coin et al., 2006; Coin et al., 2007). Accordingly, our analysis showed that trypanosome XPB-R does interact with p52, and while trypanosomes do possess a p8 ortholog that is part of the TFIIH complex (Lee et al., 2009), it remains to be determined whether it interacts with XPB-R. However, our study strongly indicates, in accordance with previous comprehensive TFIIH characterizations (Lecordier et al., 2007; Lee et al., 2009; Lee et al., 2010), that XPB-R does not assemble into a TFIIH complex. In the sedimentation analysis, tagged XPB-R was found almost exclusively near the top of the sucrose gradient and did not exhibit faster co-sedimentation with the TFIIH subunits p52-HA and TSP2. Accordingly, TSP2 did not co-immunoprecipitate with XPB-R from extract. Furthermore, isolation of XPB-R by tandem affinity purification did not co-purify any TFIIH subunit suggesting that tagged XPB-R is not assembled into a stable protein complex. Finally, while the TFIIH subunits XPD and TSP2 were predominantly localized in one or two perinucleolar spots, XPB-R exhibited a more diffuse nuclear staining and rarely co-localized with p52-HA in its perinucleolar spots, further indicating that XPD and XPB-R are not part of the same complex.
How does XPB-R function in NER in the absence of other TFIIH subunits? There are two distinct pathways of NER, namely transcription-coupled NER and global genome NER, which use different proteins to detect DNA lesions and recruit TFIIH to the sites of damage (Naegeli and Sugasawa, 2011). In the Tritryp genome projects, most of the factors involved in NER factors have been identified suggesting that both pathways are functional in trypanosomes (Ivens et al., 2005; Berriman et al., 2005); recently reviewed in Passos-Silva et al., 2010). A key factor in global genome NER is XPC (accession number for the T. brucei ortholog is Tb927.9.11930) which, by directly interacting with XPB, recruits TFIIH to sites of DNA damage. Moreover, it was shown in the human system that the R-E-D residue loop, the ThM domain and the ATPase activity of XPB are important for recruitment of TFIIH to damaged sites (Oksenych et al., 2009). All of these domains are conserved in XPB-R suggesting that it can be recruited to DNA lesions by XPC in the absence of a TFIIH complex. Transcription-coupled NER removes DNA lesions that stall RNA pol II at actively transcribed genes (Laine and Egly, 2006; Hanawalt and Spivak, 2008). A key factor in this pathway is the so-called Cockayne syndrome type B or CSB protein (Tb927.7.4080), which is important for the recruitment of TFIIH and other NER factors to damaged DNA. Although it is not entirely clear how TFIIH is recruited in this pathway, early evidence revealed a direct link between CSB and XPB (Tantin, 1998) which would allow direct recruitment of XPB-R by trypanosome CSB for transcription-coupled NER.
After recruitment to DNA lesions, the function of TFIIH in global genome NER is to open up the site of damage by XPB ATPase and XPD helicase activity. The ATPase activity of XPB apparently drives the protein like a wedge into the damaged bubble enabling XPD, whose helicase activity alone is important for NER, to unwind the DNA (Coin et al., 2007). Since trypanosome XPB-R apparently does not function together with XPD, the XPB-R ATPase-driven opening of the damaged DNA site may be sufficient intrypanosomes for nucleotide repair. Alternatively, XPB-R may be a more processive helicase than its mammalian counterpart whose activity is important for NER. Mammalian XPD has also been implicated in DNA damage recognition and verification (Mathieu et al., 2010). Since there is no evidence that trypanosome XPB-R and XPD form a common complex, this function is either absent in trypanosomes or achieved independently of XPB-R. In the absence of a stable XPB-R complex, it appears that XPB-R functions like its archaeal ortholog. In vitro assays have shown that XPB of the archaeon Sulfolobus solfataricus can open up DNA lesions by 6–8 bp which is sufficient for the endonuclease Bax1 to cleave off the damaged DNA (Rouillon and White, 2010). Bax1 is thought to be the functional homolog of the eukaryotic NER endonuclease XPG (Rouillon and White, 2010) which is encoded in the T. bruceigenome (Tb927.9.11760; E value of 8e−20 derived from a protein-protein blast with the human XPG amino acid sequence) and likely functions together with XPB-R. In sum, it appears that trypanosomes have a simplified NER machinery that is separate from the transcriptionally essential TFIIH complex.
We have been able to combine gene silencing with UV exposure to assess the function of proteins in NER. This approach clearly demonstrated that XPD silencing did not alter cell survival after UV exposure whereas XPB-R and p52 silencing clearly increased the UV susceptibility of cells (Fig. 5C). These data strongly support a model in which trypanosome TFIIH has an essential role in RNA pol II transcription while NER is carried out through a simplified, XPB-R-based machinery. This dichotomy in trypanosomes provides a unique opportunity to decipher specific DNA repair and transcription functions of XPB. For example, amino acids specifically required for DNA repair should be conserved in kinetoplastid XPB-R sequences as well as in the bifunctional XPBs of model organisms but not in kinetoplastid XPBs. Accordingly, several such positions were identified in the XPB multiple sequence alignment (Fig. S1). Moreover, the bifunctional nature of human and yeast XPB has hampered XPB analysis in vivo since all mutations affecting the transcriptional function of XPB are lethal, an obstacle absent in future XPB-R analyses.
Finally, specific NER machinery may not be restricted to Kinetoplastida. Our survey of completely sequenced genomes revealed the presence of two highly divergent XPBs in several protistan taxa that belong to different supergroups (Dacks et al., 2008). In addition to the Excavata, the supergroup of kinetoplastids, highly divergent XPBs were found in the Ciliophora of the supergroup Chromalveolata and in the genus Entamoeba which is part of the Amoebozoa supergroup. Although our initial XPB phylogenetic analysis did not provide unambiguous branching at the base of the tree, which may be the reason why we did not observe an overall dichotomy of XPB and putative XPB-R sequences, our results raise the possibility that a DNA repair-dedicated XPB helicase is an ancestral eukaryotic trait.
Experimental Procedures
DNAs
For XPB-R and p52 silencing, the coding region from position 8 to position 487 and from position 1 to 500, respectively, were integrated in a stem-loop arrangement (sense-stuffer-antisense) into the pT7-stl construct (Brandenburg et al., 2007). For C-terminal PTP tagging of XPB-R, 499 bp of the 3′ terminal XPB-R coding sequence including an internal Bsgl linearization site were cloned into the pC-PTP-NEO vector using the vector’s ApaI and NotI restriction sites (Schimanski et al., 2005). DNA oligonucleotides used for semi-quantitative RT-PCR or PCR are specified in Table S1 of the supplemental material. Plasmids used in dot blots were described previously and contained the complete coding regions of GPEET procyclin, α tubulin, actin, HSP70 and 18S rRNA or the threonine tRNA/U6 snRNA gene association (Schimanski et al., 2006). The template plasmids for in vitro transcription, GPEET-trm and SLins19, have been described previously (Günzl et al., 1997; Laufer et al., 1999).
Cells
T. brucei brucei 427 cell culture, stable transfection by electroporation, and the generation of clonal cell lines by selection and limiting dilution was carried out as described (Lee et al., 2007). In RNAi experiments, dsRNA synthesis was induced with 2 μg/ml of doxycycline. For measurement of procyclic and bloodstream form culture growth, cells were counted and diluted daily to 2 × 106 cells/ml and 2 × 105 cells/ml, respectively. The procyclic clonal XPB-R+/− cell line was generated by knocking out one of the XPB-R alleles with a PCR product in which 101 bp of XPB-R 5′ and 3′ gene flanks were fused to the hygromycin phosphotransferase coding region. The clonal cell line that exclusively expressed XPB-R-PTP was obtained by targeted integration of the linearized plasmid XPB-R-PTP-NEO into the remaining XPB-R allele of the XPB-R+/− cell line. For HA tagging of p52, the construct p52-HA-BLA (Lee et al., 2007) was linearized with StuI and transfected into a clonal cell line that exclusively expressed XPB-R-PTP. Immunoblotting was performed to confirm correct tagging of the protein in each clonal cell line. The XPB-R−/− KO-1 and KO-2 knockout cell lines were created by removing the remaining XPB-R allele in XPB-R+/− cells with a PCR product of the blasticidin-S deaminase coding region surrounded by 101 bp of XPB-R 5′ and 3′ gene flanks, and by limiting dilution of the cells immediately after transfection. Correct DNA integrations were ensured in clonal cell lines by PCR amplification of genomic DNA using oligonucleotides that hybridize outside of the transfected DNA constructs.
RNA analysis
The relative abundance of RNAs in cells was determined by preparing total RNA from 8 × 107 cells using Trizol reagent (Invitrogen), reverse transcription and semi-quantitative PCR. Reverse transcription was carried out with SuperScript II reverse transcriptase (Invitrogen) according to the manufacturer’s protocol using oligo-dT as primer. For semi-quantitative PCR, the number of cycles for the linear amplification range was determined empirically for each oligonucleotide pair. Labeling and analysis of nascent RNA using a permeabilized cell system was carried out as described previously (Schimanski et al., 2006). Quantification was performed by densitometry using ImageJ for SL RNA signals and by scintillation counting for dot blot signals.
Protein Analysis
Immunoprecipitations (IPs) were performed as described previously (Lee et al., 2007). Briefly, for reciprocal co-IPs, 100 μl of crude extract was incubated with either 40 μl settled volume of IgG beads (Amersham, Piscataway, NJ) which interact with the PTP tag, or 25 μl settled volume of anti-HA antibody (Roche) immobilized on protein G-Sepharose (Amersham). Once bound to protein G, the anti-HA antibody is unable to interact with the protein A domains of the PTP tag, ensuring that these beads precipitate HA-tagged protein only. After washing the beads six times with 700 μl of TET100 buffer (100 mM NaCl, 20 mM Tris-HCl, pH 8.0, 3 mM MgCl2, 0.05% Tween 20), PTP-tagged protein was released by AcTEV protease (Invitrogen) digestion, whereas HA-tagged protein was directly eluted into SDS loading buffer at 100°C for 5 min. Precipitated complexes were analyzed by immunoblotting; PTP- and HA-tagged proteins were detected on blots with the anti-protein C antibody HPC4 (Roche) or a monoclonal rat anti-HA antibody (Roche), and TSP2 with a polyclonal rat immune serum (Lee et al., 2009) in combination with the BM chemiluminescence blotting substrate (Roche) according to the manufacturer’s specifications.
XPB-R-PTP was tandem affinity purified according to the standard PTP purification protocol (Schimanski et al., 2005). Purified proteins were separated on a 10 to 20% SDS-polyacrylamide gradient gel and stained with SYPRO Ruby (Invitrogen) according to the manufacturer’s protocol. Proteins were trypsin digested, eluted from the gel and subjected to liquid chromatography-tandem mass spectrometry.
For sedimentation analysis, 200 μl of crude extract prepared from a clonal cell line expressing XPB-R-PTP and p52-HA was subjected to ultracentifugation in 4 ml 10–40% linear sucrose gradients at 41,000 rpm for 16 h at 4°C as described previously (Lee et al., 2010). Twenty fractions were collected from top to bottom and protein in each fraction was collected with hydrophobic StrataClean resin (Stratagene, La Jolla, CA) as previously described (Schimanski et al., 2005). Proteins were separated by denaturing PAGE and detected by immunoblotting.
In vitro transcription
Preparation of extract active in transcription and the carrying out of in vitro transcription assays were performed as described previously (Laufer et al., 1999; Laufer and Günzl, 2001). Newly synthesized RNAs of the GPEET-trm and SLins19 templates were detected by primer extension of 32P-end-labeled oligonucleotides Tag-PE and SLtag, which hybridize to unrelated oligonucleotide tags of the GPEET-trm and SLins19 RNAs. Primer extension products were resolved on 6% polyacrylamide-50% urea gels and visualized by autoradiography.
Chromatin Immunoprecipitation
ChIP assays were performed as described previously (Lee et al., 2010). XPD-PTP- and XPB-R-PTP-bound chromatin fragments were immunoprecipitated overnight at 4°C with a polyclonal rabbit antibody directed against the protein A domains of the PTP tag (Sigma). Immunoprecipitated DNA was analyzed by semiquantitative and quantitative real-time PCR of the SLRNA promoter region (positions −90 to +63 relative to the transcription initiation site), the SLRNA intergenic region (+407 to +540), and the α-tubulin-coding region (+737 to +860 relative to the translation initiation codon) using the primer pairs listed in Table S1. For quantification of immunoprecipitated DNA, qPCR was performed using the SsoFast EvaGreen Supermix (BioRad) on a CFX96 cycler (BioRad) according to the manufacturer’s recommendations. Two independent ChIP experiments were performed for both XPD-PTP and XPB-R-PTP cell lines. For each amplification and ChIP experiment, triplicate qPCR samples were analyzed using the Bio-Rad CFX Manager software package. The specificity of each amplification product was ensured by agarose gel electrophoresis and melting curve analysis. Standard curves for oligonucleotide pairs were obtained from serial dilution of input DNA. Their coefficient of determination (R2) value was above 0.98. Samples were standardized according to the starting quantities of tubulin DNA in each experiment and the fold enrichment values were calculated as the ratio between starting quantities of positive and control precipitations.
Survival assays
Sensitivity of cells towards DNA damaging agents were tested at a cell density of 2 × 106 cells/ml. For assessing UV irradiation effects, the wildtype (427) and knockout cells were exposed to doses of 300, 500 or 1000 J/m2 using the Stratalinker® UV Crosslinker (Stratagene). For Cisplatin (Sigma) and MMS (Sigma) sensitivity assays, cells were cultured with Cisplatin ranging from 0.5 to 10 μM and with MMS from 0.0001 to 0.0004% for two days. In each case, treated and untreated control cell cultures were diluted daily to a concentration of 2 × 106 cells/ml. Total cell count was determined after two days and the survival was calculated as a percentage of untreated controls. In order to assess the sensitivity of RNAi cell lines towards UV irradiation, procyclic cell culture at the density of 2 × 106 cells/ml was induced for dsRNA synthesis with 2 μg/ml of doxycycline. After 24 hours, induced and non-induced cells were diluted to a concentration 2 × 106 cells/ml and exposed to 500 and 1000 J/m2 of UV light. The cells were counted 24 hours after exposure and the survival was determined as a percentage of untreated controls.
Light Microscopy
Immunolocalizations were carried out as described previously (Luz Ambrosio D. et al., 2009). XPB-R-PTP and XPD-PTP were detected with a rabbit polyclonal anti-protein A immune serum (Sigma) followed by an Alexa 594-conjugated anti-rabbit secondary antibody (Invitrogen) whereas Tbp52-HA was visualized with a mouse monoclonal anti-HA antibody conjugated with fluorescein isothiocyanate (FITC; Sigma). Images were acquired on a Zeiss Axiovert 200 microscope equipped with DAPI (4′,6′-diamidino-2-phenylindole), EGFP, and Texas-Red filters using a 100× (1.3-numerical-aperture) oil immersion objective.
Supplementary Material
Acknowledgments
We thank Dr. Richard McCulloch (University of Glasgow) for sharing unpublished data, Dr. J. Peter Gogarten (University of Connecticut) for help with the phylogenetic analysis and our laboratory member Justin Kirkham for critical reading of the manuscript.
This work was funded by NIH grants R01 AI073300 and R01 AI059377 to A.G.
References
- Alsford S, duBois K, Horn D, Field MC. Epigenetic mechanisms, nuclear architecture and the control of gene expression in trypanosomes. Expert Rev Mol Med. 2012;14:e13. doi: 10.1017/erm.2012.7. [DOI] [PubMed] [Google Scholar]
- Balasingham SV, Zegeye ED, Homberset H, Rossi ML, Laerdahl JK, Bohr VA, Tonjum T. Enzymatic activities and DNA substrate specificity of Mycobacterium tuberculosis DNA helicase XPB. PLoS One. 2012;7:e36960. doi: 10.1371/journal.pone.0036960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedez F, Linard B, Brochet X, Ripp R, Thompson JD, Moras D, et al. Functional insights into the core-TFIIH from a comparative survey. Genomics. 2013;101:178–186. doi: 10.1016/j.ygeno.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, Bartholomeu DC, et al. The Genome of the African Trypanosome Trypanosoma brucei. Science. 2005;309:416–422. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- Biswas T, Pero JM, Joseph CG, Tsodikov OV. DNA-dependent ATPase activity of bacterial XPB helicases. Biochemistry. 2009;48:2839–2848. doi: 10.1021/bi8022416. [DOI] [PubMed] [Google Scholar]
- Brandenburg J, Schimanski B, Nogoceke E, Nguyen TN, Padovan JC, Chait BT, et al. Multifunctional class I transcription in Trypanosoma brucei depends on a novel protein complex. EMBO J. 2007;26:4856–4866. doi: 10.1038/sj.emboj.7601905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem R, Fernet M, Chapot B, Hall J. The methyl methanesulfonate induced S-phase delay in XRCC1-deficient cells requires ATM and ATR. DNA Repair (Amst) 2008;7:849–857. doi: 10.1016/j.dnarep.2008.02.002. [DOI] [PubMed] [Google Scholar]
- Coin F, Oksenych V, Egly JM. Distinct roles for the XPB/p52 and XPD/p44 subcomplexes of TFIIH in damaged DNA opening during nucleotide excision repair. Mol Cell. 2007;26:245–256. doi: 10.1016/j.molcel.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Coin F, Proietti De SL, Nardo T, Zlobinskaya O, Stefanini M, Egly JM. p8/TTD-A as a repair-specific TFIIH subunit. Mol Cell. 2006;21:215–226. doi: 10.1016/j.molcel.2005.10.024. [DOI] [PubMed] [Google Scholar]
- Compe E, Egly JM. TFIIH: when transcription met DNA repair. Nat Rev Mol Cell Biol. 2012;13:343–354. doi: 10.1038/nrm3350. [DOI] [PubMed] [Google Scholar]
- Dacks JB, Walker G, Field MC. Implications of the new eukaryotic systematics for parasitologists. Parasitol Int. 2008;57:97–104. doi: 10.1016/j.parint.2007.11.004. [DOI] [PubMed] [Google Scholar]
- Fan L, Arvai AS, Cooper PK, Iwai S, Hanaoka F, Tainer JA. Conserved XPB core structure and motifs for DNA unwinding: implications for pathway selection of transcription or excision repair. Mol Cell. 2006;22:27–37. doi: 10.1016/j.molcel.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Feaver WJ, Svejstrup JQ, Bardwell L, Bardwell AJ, Buratowski S, Gulyas KD, et al. Dual roles of a multiprotein complex from S. cerevisiae in transcription and DNA repair. Cell. 1993;75:1379–1387. doi: 10.1016/0092-8674(93)90624-y. [DOI] [PubMed] [Google Scholar]
- Fregoso M, Laine JP, guilar-Fuentes J, Mocquet V, Reynaud E, Coin F, et al. DNA repair and transcriptional deficiencies caused by mutations in the Drosophila p52 subunit of TFIIH generate developmental defects and chromosome fragility. Mol Cell Biol. 2007;27:3640–3650. doi: 10.1128/MCB.00030-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günzl A. The pre-mRNA splicing machinery of trypanosomes: complex or simplified? Eukaryot Cell. 2010;9:1159–1170. doi: 10.1128/EC.00113-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günzl A. RNA Polymerases and Transcription Factors of Trypanosomes. In: Bindereif A, editor. RNA Metabolism in Trypanosomes. Springer Press; 2012. pp. 1–27. [Google Scholar]
- Günzl A, Ullu E, Dörner M, Fragoso SP, Hoffmann KF, Milner JD, et al. Transcription of the Trypanosoma brucei spliced leader RNA gene is dependent only on the presence of upstream regulatory elements. Mol Biochem Parasitol. 1997;85:67–76. doi: 10.1016/s0166-6851(96)02816-2. [DOI] [PubMed] [Google Scholar]
- Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- Hoogstraten D, Bergink S, Ng JM, Verbiest VH, Luijsterburg MS, Geverts B, et al. Versatile DNA damage detection by the global genome nucleotide excision repair protein XPC. J Cell Sci. 2008;121:2850–2859. doi: 10.1242/jcs.031708. [DOI] [PubMed] [Google Scholar]
- Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G, Berriman M, et al. The Genome of the Kinetoplastid Parasite, Leishmania major. Science. 2005;309:436–442. doi: 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jawhari A, Laine JP, Dubaele S, Lamour V, Poterszman A, Coin F, et al. p52 Mediates XPB function within the transcription/repair factor TFIIH. J Biol Chem. 2002;277:31761–31767. doi: 10.1074/jbc.M203792200. [DOI] [PubMed] [Google Scholar]
- Kamileri I, Karakasilioti I, Garinis GA. Nucleotide excision repair: new tricks with old bricks. Trends Genet. 2012;28:566–573. doi: 10.1016/j.tig.2012.06.004. [DOI] [PubMed] [Google Scholar]
- Kartalou M, Essigmann JM. Mechanisms of resistance to cisplatin. Mutat Res. 2001;478:23–43. doi: 10.1016/s0027-5107(01)00141-5. [DOI] [PubMed] [Google Scholar]
- Laine JP, Egly JM. When transcription and repair meet: a complex system. Trends Genet. 2006;22:430–436. doi: 10.1016/j.tig.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Laufer G, Günzl A. In-vitro competition analysis of procyclin gene and variant surface glycoprotein gene expression site transcription in Trypanosoma brucei. Mol Biochem Parasitol. 2001;113:55–65. doi: 10.1016/s0166-6851(00)00380-7. [DOI] [PubMed] [Google Scholar]
- Laufer G, Schaaf G, Bollgönn S, Günzl A. In vitro analysis of alpha-amanitin-resistant transcription from the rRNA, procyclic acidic repetitive protein, and variant surface glycoprotein gene promoters in Trypanosoma brucei. Mol Cell Biol. 1999;19:5466–5473. doi: 10.1128/mcb.19.8.5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecordier L, Devaux S, Uzureau P, Dierick JF, Walgraffe D, Poelvoorde P, et al. Characterization of a TFIIH homologue from Trypanosoma brucei. Mol Microbiol. 2007;64:1164–1181. doi: 10.1111/j.1365-2958.2007.05725.x. [DOI] [PubMed] [Google Scholar]
- Lee JH, Cai G, Panigrahi AK, Dunham-Ems S, Nguyen TN, Radolf JD, et al. A TFIIH-associated mediator head is a basal factor of small nuclear spliced leader RNA gene transcription in early-diverged trypanosomes. Mol Cell Biol. 2010;30:5502–5513. doi: 10.1128/MCB.00966-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Jung HS, Günzl A. Transcriptionally active TFIIH of the early-diverged eukaryote Trypanosoma brucei harbors two novel core subunits but not a cyclin-activating kinase complex. Nucleic Acids Res. 2009;37:3811–3820. doi: 10.1093/nar/gkp236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Nguyen TN, Schimanski B, Günzl A. Spliced leader RNA gene transcription in Trypanosoma brucei requires transcription factor TFIIH. Eukaryot Cell. 2007;6:641–649. doi: 10.1128/EC.00411-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Choi WS, Gralla JD. TFIIH XPB mutants suggest a unified bacterial-like mechanism for promoter opening but not escape. Nat Struct Mol Biol. 2005;12:603–607. doi: 10.1038/nsmb949. [DOI] [PubMed] [Google Scholar]
- Logan-Klumpler FJ, De SN, Boehme U, Rogers MB, Velarde G, McQuillan JA, et al. GeneDB--an annotation database for pathogens. Nucleic Acids Res. 2012;40:D98–108. doi: 10.1093/nar/gkr1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luz Ambrosio D, Lee JH, Panigrahi AK, Nguyen TN, Cicarelli RM, Günzl A. Spliceosomal proteomics in Trypanosoma brucei reveal new RNA splicing factors. Eukaryot Cell. 2009;8:990–1000. doi: 10.1128/EC.00075-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Calvillo S, Vizuet-de-Rueda JC, Florencio-Martinez LE, Manning-Cela RG, Figueroa-Angulo EE. Gene expression in trypanosomatid parasites. J Biomed Biotechnol. 2010;2010:525241. doi: 10.1155/2010/525241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Calvillo S, Yan S, Nguyen D, Fox M, Stuart K, Myler PJ. Transcription of Leishmania major Friedlin chromosome 1 initiates in both directions within a single region. Mol Cell. 2003;11:1291–1299. doi: 10.1016/s1097-2765(03)00143-6. [DOI] [PubMed] [Google Scholar]
- Mathieu N, Kaczmarek N, Naegeli H. Strand- and site-specific DNA lesion demarcation by the xeroderma pigmentosum group D helicase. Proc Natl Acad Sci U S A. 2010;107:17545–17550. doi: 10.1073/pnas.1004339107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaeli S. Spliced leader RNA silencing (SLS) - a programmed cell death pathway in Trypanosoma brucei that is induced upon ER stress. Parasit Vectors. 2012;5:107. doi: 10.1186/1756-3305-5-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naegeli H, Sugasawa K. The xeroderma pigmentosum pathway: decision tree analysis of DNA quality. DNA Repair (Amst) 2011;10:673–683. doi: 10.1016/j.dnarep.2011.04.019. [DOI] [PubMed] [Google Scholar]
- Nguyen TN, Schimanski B, Zahn A, Klumpp B, Günzl A. Purification of an eight subunit RNA polymerase I complex in Trypanosoma brucei. Mol Biochem Parasitol. 2006;149:27–37. doi: 10.1016/j.molbiopara.2006.02.023. [DOI] [PubMed] [Google Scholar]
- Oksenych V, Bernardes de JB, Zhovmer A, Egly JM, Coin F. Molecular insights into the recruitment of TFIIH to sites of DNA damage. Embo J. 2009;28:2971–2980. doi: 10.1038/emboj.2009.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passos-Silva DG, Rajao MA, Nascimento de Aguiar PH, Vieira-da-Rocha JP, Machado CR, Furtado C. Overview of DNA Repair in Trypanosoma cruzi, Trypanosoma brucei, and Leishmania major. J Nucleic Acids. 2010;2010:840768. doi: 10.4061/2010/840768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards JD, Cubeddu L, Roberts J, Liu H, White MF. The archaeal XPB protein is a ssDNA-dependent ATPase with a novel partner. J Mol Biol. 2008;376:634–644. doi: 10.1016/j.jmb.2007.12.019. [DOI] [PubMed] [Google Scholar]
- Rouillon C, White MF. The XBP-Bax1 helicase-nuclease complex unwinds and cleaves DNA: implications for eukaryal and archaeal nucleotide excision repair. J Biol Chem. 2010;285:11013–11022. doi: 10.1074/jbc.M109.094763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouillon C, White MF. The evolution and mechanisms of nucleotide excision repair proteins. Res Microbiol. 2011;162:19–26. doi: 10.1016/j.resmic.2010.09.003. [DOI] [PubMed] [Google Scholar]
- Schaeffer L, Roy R, Humbert S, Moncollin V, Vermeulen W, Hoeijmakers JH, et al. DNA repair helicase: a component of BTF2 (TFIIH) basic transcription factor. Science. 1993;260:58–63. doi: 10.1126/science.8465201. [DOI] [PubMed] [Google Scholar]
- Schimanski B, Brandenburg J, Nguyen TN, Caimano MJ, Günzl A. A TFIIB-like protein is indispensable for spliced leader RNA gene transcription in Trypanosoma brucei. Nucleic Acids Res. 2006;34:1676–1684. doi: 10.1093/nar/gkl090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimanski B, Nguyen TN, Günzl A. Highly efficient tandem affinity purification of trypanosome protein complexes based on a novel epitope combination. Eukaryotic Cell. 2005;4:1942–1950. doi: 10.1128/EC.4.11.1942-1950.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheader K, te VD, Rudenko G. Bloodstream form-specific up-regulation of silent vsg expression sites and procyclin in Trypanosoma brucei after inhibition of DNA synthesis or DNA damage. J Biol Chem. 2004;279:13363–13374. doi: 10.1074/jbc.M312307200. [DOI] [PubMed] [Google Scholar]
- Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- Tantin D. RNA polymerase II elongation complexes containing the Cockayne syndrome group B protein interact with a molecular complex containing the transcription factor IIH components xeroderma pigmentosum B and p62. J Biol Chem. 1998;273:27794–27799. doi: 10.1074/jbc.273.43.27794. [DOI] [PubMed] [Google Scholar]
- Thoma F. Light and dark in chromatin repair: repair of UV-induced DNA lesions by photolyase and nucleotide excision repair. Embo J. 1999;18:6585–6598. doi: 10.1093/emboj/18.23.6585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirode F, Busso D, Coin F, Egly JM. Reconstitution of the transcription factor TFIIH: assignment of functions for the three enzymatic subunits, XPB, XPD, and cdk7. Mol Cell. 1999;3:87–95. doi: 10.1016/s1097-2765(00)80177-x. [DOI] [PubMed] [Google Scholar]
- Truglio JJ, Croteau DL, Van HB, Kisker C. Prokaryotic nucleotide excision repair: the UvrABC system. Chem Rev. 2006;106:233–252. doi: 10.1021/cr040471u. [DOI] [PubMed] [Google Scholar]
- Ullu E, Tschudi C. Permeable trypanosome cells as a model system for transcription and trans-splicing. Nucleic Acids Research. 1990;18:3319–3326. doi: 10.1093/nar/18.11.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen W, Bergmann E, Auriol J, Rademakers S, Frit P, Appeldoorn E, et al. Sublimiting concentration of TFIIH transcription/DNA repair factor causes TTD-A trichothiodystrophy disorder. Nat Genet. 2000;26:307–313. doi: 10.1038/81603. [DOI] [PubMed] [Google Scholar]
- Weber A, Chung HJ, Springer E, Heitzmann D, Warth R. The TFIIH subunit p89 (XPB) localizes to the centrosome during mitosis. Cell Oncol. 2010;32:121–130. doi: 10.3233/CLO-2009-0509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welburn SC, Macleod E, Figarella K, Duzensko M. Programmed cell death in African trypanosomes. Parasitology. 2006;132(Suppl):S7–S18. doi: 10.1017/S0031182006000825. [DOI] [PubMed] [Google Scholar]
- Wirtz E, Leal S, Ochatt C, Cross GAM. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol Biochem Parasitol. 1999;99:89–101. doi: 10.1016/s0166-6851(99)00002-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.