

Abstract
Expression analysis of mRNAs transcribed from certain genes can be used as important sources of biomarkers for in vitro diagnostics. While the use of reverse transcription quantitative PCR (RT-qPCR) can provide excellent analytical sensitivity for monitoring transcript numbers, more sensitive approaches for expression analysis that can report results in near real-time are needed for many critical applications. We report a novel assay that can provide exquisite limits-of-quantitation and consists of reverse transcription (RT) followed by a ligase detection reaction (LDR) with single-pair fluorescence resonance energy transfer (spFRET) to provide digital readout through molecular counting. For this assay, no PCR was employed, which enabled short assay turnaround times. To facilitate implementation of the assay, a cyclic olefin copolymer (COC) microchip, which was fabricated using hot embossing, was employed to carry out the LDR in a continuous flow format with on-line single-molecule detection following the LDR. As demonstrators of the assay's utility, MMP-7 mRNA was expression profiled from several colorectal cancer cell lines. It was found that the RT-LDR/spFRET assay produced highly linear calibration plots even in the low copy number regime. Comparison to RT-qPCR indicated a better linearity over the low copy number range investigated (10 − 10,000 copies) with an R2 = 0.9995 for RT-LDR/spFRET and R2 = 0.98 for RT-qPCR. In addition, differentiating between copy numbers of 10 and 50 could be performed with higher confidence using RT-LDR/spFRET. To demonstrate the short assay turnaround times obtainable using the RT-LDR/spFRET assay, a 2 thermal cycle LDR was carried out on amphiphysin gene transcripts that can serve as important diagnostic markers for ischemic stroke. The ability to supply diagnostic information on possible stroke events in short turnaround times using RT-LDR/spFRET will enable clinicians to treat patients effectively with appropriate time-sensitive therapeutics.
Introduction
Expression analysis of mRNA to determine gene activity has a number of important applications, one example being the diagnosis of various disease states.1, 2 mRNA molecules can be analyzed using such techniques as Northern blotting, ribonuclease protection assays (RPA), in-situ hybridization (ISH), reverse-transcription quantitative PCR (RT-qPCR), or cDNA microarrays. In many laboratories, Northern blotting continues to be the standard protocol,3 which can determine the size of a transcript and allow comparison of mRNA abundance from different samples on a single membrane. However, Northern analyses generally require significant amounts of mRNA due to its low sensitivity. RPA is a solution-based hybridization technique for expression analysis that can be used to detect mRNAs that are expressed at low levels. ISH is a powerful method especially useful for localizing and detecting the expression of mRNAs in specific regions of cells or within morphologically preserved tissue sections.4
The aforementioned methods, unfortunately, involve many laborious steps and high levels of operator expertise to achieve the desired results, which makes them problematic for producing results in a timely manner by operators not well-trained in the art.5 Therefore, mRNA expression analysis for in vitro diagnostics, where treatment decisions are predicated on securing highly quantitative and sensitive results by clinical laboratory technicians, are difficult to envision.
cDNA microarrays can simultaneously analyze a large number of gene transcripts in parallel with the capacity for automation making cDNA microarrays attractive for expression profiling on a global scale for discovery based applications.6 But, this technique is limited by its low sensitivity and accuracy when quantitative measurements of mRNA expression are desired.7, 8
RT-qPCR is a much more efficient method for mRNA expression analysis and is characterized by shorter assay turnaround times and lower limits-of-detection compared to the aforementioned techniques. It possesses a theoretical limit of detection of a single copy of an mRNA target.9 In addition, RT-qPCR has been demonstrated to possess sufficient analytical sensitivity to discern a 1.5-fold difference in expression.10, 11 However, the exponential amplification subjects it to high levels of variation because minute errors introduced at early stages of the amplification process will be compounded with increasing cycle number. Additionally, the accuracy of the results is greatly affected by sample preparation, reagent quality, primer design and skill of the operator.12
Several alternative RT-qPCR techniques have been suggested that utilize a digital readout format instead of an analog one typically employed for this technique; these include digital and droplet PCR.13-17 For these methods, a single copy of the target (Poisson distributed) is discretely amplified in individual wells or droplets and quantitation is secured through counting positive results in each well or droplet. Several papers have shown that digital PCR can provide improved ability for measuring minute expression differences compared to RT-qPCR.15, 17
There are various PCR-free techniques that have been developed to emulate the sensitivity of RT-qPCR while avoiding its amplification steps. For example, Medley et al. utilized dual-labeled molecular beacon probes to monitor the expression of multiple gene transcripts in living breast carcinoma cells.18 Chen et al. reported an electrical sensor array combined with multiple hybridization steps for mRNA analysis,19 which used a pair of nano-gapped electrodes producing a conductance change when the target hybridized to the appropriate probes. Lin and co-workers utilized a branched-DNA assay (bDNA) for mRNA quantification.20 The bDNA assay generated detectable quantities of target mRNA by enhancing the signal using multiply-labeled oligonucleotide probes. This technique was able to detect 1 fM of target mRNA in p185 BCR-ABL leukemia fusion transcripts.
Single-molecule detection (SMD) can also be used for monitoring expression levels of mRNAs with the potential to offer extraordinary sensitivity due to the digital nature of the readout. Neely et al. reported laser-induced fluorescence SMD for the quantitation of microRNA (miRNA) expression;21 a four-color laser-induced fluorescence instrument was used to count coincident events arising from fluorescently labeled locked nucleic acid DNA probes hybridized to target miRNA molecules. Rigler and co-workers developed a hybridization assay containing two oligonucleotide probes for gene expression.22 Simultaneous hybridization of dye-labeled probes to the target gene produced cross-correlation features, which were measured using two-color fluorescence correlation spectroscopy.
The commonality in many of these PCR-free approaches was that hybridization-based recognition was required, which can be challenging when single-base variations must be monitored demanding the need for stringent hybridization conditions. Also, due to the thermodynamic nature of hybridization, higher copy numbers of interfering sequences can make detection of the target problematic when it is a minority in a mixed population.
Herein, we report a unique single-molecule detection scheme for expression profiling mRNA that uses reverse transcription and the ligase detection reaction coupled to single-pair FRET (RT-LDR/spFRET) to digitally count and quantify mRNAs (i.e. molecular counting) for securing exquisite limits-of-quantitation in the low copy number regime. This assay is illustrated in Figure 1 and takes advantage of the linear amplification process associated with LDR. A similar scheme has been adopted for detecting single nucleotide polymorphisms in KRAS genes and strain specific identification of bacterial pathogens.23, 24
Figure 1.
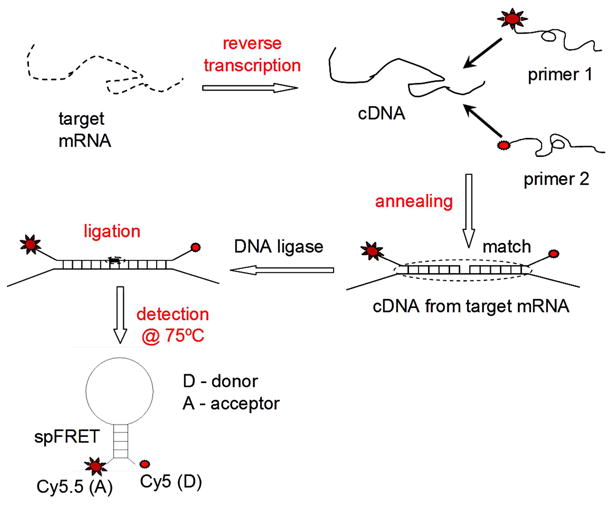
Illustration of RT-LDR/spFRET assay for the expression analysis of mRNA transcripts. The assay utilizes no PCR and instead, employs a linear amplification scheme that can provide favorable limits-of-quantitation with high analytical sensitivity due to the digital readout format (i.e., single-molecule counting). Following reverse transcription, flanking primer pairs targeting unique sequences in specific mRNAs are subjected to ligation via the LDR. Successful ligation events generate MBs that undergo FRET. Single-molecule detection of MBs can give a direct readout of the mRNA copy number.
In the assay reported in this work, mRNA molecules were first converted into their complementary DNA sequences (cDNA) by reverse transcription. A pair of oligonucleotide primers was designed to flank two adjacent sequences located within a reporter region of this transcript, which consisted of a span of sequence that did not appear in any other mRNA species found in the biological sample. These primers also contained a 10-base stem sequence complementary to each other while not complementary to the target sequence. The end of each stem sequence was attached to either a donor or acceptor dye for fluorescence readout using single-pair fluorescence resonance energy transfer (spFRET) that occurred when the stem sequences were hybridized. Successful ligation of the LDR primers occurred only if the target sequence was complementary to both LDR primers resulting in the formation of a molecular beacon (MB). The stem sequences of the MB were designed to possess a high melting temperature (Tm) by incorporating a GC-rich sequence and thus, thermodynamically more stable than the target-oligonucleotide duplex. Therefore, the MB adopted a stable stem-loop conformation following ligation forming the MB. The spFRET signal from individual MBs was reported by a SMD instrument from which the copy number of the mRNA could be deduced.
LDR thermal cycling and single-molecule readout were performed directly on a thermoplastic fluidic device to provide near real time results with excellent limits-of-quantitation and high analytical sensitivity resulting from molecular counting. The chip in this work performed the LDR using a continuous flow format and then, detected MBs on-line and in near real time.
The exceptional fidelity of LDR in recognizing matched targets even in the presence of large excesses of mismatched sequences provided high specificity. This specificity was derived from: 1) The need for two concurrent hybridization events. Therefore, primers can be designed that target a unique reporter sequence within the mRNA transcript of interest, negating the need for single-base discrimination. 2) DNA ligase is intolerant of a 3′-end mismatch. The outstanding specificity of LDR allows it to discriminate even a single base mismatch at the 3′-end.25 This specificity is especially valuable for differentiating miRNAs because some human miRNA members differ by only one or two nucleotide bases.26
The utility of this LDR/spFRET assay, when transitioned to a fluidic chip, was demonstrated by profiling MMP-7 gene activity in various colorectal cancer cell lines. Colorectal cancer has evolved into a serious health problem, especially in developed countries.27 MMP-7 has been identified as an important biomarker for colorectal cancer staging and it has been extensively reported that over-expression of this gene provides strong implications as to the extent of invasion, progression, and metastasis.28-30 In this study, mRNA transcripts from the MMP-7 gene were accurately quantified using our RT-LDR/spFRET assay. We will demonstrate the utility of RT-LDR/spFRET for the quantitation of low-copy number transcripts with comparisons to conventional RT-qPCR.
To demonstrate the utility of the RT-LDR/spFRET assay for near real time reporting, we will monitor the expression of genes whose aberrant activity can be associated with brain trauma, such as stroke. Monitoring changes in expression levels of functional genes has been used for the diagnosis of stroke as well as other brain disorders. Moore et al. conducted gene expression profiling using cDNA microarrays of peripheral blood mononuclear cells (PBMCs) from ischemic stroke patients with several genes showing altered activity as a result of a stroke event.31 Recently, Baird discovered two genes, amphiphysin (AMPH) and IL1R2, that were expressed differentially among hemorrhagic and ischemic stroke patients; the discovery of these markers were affected through cDNA microarray studies.32 Unfortunately, the extended assay turnaround time and sophisticated nature of both RT-qPCR and cDNA microarrays can make these assay strategies prohibitive for monitoring mRNA transcript levels for stroke diagnostics at the point-of-care. Time to diagnosis is critical in stroke because some treatment regimens have a short effective time window, such as tissue plasminogen activator treatment for ischemic stroke (effective treatment window = 3 h).33
Experimental
Materials and Reagents
The oligonucleotide primers for LDR and RT-qPCR were synthesized by Integrated DNA Technologies (Coralville, IA), purified by RP-HPLC and suspended in 1× TE buffer. AmpliTaq Gold polymerase was purchased from Applied Biosystems (Foster City, CA). Taq DNA ligase was purchased from New England BioLabs (Ipswich, MA). Cyclic olefin co-polymer, COC, was secured from Topas Advanced Polymers (Florence, Kentucky). The primers used for the LDR are given in Table S1 and their design considerations outlined in the Supporting Information.
Fluidic chips for LDR/spFRET measurements
The basic architecture of the chip used in these studies is shown in Figure 2. It consisted of a continuous flow thermal reactor for carrying out either 2 or 20 thermal cycles for the LDR, the output of which was directly interrogated using SMD with a laser beam focused onto a tapered channel poised at the output end of the continuous flow thermal reactors. A description of the fabrication strategy for producing this chip can be found in the Supporting Information.
Figure 2.
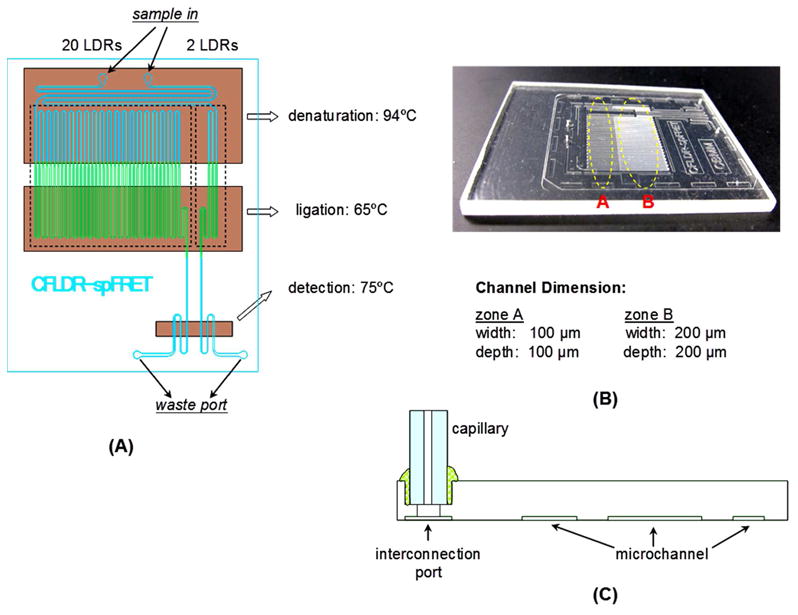
Layout of the microchannels for the hot-embossed COC chip used to carry out a continuous flow LDR with on-line single-molecule detection. (A) Schematic diagram of the microfluidic chip. (B) Photographic image of the chip. (C) Illustration for the attachment of a capillary to the chip to allow pumping of sample through the chip hydrodynamically.
mRNA extraction
For a description of the experimental procedures for mRNA extraction from biological cells, please see the Supporting Information.
Operation of the LDR/spFRET chip
A capillary tube attached to the chip was connected to a glass syringe (SGE, Australia) via a syringe-to-capillary adapter (InnovaQuartz, Phoenix, AZ). The LDR cocktail was first run through a 0.2 μm filter to remove any large particulates. Then, the reaction mixture was loaded into a glass syringe and driven by a syringe pump (Pico Plus, Harvard Apparatus, Holliston, MA) through the thermal reactor's microchannels. Thin-film kapton heaters were placed at the appropriate zones of the chip to provide the desired temperatures for the LDR/spFRET measurements. The denaturation and renaturation/ligation zones are shown in Figure 2A. A 3.5 mm gap was placed between the denaturation and renaturation/ligation zones to minimize thermal crosstalk. The volume flow rate was set between 0.1− 8 μL/min depending on the required ligation time. For a description of the experimental conditions used for performing on-chip continuous flow LDR, see the Supporting Information.
Results and Discussion
LDR using continuous flow thermal cycling in a COC chip
Thermal cycling using continuous flow reactors have been found to provide better thermal management characteristics compared to batch-type thermal cyclers because the heating/cooling of large thermal masses are not required for the continuous flow format providing the ability to perform reactions at rates limited by enzyme kinetics.34-37 However, because continuous flow reactors are characterized by large surface-to-volume ratios, the reaction can be passivated due to enzyme deactivation induced by adsorption to channel walls. To mitigate this artifact, BSA can be used as a dynamic coating material because of its propensity to adsorb to many surfaces and thus, maintain the efficiency of the bio-reaction.38 The surface of many thermoplastics typically used for microfluidics can be coated with BSA to prevent bio-enzyme deactivation.39
The proper selection of the substrate used for the LDR continuous flow thermal reaction with onboard single-molecule detection is critical because the Tg of the polymer must be higher than the cDNA denaturation temperature used for the LDR to minimize microstructure deformation during operation and also, possess good optical properties. Based on these criteria, we selected cyclic olefin copolymer, COC, as the substrate.40 When treated with BSA, COC showed a significant reduction in its sessile water contact angle (see Supporting Information for experimental details), which was 90° prior to BSA treatment and 48° after BSA treatment. This surface coating was also found to be stable for extended periods of operation irrespective of the temperature supplied to the chip as long as it was below the Tg of COC (Tg ∼130°C).
To determine that the appropriate LDR products were generated as well as optimizing reaction efficiency, the output of the continuous flow LDR reactor was analyzed using capillary gel electrophoresis off-line (Figure S1). As seen in Figure S1B, very little LDR product was formed when the reaction was carried out in a pristine COC chip due to ligation enzyme deactivation generated by non-specific adsorption as noted previously. Figure S1A shows electrophoresis results of LDR products obtained using the continuous flow COC reactor treated with BSA. Significantly higher amounts of LDR product were observed suggesting that treatment with BSA reduces the amount of enzyme deactivation.
Improving sampling efficiency for on-chip SMD
For detection of single molecules using laser-induced fluorescence, it is desirable to create a diffraction-limited probe volume to improve the SNR in the measurement by minimizing background due to scattering or impurity fluorescence.41 For quantitative analysis, however, diffraction-limited sampling volumes are challenged by poor sampling efficiency (defined as the ratio of the 1/e2 laser beam cross-section to the cross-sectional area of the microchannel), due to the fact that only a small portion of the target molecules actually traverse through the 1/e2 defined sampling volume.
Sampling efficiency in SMD can be improved by confining the molecules to travel through the center of the fluid channel using hydrodynamic focusing, electrokinetic focusing, or reducing the flow channel dimensions.42-46 In this study, a microfluidic channel with a tapered detection region of 25 × 25 μm cross section was fabricated to improve sampling efficiency of single molecules. In this design, one section of the channel had a cross section of 100 × 100 μm to accommodate the thermal reactor while the other section had a reduced cross section to confine more analyte molecules to flow through the laser-defined probe volume. With a probe volume defined by the 1/e2 laser beam size (2 × 5 μm) and the depth of focus on the collection objective (∼1 μm), the estimated sampling efficiency for the 25 × 25 μm channel was 0.8%, and 0.05% for the 100 × 100 μm channel, assuming the laser spot was 2 × 5 μm, and the depth of focus for the objective was ∼1 μm.
Figure S2A shows photon bursts from single fluorescent microspheres flowing through a channel with a cross section of 100 × 100 μm. The background fluorescence for this microchannel was ∼1,000 cps. Thirty-two bursts were revealed above the threshold (3,000 cps), corresponding to a sampling efficiency of 0.05%.
The background fluorescence of the microchannel with a tapered detection section is shown in Figure S2B and was ∼2,000 cps. Using a threshold level of 6,000 cps, a total of 312 events were observed, corresponding to a sampling efficiency of 0.8%, close to that predicted based on the cross-sectional size of the fluidic channel and the size of the laser beam. It was also noted that the average peak height in Figure S2B was higher than that in Figure S2A, most likely resulting from an increased number of microspheres traveling through the beam center where they would experience higher excitation rates. In the remaining experiments, the 25 × 25 μm tapered channel was used.
Effects of flow rate on the LDR/spFRET assay
In photon burst detection, high photon burst peak amplitudes are favored due to better SNRs, which can be produced through the use of longer residence times of the fluorophore within the excitation laser beam as long as significant amounts of photobleaching do not occur. In our case, the flow rate also dictated the efficiency and time for the LDR because the sample is continuously fed through the thermal reactor and into the single-molecule detector. Therefore, the assay efficiency was evaluated at different volumetric flow rates ranging from 0.35 − 6.24 μL/min (see Figure S3A for photon burst data). Figure S3B shows the average peak height of photon bursts as a function of sample flow rate and Figure S3C shows the number of counted bursts as a function of flow rate. From this data, to keep assay time short and maximize the SNR for single molecules, a flow rate of 1 μL/min was used.
At this optimal flow rate, an assay turnaround time of 22.3 min for 20 LDR thermal cycles would result. We should note that even at the low flow rate, we found that photobleaching was of a minimal consequence (see Supporting Information). Because our COC chip also contained a 2 cycle device, the assay turnaround time in this case would be 2.2 min. However, due to the linear amplification associated with LDR, the 2 cycle LDR would reduce the number of the resultant products by 10-fold and thus, reduce assay analytical sensitivity.
Quantitative measurements of MMP-7 transcripts using RT-qPCR
To quantify the MMP-7 mRNA expression level in different cell lines, a standard curve was constructed using RT-qPCR. As seen in Figure 3A, the fluorescence signal was very reproducible for each input copy number (the copy number was altered by changing the number of cells used for RT-qPCR). The CT value was plotted against the log of the copy number in each sample and fit to a linear function as shown in Figure 3B. The fitted line had a R2 value of 0.98. In Figure 3A, we can see that the fluorescence from the no-template control (NTC) and no-RT control (NRTC) were very weak. In Table S2, the copy number of MMP-7 in different cells lines as determined by RT-qPCR is provided.
Figure 3.
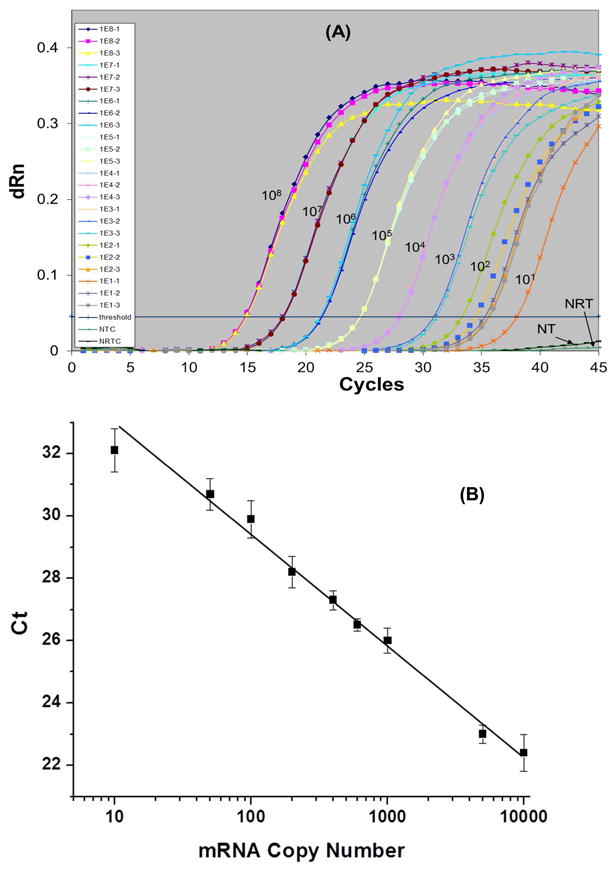
Standard curve for quantification of MMP-7 transcripts using RT-qPCR. (A) Raw fluorescence data. (B) Linear calibration plot of CT, which represents the crossing threshold value, versus input log copy number over the range of 10 − 10,000 copies. Purified HT29 mRNAs were used as the input material for the RT-qPCR assay. The reaction was incubated at 50°C for 30 min followed by 40 thermal cycles at 95°C for 30 s, 59°C for 1 min, 72°C for 30 s. Each experiment was run in replicates of three. For further description of the RT-qPCR method and mRNA purification, see the Supporting Information.
Figure S4 shows the melting curves for the PCR amplicons generated from the above experiment. The peak at 84°C corresponded to the 395 bp amplicon expected. In this plot, there were no other peaks identified suggesting that the primers designed to amplify the MMP-7 cDNA were highly specific to this target sequence.
On-line single-molecule detection of MMP-7 mRNA using RT-LDR/spFRET
The aim of these studies was to quantitatively measure the expression level of specific mRNA transcripts using LDR performed in a continuous flow thermal reactor with the resultant LDR products digitally counted via spFRET. As RNA cannot typically serve as a template for LDR due to low yields, the target mRNAs were first reverse transcribed into cDNAs and then directly subjected to LDR with no intervening PCR, which reduced time overhead in securing results and potentially could improve the quantitative capabilities of the assay. In this study, HT29 and SW620 colorectal cancer cell lines were used as models for expression profiling MMP-7 transcripts. HEL299 was used as a negative control because of its negligible expression of MMP-7 (see Table S2). In this negative control, all of the LDR reagents were present, including unligated primers, which should account for direct excitation of the spFRET acceptor. The sensitivity and quantification capabilities of RT-LDR/spFRET for expression profiling were evaluated and compared to our RT-qPCR results.
The generated single-molecule photon bursts are shown in Figure 4. To exclude false positives, a threshold level of 3,000 cps was set as a discriminator threshold, which produced a false positive rate of 0 for negative controls.24 In Figure 4, a total of 97 photon bursts were identified from HT29 cells over a 1 min sampling time (t) and a volumetric flow rate (ν) of 1 μL/min. This number corresponds to 199 copies/cell of MMP-7 transcripts in the original sample after being corrected for reverse transcription efficiency (ηRT, 0.76), LDR efficiency (ηLDR, 0.4), the number of LDR cycles (κLDR, 20), the number of cells analyzed (ηcell; ηcell = [cells] × v × t; [cells] = cell concentration, ∼10 cells μL-1) and a sampling efficiency (ηsampling, 0.8%) according to the following equation;
Figure 4.
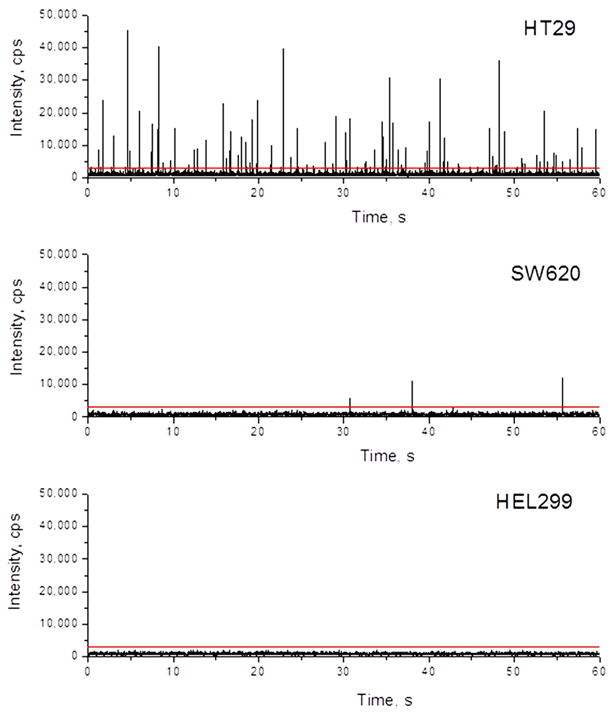
Photon bursts generated from MBs produced as a result of successful LDR events for expression profiling of MMP-7 transcripts. Following reverse transcription, the cDNA was mixed with the LDR reaction cocktail and loaded into a 50 μL glass syringe that was affixed to the COC chip. The LDR mixture contained 10 pM of upstream and downstream LDR primers, 2 U/μL of DNA ligase, 0.5 mg/mL of BSA and mRNA from HT29, SW620 and HEL299 cell lines. The LDR was subjected to 20 thermal cycles in the continuous-flow reactor that was BSA-treated and the reactants were driven at a flow rate of 1.0 μL/min. The data was collected for 1 min. A threshold of 6,000 cps was set to eliminate false positive events. Two hundred ng of total RNA was extracted from each of these cell lines and converted into cDNA by AffinityScript reverse transcriptase.
| (1) |
This value is consistent with the result of 178 copies/cell obtained by RT-qPCR. There were 3 photon bursts identified using cDNA from the SW620 cell line during the 1 min sampling time, which corresponded to 6.2 ±0.7 copies/cell. This agreed favorably with the result of 3.2 ±0.9 copies/cell obtained by RT-qPCR. There were no photon bursts identified from the HEL299 cell line consistent with our RT-qPCR results (see Table S2).
Quantitative measurements of MMP-7 transcripts using RT-LDR/spFRET
Figure 5 shows a calibration plot for the number of photon bursts detected as a function of the input copy number of MMP-7 transcripts using the LDR/spFRET assay. The calibration plot was linear with a R2 value of 0.9995 over a copy number range of 0 − 10,000, which was better than that compared to RT-qPCR, which generated an R2 value of 0.98. Careful inspection of the calibration plots at the low copy number regime (<500 copies) indicated that the linearity was better for the RT-LDR/spFRET assay with better sensitivity as well. For example, the difference in the means for the burst numbers at 10 and 50 mRNA copies was significant at p = 0.005 (two-sided t test with n = 6), while for the RT-qPCR assay, p = 0.05 for the same copy numbers.
Figure 5.
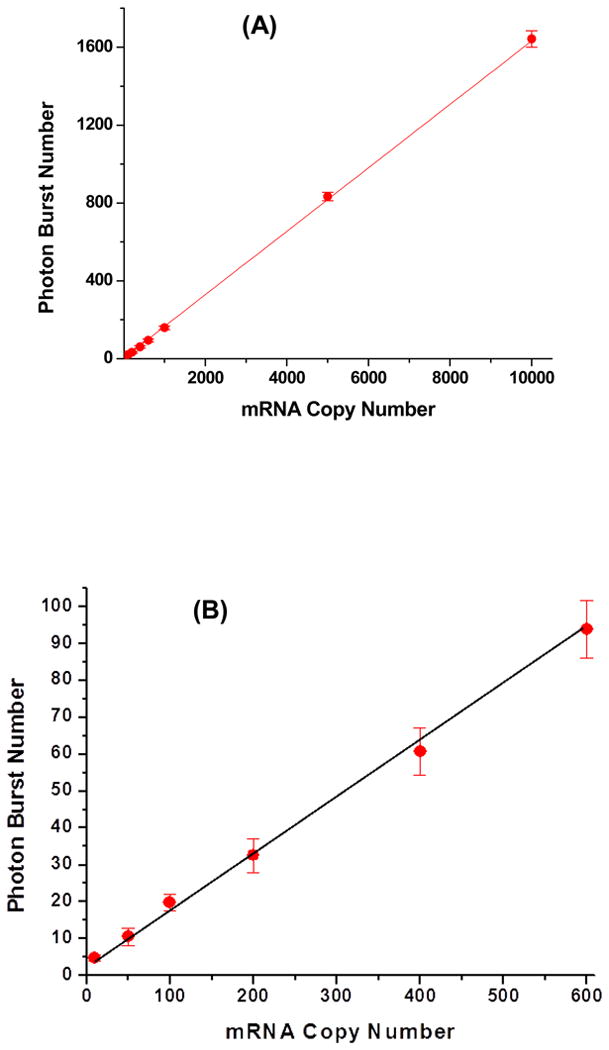
Calibration plot of photon bursts as a function of input copy number of MMP-7 transcripts. The LDR cocktail contained 10 pM of upstream and downstream LDR primers, 2 U/μL of DNA ligase, 0.5 mg/mL of BSA, and cDNAs that were reverse transcribed from MMP-7 transcripts with input copy numbers ranging from 10 − 10,000 (A) with an expanded view in the 10-600 copy range shown in (B). The reaction mixture was driven at a flow rate of 1.0 μL/min and subjected to 20 thermal cycles through the 65°C ligation zone and 94°C denaturation zone.
Measurements of AMPH transcripts using RT-LDR/spFRET
To demonstrate the rapid reporting capabilities of the RT-LDR/spFRET assay, expression analysis of AMPH transcripts for stroke diagnosis were evaluated. The LDR cocktail was shuttled through the reactor at a flow rate of 1.0 μL/min using only 2 thermal cycles to keep the assay time short (∼2 min). The generated MBs were directly observed downstream of the LDR reactor, where a 25 × 25 μm detection window was poised. A calibration plot of the number of detected photon bursts as a function of the input copy number of AMPH mRNAs was constructed. The number of photon burst events was linear with the input copy number of AMPH mRNA (R2 = 0.9991) and the slope of the calibration plot was 0.0433 photon bursts per AMPH mRNA, which was a factor or 10 less than the sensitivity observed for the 20-cycle assay (see Figure 5).
Conclusions
A unique assay that can provide near real-time reporting of mRNA expression with limits-of-quantitation superior to that of RT-qPCR, especially in the low copy number regime, was presented. The short assay turnaround time was affected by eliminating the need for PCR and instead, using a linear amplification of cDNAs with digital readout (single-molecule detection). To facilitate sample processing by providing automation, a COC microchip was employed that was able to carry out the LDR as well as the single-molecule detection requiring no operator intervention. The assay turnaround time was primarily determined by the processing flow rate, which must be balanced by optimizing the performance of the LDR and the single-molecule detection efficiency. Using the optimized flow rate, we could carry out the RT-LDR/spFRET assay in either 22 min or 2 min, depending on the number of thermal cycles required. Expression analysis of the MMP-7 gene was performed to analyze the transcriptional level from different cell lines to understand copy number effects on the limits-of-quantitation. For the spFRET readout, we could distinguish between mRNA copy numbers of 10 and 50 at a higher confidence level compared to RT-qPCR. Expression of AMPH transcripts was also analyzed using this assay and could produce results in ∼2 min, which will be an important assay metric when considering treatment decisions for ischemic versus hemorrhagic stroke patients using recombinant tissue plasminogen activator therapy.
Future work will focus on including into the fluidic chip architecture steps for carrying out cell selection from whole blood, cell lysis, solid-phase extraction/purification of mRNA and reverse transcription integrated to the LDR/spFRET steps reported herein. This will provide an autonomous system for the point-of-care analysis of clinical samples to expression profile certain genes that can provide important diagnostic information in near real time with exquisite quantitative capabilities, even at the point-of-care. In addition, integrating this chip-based system to the compact laser instrumentation we have developed for single-molecule detection will enable single-molecule detection will enhance the prospects of field monitoring.47
Supplementary Material
Acknowledgments
The authors would like to thank the National Institutes of Health (NIBIB; EB010087) for support of this work. The authors would also like to thank the World Class University (WCU) Program of South Korea for partial support of this work.
References
- 1.Cohen CD, Frach K, Schlondorff D, Kretzler M. Kidney Inter. 2002;61:133–140. doi: 10.1046/j.1523-1755.2002.00113.x. [DOI] [PubMed] [Google Scholar]
- 2.Tenedini E, Roncaglia E, Ferrari F, Orlandi C, Bianchi E, Bicciato S, Tagliafico E, Ferrari S. Cell Death & Dis. 2010;1 doi: 10.1038/cddis.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rapley R, Walker JM. Molecular Biomethods Handbook. 2. Humana Press; Totowa, NJ: 2008. [Google Scholar]
- 4.Jin L, Lloyd RV. J Clin Lab Anal. 1997;11:2–9. doi: 10.1002/(SICI)1098-2825(1997)11:1<2::AID-JCLA2>3.0.CO;2-F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamaguchi Y, Yatsushiro S, Yamamura S, Abe H, Abe K, Watanabe M, Kajimoto K, Shinohara Y, Baba Y, Kataoka M. Analyst. 2011;136:2247–2251. doi: 10.1039/c0an01044h. [DOI] [PubMed] [Google Scholar]
- 6.Schulze A, Downward J. Nat Cell Biol. 2001;3:E190–E195. doi: 10.1038/35087138. [DOI] [PubMed] [Google Scholar]
- 7.Wang E, Miller LD, Ohnmacht GA, Liu ET, Marincola FM. Nat Biotechnol. 2000;18:457–459. doi: 10.1038/74546. [DOI] [PubMed] [Google Scholar]
- 8.Nelson PT, Baldwin DA, Scearce LM, Oberholtzer JC, Tobias JW, Mourelatos Z. Nat Methods. 2004;1:155–161. doi: 10.1038/nmeth717. [DOI] [PubMed] [Google Scholar]
- 9.Palmer S, Wiegand AP, Maldarelli F, Bazmi H, Mican JM, Polis M, Dewar RL, Planta A, Liu S, Metcalf JA, Mellors JW, Coffin JM. J Clin Microbiol. 2003;41:4531–4536. doi: 10.1128/JCM.41.10.4531-4536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiu RW, Murphy MF, Fidler C, Zee BC, Wainscoat JS, Lo YM. Clin Chem. 2001;47:667–672. [PubMed] [Google Scholar]
- 11.Zimmermann B, Holzgreve W, Wenzel F, Hahn S. Clin Chem. 2002;48:362–363. [PubMed] [Google Scholar]
- 12.Wong ML, Medrano JF. Biotechniques. 2005;39:75–85. doi: 10.2144/05391RV01. [DOI] [PubMed] [Google Scholar]
- 13.Bizouarn F. Gen Eng & Biotech News. 2012;32:32–33. [Google Scholar]
- 14.Frampton J, Thorne C, Spivey V, Goodall J, Lowe C. Virchows Archiv. 2012;461:S30–S30. [Google Scholar]
- 15.Hayden RT, Gu Z, Ingersoll J, Abdul-Ali D, Shi L, Pounds S, Caliendo AM. J Clin Microbiol. 2013;51:540–546. doi: 10.1128/JCM.02620-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin CQ, Yao B. Pro in Chem. 2012;24:2415–2423. [Google Scholar]
- 17.Whale AS, Huggett JF, Cowen S, Speirs V, Shaw J, Ellison S, Foy CA, Scott DJ. Nucleic Acids Res. 2012;40 doi: 10.1093/nar/gks203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Medley CD, Drake TJ, Tomasini JM, Rogers RJ, Tan WH. Anal Chem. 2005;77:4713–4718. doi: 10.1021/ac050881y. [DOI] [PubMed] [Google Scholar]
- 19.Chen XJ, Roy S, Peng YF, Gao ZQ. Anal Chem. 2010;82:5958–5964. doi: 10.1021/ac1003135. [DOI] [PubMed] [Google Scholar]
- 20.Lee AC, Dai ZY, Chen BW, Wu H, Wang J, Zhang AG, Zhang LR, Lim TM, Lin YH. Anal Chem. 2008;80:9402–9410. doi: 10.1021/ac801263r. [DOI] [PubMed] [Google Scholar]
- 21.Neely LA, Patel S, Garver J, Gallo M, Hackett M, McLaughlin S, Nadel M, Harris J, Gullans S, Rooke J. Nat Methods. 2006;3:41–46. doi: 10.1038/nmeth825. [DOI] [PubMed] [Google Scholar]
- 22.Korn K, Gardellin P, Liao B, Amacker M, Bergstrom A, Bjorkman H, Camacho A, Dorhofer S, Dorre K, Enstrom J, Ericson T, Favez T, Gosch M, Honegger A, Jaccoud S, Lapczyna M, Litborn E, Thyberg P, Winter H, Rigler R. Nucleic Acids Res. 2003;31 doi: 10.1093/nar/gng089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wabuyele MB, Farquar H, Stryjewski W, Hammer RP, Soper SA, Cheng YW, Barany F. JACS. 2003;125:6937–6945. doi: 10.1021/ja034716g. [DOI] [PubMed] [Google Scholar]
- 24.Peng ZY, Soper SA, Pingle MR, Barany F, Davis LM. Anal Chem. 2010;82:9727–9735. doi: 10.1021/ac101843n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barany F. Proc Natl Acad Sci USA. 1991;88:189–193. doi: 10.1073/pnas.88.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miska EA, Alvarez-Saavedra E, Townsend M, Yoshii A, Sestan N, Rakic P, Constantine-Paton M, Horvitz HR. Gen Biol. 2004;5 doi: 10.1186/gb-2004-5-9-r68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levin B, Lieberman DA, McFarland B, Andrews KS, Brooks D, Bond J, Dash C, Giardiello FM, Glick S, Johnson D, Johnson CD, Levin TR, Pickhardt PJ, Rex DK, Smith RA, Thorson A, Winawer SJ, Ad ACSCC, Canc ACRC. Gastroenterology. 2008;134:1570–1595. doi: 10.1053/j.gastro.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 28.Barrier A, Lemoine A, Brault D, Houry S, Flahault A, Dudoit S. Clin Cancer Res. 2005;11:8999s–8999s. [Google Scholar]
- 29.Luo HZ, Zhou ZG, Yang L, Yu YY, Tian C, Zhou B, Zheng XL, Xia QJ, Li Y, Wang R. Japanese J Clin Oncol. 2005;35:739–744. doi: 10.1093/jjco/hyi195. [DOI] [PubMed] [Google Scholar]
- 30.Zeng ZS, Shu WP, Cohen AM, Guillem JG. Clin Cancer Res. 2002;8:144–148. [PubMed] [Google Scholar]
- 31.Moore DF, Li H, Jeffries N, Wright V, Cooper RA, Elkahloun A, Gelderman MP, Zudaire E, Blevins G, Yu H, Goldin E, Baird AE. Circulation. 2005;111:212–221. doi: 10.1161/01.CIR.0000152105.79665.C6. [DOI] [PubMed] [Google Scholar]
- 32.Alison E. Baird, D. F. M., Ehud Goldin, Kory Johnson:US, 2010, pp143.
- 33.Group, N. r.-P. S. S. N Engl J Med. 1995;333:1581–1487. [Google Scholar]
- 34.Hashimoto M, Chen PC, Mitchell MW, Nikitopoulos DE, Soper SA, Murphy MC. LOC. 2004;4:638–645. doi: 10.1039/b406860b. [DOI] [PubMed] [Google Scholar]
- 35.Hashimoto M, Barany F, Soper SA. Biosen & Bioelec. 2006;21:1915–1923. doi: 10.1016/j.bios.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 36.Hashimoto M, Hupert ML, Murphy MC, Soper SA, Cheng YW, Barany F. Anal Chem. 2005;77:3243–3255. doi: 10.1021/ac048184d. [DOI] [PubMed] [Google Scholar]
- 37.Hashimoto M, Barany F, Xu F, Soper SA. Analyst. 2007;132:913–921. doi: 10.1039/b700071e. [DOI] [PubMed] [Google Scholar]
- 38.Smith JR, Cicerone MT, Meuse CW. Langmuir. 2009;25:4571–4578. doi: 10.1021/la802955w. [DOI] [PubMed] [Google Scholar]
- 39.Kricka LJ, Wilding P. Anal Bioanal Chem. 2003;377:820–825. doi: 10.1007/s00216-003-2144-2. [DOI] [PubMed] [Google Scholar]
- 40.Shadpour H, Musyimi H, Chen JF, Soper SA. J Chromatog A. 2006;1111:238–251. doi: 10.1016/j.chroma.2005.08.083. [DOI] [PubMed] [Google Scholar]
- 41.Eigen M, Rigler R. Proc Nat Acad Sci USA. 1994;91:5740–5747. doi: 10.1073/pnas.91.13.5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Mello AJ, Edel JB. J Appl Phys. 2007;101 [Google Scholar]
- 43.Foquet M, Korlach J, Zipfel WR, Webb WW, Craighead HG. Anal Chem. 2004;76:1618–1626. doi: 10.1021/ac035088o. [DOI] [PubMed] [Google Scholar]
- 44.Haab BB, Mathies RA. Anal Chem. 1999;71:5137–5145. doi: 10.1021/ac990644t. [DOI] [PubMed] [Google Scholar]
- 45.Lyon WA, Nie SM. Anal Chem. 1997;69:3400–3405. [Google Scholar]
- 46.Wang TH, Peng YH, Zhang CY, Wong PK, Ho CM. JACS. 2005;127:5354–5359. doi: 10.1021/ja042642i. [DOI] [PubMed] [Google Scholar]
- 47.Emory JM, Peng ZY, Young B, Hupert ML, Rousselet A, Patterson D, Ellison B, Soper SA. Analyst. 2012;137:87–97. doi: 10.1039/c1an15658f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.