

Abstract
The driving theoretical framework of Alzheimer’s disease (AD) has been built around the Aβ cascade in which amyloid pathology precedes and drives tau pathology. Other evidence has suggested that tau and amyloid pathology may arise independently. Both lines of research suggest that there may be epistatic relationships between genes involved in amyloid and tau pathophysiology. In the current study, we hypothesized that genes coding GSK-3 and comparable tau kinases would modify genetic risk for amyloid plaque pathology. Quantitative amyloid PET data from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) served as the quantitative outcome in regression analyses, covarying for age, gender and diagnosis. Three interactions reached statistical significance, all involving the GSK3β SNP rs334543—two with APBB2 (rs2585590, rs3098914) and one with APP (rs457581). These interactions explained 1.2%, 1.5%, and 1.5% of the variance in amyloid deposition respectively. Our results add to a growing literature on the role of GSK-3 activity in amyloid processing and suggest that combined variation in GSK3β and APP-related genes may result in increased amyloid burden.
Keywords: Imaging Genetics, Alzheimer’s Disease, Amyloid, Tau, PET, ADNI
INTRODUCTION
The pathologic cascade in Alzheimer’s disease (AD) involves two primary lesions: amyloid-β (Aβ) plaques and neurofibrillary tangles made up of hyper-phosphorylated tau. Genes involved in the production of the Aβ cause autosomal dominantly inherited forms of AD (Price & Sisodia, 1998). The genetic etiology of late-onset AD is more complex and includes a great deal of missing heritability based on current approximations (Bertram, Lill, & Tanzi, 2010).
The driving theoretical framework of AD over the past decade has been built around the Aβ cascade. The amyloid cascade hypothesis suggests that the disease process is initiated by Aβ formation leading to downstream pathologies and neurodegeneration (Hardy & Selkoe, 2002). Within such a framework, amyloid pathology precedes tau pathology and would have to drive its formation in some way. As nicely reviewed by Itner and Götz (2010), there is substantial evidence that such a causal relationship does exist. However, other evidence has suggested that tau and amyloid pathology may arise independently, with upstream genetic interactions causing both pathologies through separate defects in distinct molecular pathways (Small & Duff, 2008). In either scenario, it may be that genes which confer risk for tau pathology also confer risk for amyloid pathology through complex epistatic relationships. The current project sought to identify such interaction effects, primarily focusing on the tau kinases which have recently been implicated in both pathologic pathways.
One such kinase that has been implicated in both tau and amyloid pathology is glycogen synthase kinase 3 (GSK-3). GSK-3 has been implicated in tau hyper-phosphorylation, subsequent neurodegeneration (Lucas et al., 2001) and amyloid accumulation (Martin et al., 2013). Moreover, GSK-3 appears to regulate Aβ production (Phiel, Wilson, Lee, & Klein, 2003), and silencing GSK-3 leads to reduced plaque and tangle formation in transgenic mouse models of AD (Hurtado et al., 2012). These findings have led to the GSK-3 hypothesis which suggests over-activity of GSk-3 can account for cognitive impairments, the pathological cascade, and the neuroinflammatory response characteristic of AD (Hooper, Killick, & Lovestone, 2008).
In addition to GSK-3, two other tau kinases have been implicated in both amyloid and tau pathology. As reviewed previously (Martin et al., 2013), knock-down of cyclin-dependent kinase 5 (CDK5) results in reduced tau pathology in transgenic AD models (Piedrahita et al., 2010), and the CDK5-related tau cascade appears to be activated by Aβ (Lopes, Oliveira, & Agostinho, 2010). Dual specificity tyrosine-phosphorylation-regulated kinase 1A (DYRK1A) phosphorylates both tau and APP (Martin et al., 2013; Ryoo et al., 2008), and has been related to the pathologic cascades of tau and Aβ (Wegiel, Gong, & Hwang, 2011). In addition, there has been some evidence that phosphorylation of tau by DYRK1A leads to additional tau phosphorylation by GSK-3, ultimately resulting in hyper-phosphorylation (Liu et al., 2008).
The aim of the current study was to identify epistatic relationships between genes coding tau kinases and genes previously associated with amyloid deposition. We hypothesized that genes coding GSK-3 and comparable tau kinases would modify genetic risk for amyloid plaque pathology.
MATERIALS AND METHODS
Data used in the preparation of this article were obtained from the ADNI database (adni.loni.ucla.edu). The ADNI was launched in 2003 by the National Institute on Aging (NIA), the National Institute of Biomedical Imaging and Bioengineering (NIBIB), the Food and Drug Administration (FDA), private pharmaceutical companies and non-profit organizations, as a $60 million, 5-year public-private partnership. The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), Positron Emission Tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of MCI and early AD. Determination of sensitive and specific markers of very early AD progression is intended to aid researchers and clinicians to develop new treatments and monitor their effectiveness, as well as lessen the time and cost of clinical trials.
The Principal Investigator of this initiative is Michael W. Weiner, MD, VA Medical Center and University of California – San Francisco. ADNI is the result of efforts of many co-investigators from a broad range of academic institutions and private corporations, and subjects have been recruited from over 50 sites across the U.S. and Canada. The initial goal of ADNI was to recruit 800 adults, ages 55 to 90, to participate in the research, approximately 200 cognitively normal older individuals to be followed for 3 years, 400 people with MCI to be followed for 3 years and 200 people with early AD to be followed for 2 years. For up-to-date information, see ww.adni-info.org.
Subjects
Demographic data are presented in Table 1. Participants were enrolled based on the criteria outlined in the ADNI protocol (http://www.adni-info.org/Scientists/AboutADNI.aspx) and the ADNI2/ADNI-GO protocols (http://adni.loni.ucla.edu/wp-content/uploads/2008/07/ADNI_Go_Protocol.pdf; http://adni.loni.ucla.edu/wp-content/uploads/2008/07/ADNI2_Protocol_FINAL_20100917.pdf). For the present project, analyses were restricted to Caucasian subjects who had both genotype data and PET data.
Table 1.
Demographic Information
| Baseline Clinical Diagnosisa | ||||
|---|---|---|---|---|
| Normal Control | Mild Cognitive Impairment | Alzheimer’s Disease | ||
| ADNI-1 Dataset | Number of Patients | 68 | 54 | 41 |
| Number of APOE- ε4 Carriers | 15 | 18 | 26 | |
| Number of Females | 32 | 17 | 15 | |
| Mean Baseline Age (SD) | 81.10 (5.01) | 79.41 (7.35) | 77.05 (6.54) | |
| Mean Years of Education (SD) | 16.07 (3.03) | 15.54 (3.19) | 16.15 (2.88) | |
| Mean SUVRb AV-45 (SD) | 1.07 (0.16) | 1.19 (0.25) | 1.32 (0.25) | |
| ADNI-2/GO Dataset | Number of Patients | 109 | 239 | 25 |
| Number of APOE- ε4 Carriers | 26 | 104 | 17 | |
| Number of Females | 53 | 103 | 9 | |
| Mean Baseline Age (SD) | 74.83 (5.57) | 71.82 (7.44) | 74.20 (10.06) | |
| Mean Years of Education (SD) | 16.45 (2.59) | 16.04 (2.64) | 15.80 (2.77) | |
| Mean SUVRb AV-45 (SD) | 1.11 (0.20) | 1.19 (0.22) | 1.38 (0.21) | |
| Combined Dataset | Number of Patients | 177 | 293 | 66 |
| Number of APOE- ε4 Carriers | 41 | 122 | 43 | |
| Number of Females | 85 | 120 | 24 | |
| Mean Baseline Age (SD) | 77.24 (6.16) | 73.22 (7.98) | 75.97 (8.09) | |
| Mean Years of Education (SD) | 16.31 (2.76) | 15.95 (2.75) | 16.02 (2.82) | |
| Mean SUVRb AV-45 (SD) | 1.10 (0.19) | 1.19 (0.23) | 1.34 (0.23) | |
Normal Control subjects had a Mini-Mental Status Examination (MMSE) score between 24 and 30, a Clinical Dementia Rating (CDR) score of 0, and were not depressed (Geriatric Depression Scale score < 6).
Mild Cognitive Impairment subjects had a MMSE score between 24 and 30, objective memory impairment, subjective memory impairment, and a CDR score of 0.5.
Alzheimer’s Disease subjects met clinical criteria for dementia, had an MMSE of between 20 and 26, and had CDR score of .5 or 1.
SUVR - Standardized uptake value ratio for amyloid tracer
Genotyping
We used data from all subjects who received a PET scan in the ADNI-2/GO protocol. Some of those subjects were genotyped in ADNI-1 on the Illumina Infinium Human-610-Quad BeadChip (Shen et al., 2010) and some were genotyped in ADNI-2/GO on the Illumina OmniQuad array (Potkin et al., 2009). For the present analyses we looked at candidate SNPs that were present on both chips, and combined both datasets to maximize our power.
Quality control (QC) was performed using PLINK software (version 1.07; (Purcell et al., 2007) excluding SNPs with a genotyping efficiency < 98%, a minor allele frequency of < 10%, or deviation from Hardy-Weinberg Equilibrium (HWE) < 1e−6. Subjects were excluded if they had a call rate < 90%, if there was a reported versus genetic sex inconsistency, or if relatedness to another sample was established (PI_HAT > 0.5).
SNP Selection
For tau genes, we chose to focus on tau kinase genes that had been implicated in amyloid processing, as outlined in the introduction. These included GSK3β, GSK3A, CDK5, and DYRK1A. For amyloid genes, we chose to focus on the three genes involved in dominantly inherited forms of AD (APP, PSEN1, PSEN2) as well as those genes which had previously shown either SNP or gene level associations with amyloid deposition measured with PET, including ABCG1, APBB2, DHCR24, SOAT1, and BCHE (Ramanan et al., 2013; Swaminathan et al., 2012).
SNPs that annotated to these genes were selected using the Illumina annotation file, which is freely available at http://www.switchtoi.com/annotationfiles.ilmn. We only used SNPs that were genotyped in both ADNI-1 and ADNI-2/GO and were annotated to these genes, resulting in a total of 193 SNPs used in analyses (Supplementary Table 1). Of note, there were no SNPs that passed QC and were annotated to GSK3A or SOAT1.
Quantification of Amyloid Deposition
Amyloid deposition was quantified using an 18F-AV-45 tracer and have been described extensively elsewhere (Landau & Jagust, 2011). The mean SUVR measure was calculated across the cingulate (including anterior and posterior regions), frontal, temporal (including middle and lateral regions), and lateral parietal (including the precuneus and supramarginal gyrus) cortices, and divided by the reference region (cerebellar grey matter).
SNP-SNP Interaction Analysis
Interaction analyses were run using SAS version 9.3 (http://www.sas.com/). Mean SUVR was set as the quantitative outcome measure in a general linear regression model (PROC GLM). Covariates included age, gender, and diagnosis. We included the main effect of each SNP (one from an amyloid-related gene and one from a tau-kinase gene) and the interaction term. A full additive model was used for SNP terms, meaning each SNP was coded as 0, 1, 2 based on the number of minor alleles. A total of 4,175 tests were run, evaluating all SNP – SNP interactions between tau and amyloid genes. Correction for multiple comparisons using the false discovery rate procedure (FDR < .05, PROC MULTTEST) and the Bonferroni procedure (FWE < 0.05, PROC MULTTEST) was performed across all 4,175 analyses.
Posthoc Hierarchical Linear Regression
Following the identification of significant interactions, we used hierarchical linear regression in IBM SPSS 20 (http://www-01.ibm.com/software/analytics/spss/) to quantify the amount of variance in amyloid deposition accounted for by these interaction terms. Our first step included age, diagnosis, and gender. Next we included APOE status and the SNP main effects from the two candidate genes. Finally we included the SNP – SNP interaction term to see how much additional variance was explained by the interaction term beyond these known predictors of amyloid deposition.
Posthoc Binary Logistic Regression
The variable quantifying amyloid load in the current analyses was not normally distributed within or across diagnostic groups. Although linear regression is known to be fairly robust to deviations from normality, we chose to validate our findings using binary logistic regression. A binary variable differentiating amyloid positive v. amyloid negative individuals was derived using a previously identified and accepted cut-point of mean SUVR > 1.11 (Landau & Jagust, 2011). This variable was set as a binary outcome measure in a logistic regression model using the same parameters as those in the original SNP-SNP interaction analysis above. Binary logistic regression was only run as a posthoc examination of the significant interactions identified in the primary analysis.
RESULTS
SNP-SNP Interaction Results
Three SNP-SNP interactions reached statistical significance when correcting for multiple comparisons (Table 2). One GSK3β SNP (rs334543) was involved in all three of interactions, two with SNPs annotated to APBB2 (rs2585590, rs3098914) and one with a SNP annotated to APP (rs457581). We also evaluated whether the observed effects were consistent across the two genotyping platforms. All interactions showed an effect across the two chips, although the APP x GSK3β interaction only showed a trend level association in the ADNI-1 sub-sample (Table 2).
Table 2.
SNP-SNP Interaction Analysis
| Gene | SNP | Combined Dataset | ADNI-1 Dataset | ADNI2/GO Dataset | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||
| MAF | a T | b R2 | p-value | c FWE | d FDR | eχ2 | p-value | Ta | p-value | Ta | p-value | ||
| APPB2 x GSK3β | rs2585590 x rs334543 | 0.46 | 4.41 | 0.015 | 1.2 × 10−5 | 0.052 | 0.036 | 11.14 | 1.0 × 10−3 | 3.21 | 0.002 | 3.17 | 0.002 |
| 0.31 | |||||||||||||
|
| |||||||||||||
| APPB2 x GSK3β | rs3098914 x rs334543 | 0.24 | −4.25 | 0.012 | 2.5 × 10−5 | 0.107 | 0.036 | 10.63 | 1.0 × 10−3 | −2.51 | 0.013 | −3.37 | 0.001 |
| 0.31 | |||||||||||||
|
| |||||||||||||
| APP x GSK3β | rs457581 x rs334543 | 0.25 | 4.25 | 0.015 | 2.5 × 10−5 | 0.107 | 0.036 | 13.86 | 2.0 × 10−4 | 1.85 | 0.066 | 3.51 | 0.001 |
| 0.31 | |||||||||||||
T value for SNP x SNP interaction term
R2 value represents the change in R2 when introducing the SNP x SNP interaction term into the post-hoc hierarchical linear regression model.
FWE represents the p-value when correcting for multiple comparisons using the Bonferroni procedure (4,175 tests)
FDR represents the p-value when correcting for multiple comparisons using the false discovery rate procedure
χ2 value for SNP x SNP interaction term in the post-hoc binary logistic regression model using amyloid positive/negative status as outcome
Posthoc Hierarchical Linear Regression
Gender, age, and diagnosis were entered into the model first and accounted for 12% of variance in amyloid deposition. Next, APOE status was entered into the model and accounted for an additional 18% of variance. Four separate hierarchical linear regression models were run across the four significant interactions. (We did not include all interactions in one model). In each case, we added in the genetic main effects first and then the genetic interaction term to determine the variance associated with the interaction term alone. For APPB2 (rs3098914) x GSK3β (rs334543) the non-significant (p > 0.05) SNP main effects accounted for 0.5% of variance, and the interaction term accounted for 1.5% of variance (2% of variance for the main effects and interaction combined). For APBB2 (rs2585590) x GSK3β (rs334543) the non-significant (p > 0.05) SNP main effects accounted for 0.4% of variance, and the interaction accounted for 1.2% of variance (1.7% for the main effects and interaction combined). For APP (rs457581) x GSK3β (rs334543) the non-significant (p > 0.05) SNP main effects accounted for 0.4% of variance, and the interaction term accounted for 1.5% of variance (1.9% for the main effect and interaction combined). Finally, all three interactions remained statistically significant when performing binary logistic regression as outlined in the methods section above (Table 2).
DISCUSSION
The current project has identified three interactions with one GSK3β SNP (rs334543) that suggest GSK3β may indeed modify risk for amyloid deposition within specific genetic contexts. Given the role of GSK-3 in the neuroinflammatory response system and its suggested role in both amyloid and tau phosphorylation, it is not surprising that the genetic relationship to amyloid load in the present cohort is quite complex. Our results suggest that combined variation in GSK3β and APP-related genes may result in increased amyloid burden.
GSK3β (rs334543) SNP Function
All interactions in the current analyses involved rs334543. As reported in Haploreg (Ward & Kellis, 2012), this SNP is 20kb 5′ from GSK3β; it acts a strong enhancer in a variety of cell lines including epithelial cells, skeletal muscle myoblasts, and lung fibroblasts among others; it has been shown to bind with four transcription factors in ENCODE tracks including FOXA, POL2, and STAT3, and has been shown to alter the p300 regulatory motif p300_known1 identified using position weigh matrix techniques. This suggests that this SNP is in highly active genetic region and may regulate gene expression or otherwise play an active role in GSK3β function. In addition, rs334543 is in a DNase-I hypersensitivity uniform peak in an astrocyte cell line, suggesting this SNP may be functionally active in the brain (Rosenbloom et al., 2013).
GSK3B and Amyloid Burden
The first interaction in which the minor allele in GSK3β (rs334543) was related to high levels of amyloid deposition was a GSK3β x APP interaction. Although only six subjects were homozygote carriers of both the APP and GSK3β minor alleles (Figure 1), none of the subjects were statistical outliers in amyloid deposition with each falling between 1.4 and 1.8 mean SUVR. Previous research has suggested that GSK3β might play a role in APP processing. A GSK-3 blocker, lithium, has been shown to decrease amyloid burden in APP mice, and the effect appears to be driven by GSK3β, as genetic modification of GSK3β mimics this effect (Su et al., 2004). Additional evidence has suggested that modulation of GSK3β activity reduces APP phosphorylation and amyloid load (Rockenstein et al., 2007). In the present result, the effect of GSK3β was only present in carriers of the APP (rs457581) minor allele, and neither SNP showed a main effect in conferring risk for amyloid when the interaction term was excluded. This may suggest that slight increases in GSK3β activity are only related to a negative outcome when APP is overexpressed as well, perhaps via the increased phosphorylation of APP suggested previously (Rockenstein et al., 2007). Regardless of the exact mechanism, the observed interaction adds additional support to a GSK3β – APP relationship that appears to meaningfully impact risk for amyloid burden in vivo.
Figure 1. GSK3β x APP on Amyloid Deposition.
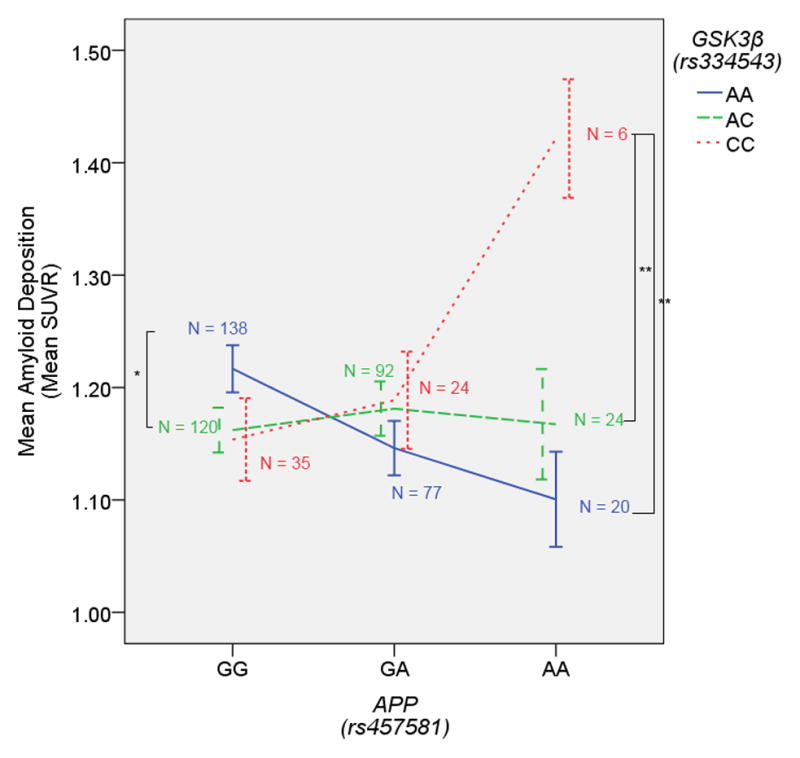
The GSK3β (rs334543) minor allele is associated with higher amyloid burden in homozygous carriers of the APP (rs457581) minor allele. Error bars represent the standard error. **p < 0.005 (one-tailed)
The other two significant interactions were between GSK3β and APBB2. In both cases, the strongest effect of GSK3β was present in homozygous carriers of the A allele for these SNPs (Figures 2 and 3), although the A allele was actually the major allele for rs2585590 (54% frequency). These two APBB2 SNPs are in low linkage disequilibrium in the 1000 Genomes dataset (r2 = 0.27, D′ = 1) leaving open the possibility that the same signal is driving both effects. However, the two SNPs did appear to differ slightly in terms of their interaction effect (Figures 2 and 3). The interaction with rs2585590 was particularly interesting because it appeared that GSK3β homozygous minor allele carriers showed especially low amyloid burden in homozygous carriers of the APBB2 A allele and especially high amyloid burden in homozygous carriers of the G allele. We would suggest that this may be due to the role GSK-3 plays in both APP processing and the neuroinflammatory response system. In certain scenarios, the increased cytokine production and microglial response driven by GSK-3 (Woodgett & Ohashi, 2005) might have beneficial effects by decreasing amyloid load through microglial phagocytosis (Rogers, Strohmeyer, Kovelowski, & Li, 2002). However, when GSK-3 activity is over active in the presence of over-expressed APP, any beneficial effects of an early pro-inflammatory response fail to clear amyloid fast enough, ultimately resulting primarily in the damaging side effects of neuroinflammation and failure to substantially reduce the aggregation of amyloid deposits.
Figure 2. GSK3β x APBB2(rs2585590) on Amyloid Deposition.
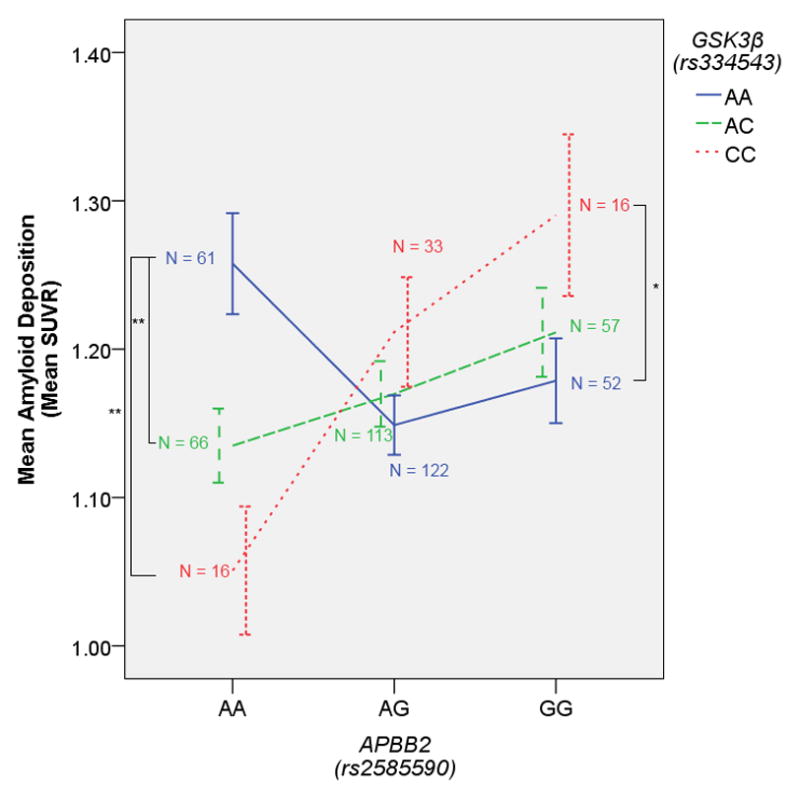
The GSK3β (rs334543) minor allele is associated with higher amyloid burden in homozygous carriers of the APBB2 (rs2585590) minor allele, and lower amyloid burden in homozygous carriers of the APBB2 major allele. Error bars represent the standard error. *p < 0.05 (one-tailed), **p < 0.005 (one-tailed)
Figure 3. GSK3β x APBB2(rs3098914) on Amyloid Deposition.
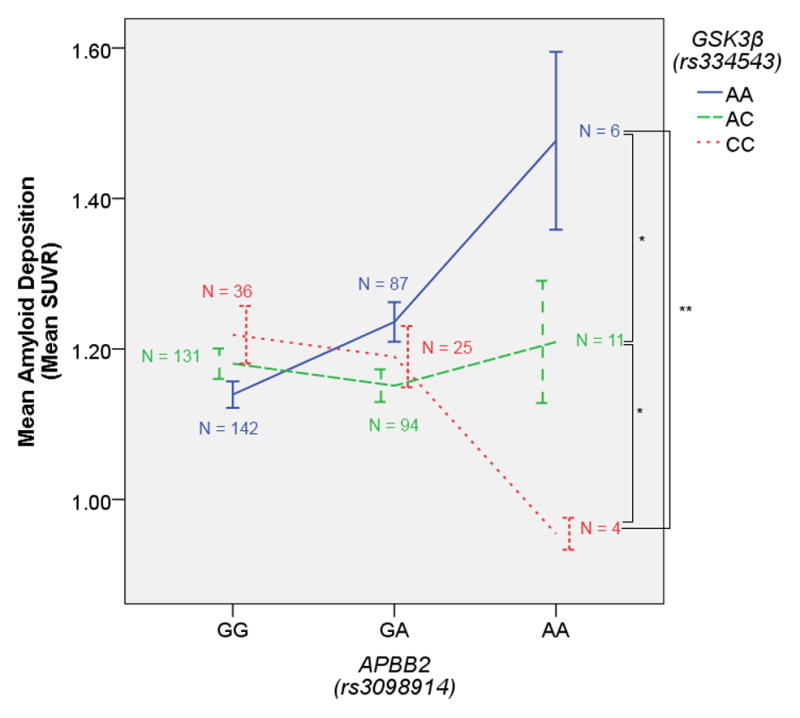
The GSK3β (rs334543) minor allele is associated with lower amyloid burden in homozygous carriers of the APBB2 (rs3098914) minor allele. Error bars represent the standard error. *p < 0.05 (one-tailed), **p < 0.005 (one-tailed)
Such a hypothetical model is particularly relevant to the APBB2 x GSK3β interaction because APBB2 appears to drive the intracellular production of both APP and GSK-3. The gene product of APBB2 is a member of the FE65 protein family, which interacts with the amyloid intracellular domain (AICD) and ultimately has an effect on APP processing (McLoughlin & Miller, 2008). Moreover, the AICD – FE65 interaction appears to have an effect on GSK3 activity in that the AICD modulates (increases) GSK-3 activity, but only when bound by FE65 (Ryan & Pimplikar, 2005). In the present result, variation in APBB2 may ultimately influence the probability of FE65 binding to the AICD and thus influence whether APP becomes overexpressed and whether GSK-3 becomes overactive. When FE65 binding to AICD is reduced (perhaps in homozygous carriers of the A allele in either of these two APBB2 SNPs), the slightly increased GSK3β expression related to minor allele status in rs334543 is actually beneficial. However, when the FE65-AICD complex is more prevalent, perhaps in rs2585590 G/G carriers, the slightly increased GSK3β expression becomes damaging in the presence of increased APP and the additional GSK-3 activity driven by the FE65-AICD complex.
Strengths and Limitations
The current manuscript has highlighted a potential gene-gene interaction in support of the GSK-3 hypothesis of Alzheimer’s disease. The strengths of the current analyses include the well characterized sample, a validated, disease relevant quantitative phenotype, and consistent interaction effects observed across two independent cohorts.
However, this study is not without limitations. Although consistent effects were observed across independent subsets in ADNI, a true replication sample from an additional data source with GWAS and PET data will be necessary to confirm our findings. This is especially relevant given the relative low cell counts present when performing interaction analyses, although the consistency in the present analysis in the fewer minor allele homozygotes is encouraging. Our two subsamples also included slight differences in clinical characteristics. The ADNI-1 sample in our analysis included fewer MCI and mild MCI subjects relative to ADNI-2/GO. In the case of the APP x GSK3β interaction, the trend level effect in ADNI-1 may indeed be driven by the discrepancy in MCI subjects relative to the other categories. The observed effects do appear to be driven by an effect in the MCI cohort in all cases when stratifying across diagnostic categories, but a larger sample of each group is necessary to test for a three-way interaction (diagnosis x APP x GSK3β). Moreover, because all AV-45 PET scans were conducted as part of ADNI-GO (about six years after ADNI-1 was initiated), subjects who were genotyped in ADNI-1 and received an AV-45 PET scan had been in the study longer and might have introduced a form of survivor bias into the ADNI-1 subsample.
The other main weakness of the current manuscript is the lack of data demonstrating the function of the implicated SNPs. Although bioinformatics methods were used to annotate findings based on known or predicted function, additional molecular experiments verifying the functional relationships between these genes, and these SNPs more specifically, is warranted.
Future Directions
The biologically plausible mechanism implicated in the current manuscript suggests some possible avenues for future exploration. Functional analyses focusing on rs334543 may help clarify the role this SNP plays in GSK-3 activity, and ultimately better elucidate the role of GSK-3 activity in APP processing and the neuroinflammatory response to amyloid deposition. Additional genetic analyses incorporating tau protein levels as measured in cerebrospinal fluid may also shed some light on the complex relationship between amyloid and tau pathology. The present work has identified a candidate genetic interaction between GSK3β and two genes involved in amyloid pathophysiology—APP and APBB2. Our results were consistent across the sub-datasets of ADNI, but future work replicating these interactions in an independent data source is warranted.
Supplementary Material
Acknowledgments
The authors declare no competing financial interests. This research was supported in part by the Vanderbilt NIMH Neurogenomics Training grant (T32 MH65215), the Vanderbilt Medical Scientist Training Program (T32 GM07347), and the Recruitment for Genetic Aging Research (P30 AG036445), and the Pharmaceutical Research and Manufacturers of America Foundation Fellowship in Translational Medicine and Therapeutics. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Data collection and sharing for this project was funded by ADNI (National Institutes of Health Grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Abbott; Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Amorfix Life Sciences Ltd;AstraZeneca; Bayer HealthCare; BioClinica, Inc; Biogen Idec Inc; Bristol-Myers Squibb Company; Eisai Inc; Elan Pharmaceuticals Inc; Eli Lilly and Company; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc; GE Healthcare; Innogenetics, N.V.; IXICO Ltd; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Medpace, Inc; Merck & Co, Inc; Meso Scale Diagnostics, LLC.; Novartis Pharmaceuticals Corporation; Pfizer Inc; Servier; Synarc Inc; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation or the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for NeuroImaging at the University of California, Los Angeles. This research was also supported by NIH grants P30 AG010129 and K01 AG030514.
Footnotes
Verification
The authors have no conflicts of interest to report. The data contained have not been previously published and will not be submitted elsewhere while under consideration at Neurobiology of Aging. Appropriate approvals were received from the local institutional review boards. All authors have reviewed the contents of the manuscript, approve of its contents, and verify the accuracy of the data.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bertram L, Lill CM, Tanzi RE. The genetics of Alzheimer disease: back to the future. Neuron. 2010;68:270–281. doi: 10.1016/j.neuron.2010.10.013. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer’s disease. Journal of neurochemistry. 2008;104:1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado DE, Molina-Porcel L, Carroll JC, MacDonald C, Aboagye AK, Trojanowski JQ, et al. Selectively silencing GSK-3 isoforms reduces plaques and tangles in mouse models of Alzheimer’s disease. The Journal of Neuroscience. 2012;32:7392–7402. doi: 10.1523/JNEUROSCI.0889-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner LM, Götz J. Amyloid-beta and tau– toxic pas de deux in Alzheimer’s disease. Nature Reviews Neuroscience. 2010;12:67–72. doi: 10.1038/nrn2967. [DOI] [PubMed] [Google Scholar]
- Landau S, Jagust W. Florbetapir processing methods. 2011 http://adni.bitbucket.org/docs/UCBERKELEYAV45/ADNI_UC_Berkeley_AV45_Methods_20121026.pdf.
- Liu F, Liang Z, Wegiel J, Hwang YW, Iqbal K, Grundke-Iqbal I, et al. Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. The FASEB Journal. 2008;22:3224–3233. doi: 10.1096/fj.07-104539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes JP, Oliveira CR, Agostinho P. Neurodegeneration in an A-beta induced model of Alzheimer’s disease: the role of Cdk5. Aging Cell. 2010;9:64–77. doi: 10.1111/j.1474-9726.2009.00536.x. [DOI] [PubMed] [Google Scholar]
- Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J. Decreased nuclear [beta]-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3[beta] conditional transgenic mice. EMBO J. 2001;20:27–39. doi: 10.1093/emboj/20.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L, Latypova X, Wilson CM, Magnaudeix A, Perrin ML, Yardin C, et al. Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Research Reviews. 2013;12:289–309. doi: 10.1016/j.arr.2012.06.003. [DOI] [PubMed] [Google Scholar]
- McLoughlin DM, Miller CCJ. The FE65 proteins and Alzheimer’s disease. Journal of Neuroscience Research. 2008;86:744–754. doi: 10.1002/jnr.21532. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Wilson CA, Lee VMY, Klein PS. GSK-3[alpha] regulates production of Alzheimer’s disease amyloid-[beta] peptides. Nature. 2003;423:435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- Piedrahita D, Hernández I, López-Tobón A, Fedorov D, Obara B, Manjunath BS, et al. Silencing of CDK5 reduces neurofibrillary tangles in transgenic alzheimer’s mice. The Journal of Neuroscience. 2010;30:13966–13976. doi: 10.1523/JNEUROSCI.3637-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potkin SG, Guffanti G, Lakatos A, Turner JA, Kruggel F, Fallon JH, et al. Hippocampal atrophy as a quantitative trait in a genome-wide association study identifying novel susceptibility genes for Alzheimer’s disease. PloS one. 2009;4:e6501. doi: 10.1371/journal.pone.0006501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DL, Sisodia SS. Mutant genes in familial Alzheimer’s disease and transgenic models. Annual review of neuroscience. 1998;21:479–505. doi: 10.1146/annurev.neuro.21.1.479. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. The American Journal of Human Genetics. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanan VK, Risacher SL, Nho K, Kim S, Swaminathan S, Shen L, et al. APOE and BCHE as modulators of cerebral amyloid deposition: a florbetapir PET genome-wide association study. Mol Psychiatry. 2013 doi: 10.1038/mp.2013.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E, Torrance M, Adame A, Mante M, Bar-on P, Rose JB, et al. Neuroprotective effects of regulators of the glycogen synthase kinase-3B signaling pathway in a transgenic model of Alzheimer’s disease are associated with reduced amyloid precursor protein phosphorylation. The Journal of Neuroscience. 2007;27:1981–1991. doi: 10.1523/JNEUROSCI.4321-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J, Strohmeyer R, Kovelowski CJ, Li R. Microglia and inflammatory mechanisms in the clearance of amyloid-beta peptide. Glia. 2002;40:260–269. doi: 10.1002/glia.10153. [DOI] [PubMed] [Google Scholar]
- Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, et al. ENCODE Data in the UCSC Genome Browser: year 5 update. Nucleic Acids Research. 2013;41:D56–D63. doi: 10.1093/nar/gks1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan KA, Pimplikar SW. Activation of GSK-3 and phosphorylation of CRMP2 in transgenic mice expressing APP intracellular domain. The Journal of cell biology. 2005;171:327–335. doi: 10.1083/jcb.200505078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryoo SR, Cho HJ, Lee HW, Jeong HK, Radnaabazar C, Kim YS, et al. Dual-specificity tyrosine(Y)-phosphorylation regulated kinase 1A-mediated phosphorylation of amyloid precursor protein: evidence for a functional link between Down syndrome and Alzheimer’s disease. Journal of neurochemistry. 2008;104:1333–1344. doi: 10.1111/j.1471-4159.2007.05075.x. [DOI] [PubMed] [Google Scholar]
- Shen L, Kim S, Risacher SL, Nho K, Swaminathan S, West JD, et al. Whole genome association study of brain-wide imaging phenotypes for identifying quantitative trait loci in MCI and AD: a study of the ADNI cohort. Neuroimage. 2010;53:1051. doi: 10.1016/j.neuroimage.2010.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small SA, Duff K. Linking Abeta and tau in late-onset Alzheimer’s disease: a dual pathway hypothesis. Neuron. 2008;60:534–542. doi: 10.1016/j.neuron.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Ryder J, Li B, Wu X, Fox N, Solenberg P, et al. Lithium, a Common Drug for Bipolar Disorder Treatment, Regulates Amyloid-beta Precursor Protein Processing. Biochemistry. 2004;43:6899–6908. doi: 10.1021/bi035627j. [DOI] [PubMed] [Google Scholar]
- Swaminathan S, Shen L, Risacher S, Yoder K, West J, Kim S, et al. Amyloid pathway-based candidate gene analysis of [11C]PiB-PET in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort. Brain Imaging and Behavior. 2012;6:1–15. doi: 10.1007/s11682-011-9136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Research. 2012;40:D930–D934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegiel J, Gong CX, Hwang YW. The role of DYRK1A in neurodegenerative diseases. FEBS Journal. 2011;278:236–245. doi: 10.1111/j.1742-4658.2010.07955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodgett JR, Ohashi PS. GSK3: an in-Toll-erant protein kinase? Nature immunology. 2005;6:751. doi: 10.1038/ni0805-751. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.